
Abstract
DNA methylation and histone modification are epigenetic mechanisms that result in altered gene expression and cellular phenotype. The exact role of methylation in myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) remains unclear. However, aberrations (e.g. loss-/gain-of-function or up-/down-regulation) in components of epigenetic transcriptional regulation in general, and of the methylation machinery in particular, have been implicated in the pathogenesis of these diseases. In addition, many of these components have been identified as therapeutic targets for patients with MDS/AML, and are also being assessed as potential biomarkers of response or resistance to hypomethylating agents (HMAs). The HMAs 5-azacitidine (AZA) and 2′-deoxy-5-azacitidine (decitabine, DAC) inhibit DNA methylation and have shown significant clinical benefits in patients with myeloid malignancies. Despite being viewed as mechanistically similar drugs, AZA and DAC have differing mechanisms of action. DAC is incorporated 100% into DNA, whereas AZA is incorporated into RNA (80–90%) as well as DNA (10–20%). As such, both drugs inhibit DNA methyltransferases (DNMTs; dependently or independently of DNA replication) resulting in the re-expression of tumor-suppressor genes; however, AZA also has an impact on mRNA and protein metabolism via its inhibition of ribonucleotide reductase, resulting in apoptosis. Herein, we first give an overview of transcriptional regulation, including DNA methylation, post-translational histone-tail modifications, the role of micro-RNA and long-range epigenetic gene silencing. We place special emphasis on epigenetic transcriptional regulation and discuss the implication of various components in the pathogenesis of MDS/AML, their potential as therapeutic targets, and their therapeutic modulation by HMAs and other substances (if known). The main focus of this review is laid on dissecting the rapidly evolving knowledge of AZA and DAC with a special focus on their differing mechanisms of action, and the effect of HMAs on transcriptional regulation.
Introduction
Targeting the methylation machinery has been assigned to the third of three waves of anticancer drug development (Dobbelstein & Moll, Citation2014). The “first wave” encompasses classical chemotherapeutics that target DNA integrity and cell division, and go back as far as 1943. Following these, evolving knowledge revolutionized cancer treatment with the advent of drugs with a high precision of molecular targeting of specific oncogenic signaling intermediates. These “second wave” anticancer drugs have been termed “clean drugs”, as, in theory, they target only malignant cells by interacting with a single tumor-specific molecule governing a single downstream pathway. However, apart from tyrosine kinase inhibitors targeting the fusion protein Bcr-Abl, drugs with such a narrow target spectrum have resulted in limited clinical benefits. Even for drugs thought to selectively target a certain oncogenic protein, such as Bcr-Abl or janus kinase 2 (Jak2), many “off-target” molecules are being found, as scientists dig deeper into their mechanisms of action. In particular, Jak2 inhibitors are effective, even in patients not harboring these mutations (Cervantes, Citation2014; de Lavallade et al., Citation2013; Ferreira & Harrison, Citation2014; Rosenthal & Mesa, Citation2014). Furthermore, various mechanisms of acquired resistance to these small molecule inhibitors have led to the return of a broader approach. “Third wave” anticancer drugs re-expand the target spectrum by addressing multicomponent cellular machinery such as that of ribosome assembly, protein folding, chromatin modifications, or proteasome modifications. These have been referred to as “dirty drugs”, because they often target: (i) several structurally related proteins, (ii) a cellular complex composed of several proteins, and/or (iii) molecules that govern several downstream pathways.
In 1964, the azanucleosides 5-azacitidine (AZA) and 2′-deoxy-5-azacitidine (decitabine, DAC) were developed as classical cytostatic agents (Sorm et al., Citation1964). Sixteen years later, these drugs were shown to inhibit DNA methylation, and thus are often referred to as hypomethylating agents (HMAs) (Jones & Taylor, Citation1980). This resulted in the initiation of their development as epigenetic drugs and the substantial refinement of clinical dosing schedules. Almost 35 years later, both drugs have shown significant clinical benefits with high overall response rates and improved overall survival in patients with myeloid malignancies (Fenaux et al., 2009, Citation2010; Kantarjian et al., Citation2012; Pleyer et al., Citation2013, Citation2014a, Citationb).
Indeed, although both drugs have been in clinical use for more than half a century, and despite decades of research to delineate the mechanisms of antileukemic activity, little is known about the precise mechanisms of action of azanucleosides, and the correlation of the latter with clinical response and patient outcomes remains elusive. Although both drugs prolong overall survival in patients with chronic and acute myeloid malignancies, they are not curative and continuous treatment is necessary.
At low concentrations, both AZA and DAC cause DNA demethylation, thereby reactivating tumor suppressor genes. At high concentrations, hypomethylating activity is lost and replaced by direct cytotoxicity (Hollenbach et al., Citation2010). Most of the early clinical trials used dosages based on maximum tolerated doses and were designed to test the potential cytotoxic effect, rather than whether DNA methyltransferase (DNMT) inhibition would be an effective target for anticancer therapies. We will focus on the mechanisms of action observed with the use of low-dose HMAs, which results in clinically relevant response rates and prolongation of overall survival in patients with myelodysplastic syndrome (MDS), chronic myelomonocytic leukemia (CMML) and acute myeloid leukemia (AML) (Fenaux et al., Citation2009, Citation2010; Pleyer et al., Citation2013, Citation2014a, Citationb).
The aim of this review is to summarize the wealth of rapidly evolving knowledge on these drugs, starting with a general introduction to transcriptional regulation in MDS and AML, followed by an overview of therapeutic targeting options for components of transcriptional regulation, before digging deep into pharmacokinetics, pharmacodynamics and current concepts of the molecular mechanisms of action of azanucleosides, with a special focus on similarities and differences between AZA and DAC. We summarize current knowledge on the involvement of the epigenetic machinery in the development of MDS/AML and their potential modulation by azanucleosides (if known); we also discuss which of these molecules may be additional therapeutic targets in MDS/AML, thus representing potential novel combination partners for HMAs.
Overview of transcriptional regulation in MDS/AML
The precise regulation of gene transcription is controlled by a complex and only partially understood system of interactions between transcription factors, histone modifications, histone and DNA modifying enzymes, and their concerted action on chromatin conformation. Molecular mechanisms of epigenetic regulation can be broken down into three main pillars discussed in detail below: (i) DNA methylation, (ii) post-translational histone tail modifications, and (iii) micro-RNA expression. In addition, the current conception of the incompletely understood phenomenon of long range gene silencing is summarized.
DNA methylation in MDS/AML
The term “epigenetics” refers to heritable changes in gene expression and cellular phenotype that are not caused by changes in the DNA sequence. Mechanisms that produce such changes include DNA methylation and histone modifications. These epigenetic changes persist through cell divisions for the duration of the cell’s life, and may be inherited over multiple generations (Bird, Citation2007). The error-rate in maintenance of DNA methylation is several orders of magnitude higher than DNA replication fidelity (Timp & Feinberg, Citation2013). Epigenetic patterns seemingly drift randomly over time and age, creating diversity and enabling Darwinian selection for patterns most permissive of neoplastic outgrowth and development of MDS/AML (Maegawa et al., Citation2014). This epigenetic instability or infidelity is a recently recognized feature of malignant cells, thought to confer enhanced adaptability as well as a competitive advantage to leukemic stem cells (LSCs) by limiting age-related “methylation-mediated loss of fitness”, and contributing to oligoclonality frequently observed in MDS and AML (Itzykson & Fenaux, Citation2014).
Global DNA hypomethylation, as well as localized hypermethylation of genes, particularly of those encoding tumor suppressor genes, is a hallmark of most types of cancer (Feinberg & Tycko, Citation2004; McCabe et al., Citation2009). Short CpG-rich regions, called CpG-islands, are found in the proximal promoter regions of nearly half of these genes. Methylation of DNA at position 5 of the pyrimidine ring of cytosine residues (5-mC) within these CpG dinucleotides is the most common covalent modification in the eukaryotic genome (Jaenisch & Bird, Citation2003). These regions are usually non-methylated in normal cells, whereas aberrant hypermethylation is found in approximately 1.5% of the CpG-islands (on average 600 of 45 000) in the genome of malignant cells, and may show non-random and tumor-type specific patterns (Costello et al., Citation2000). In this respect, MDS and AML display unique patterns and an abundance of aberrant DNA methylation (Figueroa et al., Citation2009). Recently, a core set of (aberrantly) methylated genes predictive of survival and specific for distinct biological AML subtypes was identified (Figueroa et al., Citation2010). Gene promoter hypermethylation leads to recruitment of co-repressors, altered chromatin structure, and ultimately transcriptional gene silencing (Lopez-Serra & Esteller, Citation2008). Silencing of tumor suppressor genes via DNA methylation has been proposed as a “second hit” for cellular transformation, equivalent to mutations or translocations (Herman & Baylin, Citation2003). However, the precise role of methylation in the development of MDS and leukemic progression remains unclear. Several single locus studies have demonstrated promoter hypermethylation and silencing of isolated genes in MDS and AML, often in association with progressive disease, with e.g. p15(INK4B) being a prominent example (Christiansen et al., Citation2003; Tien et al., Citation2001). Tumor cells characterized by a relatively high frequency of methylation of certain CgP-islands are considered “CgP-island hypermethylator pheonotypes” (CIMP). Quantitative analysis of the DNA methylation status of 10 selected genes identified a CIMP in solid tumors (Toyota et al., Citation1999). This simultaneous inactivation of several genes occurs via an as yet unknown mechanism has also been found in MDS, where the presence of CIMP was associated with rapid progression to AML and shorter survival (Shen et al., Citation2010). It is likely that all studies of individual genes are in fact a common subset of cases affected by CIMP, which explains why the methylation status of so many single genes has been reported to have prognostic relevance. In fact, genome-wide studies have demonstrated that hundreds of genes (10–15% of all analyzed genes) are frequently hypermethylated in MDS, and that hypermethylation across the genome correlates with poor prognosis and transformation to AML, as well as AML relapse (Figueroa et al., Citation2009; Jiang et al., Citation2009; Kroeger et al., Citation2008; Maegawa et al., Citation2014; Zhao et al., Citation2014).
Global hypomethylation is common in solid tumors and amounts to a 10–20% reduction in methylation, as compared to non-malignant tissue. In contrast, global hypomethylation is much less pronounced in AML, with leukemic DNA being only 2.7% less methylated than in healthy controls (Saied et al., Citation2012). When taking a closer look, and differentiating between methylation of promoters, gene bodies, CgP-islands, CgP-island shores, as well as various repetitive sequence classes, very distinct patterns of hypomethylation in non-promoter interspersed repeat DNA elements were associated with AML, and have been proposed as discriminating biomarkers for use in AML diagnosis (Saied et al., Citation2012); prognostic relevance remains to be established. In MDS, global hypermethylation (rather than hypomethylation) has been reported (Romermann et al., Citation2008).
DNA methylation silences genes by physically impeding the binding of transcription factors to DNA, as well as by enabling transcriptional repressors such as methyl-CpG-binding proteins (MBPs) to bind to the methylated CpG bases. In general, DNA methylation is considered as a silencing event that is more permanent than that imposed by repressive histone modifications discussed in detail below. Once DNA methylation has been established, it can be perpetuated without the original initiating signal. It still remains unclear however, whether hypermethylation is an initiating event, or the end result of gene shut down, and whether hypermethylation is cause or consequence of disease progression. Recent evidence suggests the former scenario (Santini et al., Citation2013).
Recently, 5-hydroxy-methylcytosine (5-hmC) was discovered as an enzymatic oxidation product of 5-methylcytosine (5-mC). This reaction is catalyzed by ten-eleven translocation (TET) proteins that are often mutated in cancer, coinciding with a broad loss of 5-hmC patterns across many types of malignancies including MDS and AML. It has been proposed that 5-hmC loss may correlate better with gene expression patterns than 5-mC gain (Jin et al., Citation2011). As such, 5-hmC has been proposed as an alternate biomarker for cancer. As bisulfite conversion does not distinguish between 5-mC and 5-hmC, other methodologies need to be applied to detect changes in 5-hmC patterns. These have been reviewed by others (Gronbaek et al., Citation2012).
Post-translational histone tail modifications in MDS/AML
Accumulating evidence has revealed that modifications of DNA methylation and histone (H) structure cooperate to regulate gene expression. While DNA methylation is abnormal in early stage MDS, disease progression seems to coincide with the acquisition of additional epigenetic events. Nucleosomes are the basic units of DNA packaging, consisting of 147 base-pairs of DNA wound around histone octamers that are connected by stretches of “linker-DNA” approximately 80 base-pairs in length, reminiscent of a string wrapped around beads. Histone octamers consist of eight proteins, consisting of two copies of each of the core histones H2A, H2B, H3 and H4 (Luger et al., Citation1997). So-called linker-histones (H1) are involved in chromatin compaction. The tail structures of histones are sites of post-translational modifications, which function as master switches determining chromatin packing density: Euchromatin is the lightly packed, unraveled, loose form of DNA without linker histones. It is easily accessible for transcription factors, RNA polymerase complexes and other regulatory proteins, thus enabling gene transcription. In contrast, heterochromatin represents the tightly packed, inaccessible, non-transcribed form of DNA.
Nucleosomes are mobile structures that can “slide” and reposition themselves along DNA without disruption of the histone octamer (Pennings et al., Citation1989). Nucleosome repositioning is thought to be one of the mechanisms by which large-scale, cell-specific expression of genes is regulated. Nucleosome depletion results in a transcriptionally permissive chromatin architecture. Histone variants and/or post-translational histone modifications strongly correlate with transcriptional status. Whereas histone variants such as H2A.X were found to be enriched at transcriptionally silent genes, the histone variant H2A.Z was shown to be enriched at active genes (Dalmasso et al., Citation2009). Histone modifications can have varying effects on gene transcription, depending on the type of modification, as well as the site of modification. This process is referred to as “histone coding” (de la Cruz et al., Citation2005). In general, histone remodeling enzymes (discussed in detail below), loosen or tighten histone-DNA interactions, thus either allowing or inhibiting transcription factors to readily access the DNA. Acetylation is mediated by histone acetyltransferases (HATs) and results in activation of gene transcription, whereas deacetylation by histone deacetylases (HDACs), prevents transcription. Histone methylation may activate or repress gene transcription, depending on the residue modified, as well as the degree of modification, i.e. mono-, di- or tri-methylation. In particular, methylation of histone H3 at lysines (K) 4, 36 and 79 (H3K4, H3K36 and H3K79) by histone methyltransferases (HMTs) and acetylation of lysine residues on H3K9 and H3K14 represents a permissive transcriptional state associated with gene activation, whereas (tri)-methlyation of H3K9, H3K27 and H4K20 results in a repressive chromatin architecture associated with gene silencing. Three classes of enzymes are known to antagonize histone methylation: (i) peptidylarginine deiminase, (ii) lysine specific demethylase 1 (LSD1), and the Jumonji C domain family of histone demethylases (JHDMs) (Anand & Marmorstein, Citation2007). Other histone modifications such as lysine ubiquitination, arginine methylation, serine phosphorylation, ubiquitinylation, and/or sumoylation are less well studied. Compared with DNA methylation, histone modifications are much more dynamic and have a shorter “half-life”. Whether they are autonomously transmitted through cell division remains unclear, and the issue of stable versus dynamic histone modifications remains an area in need of further research.
Genome-wide studies of histone modifications are rare in MDS/AML, but distinct patterns have been described (Muller-Tidow et al., Citation2010). MDS and AML are diseases that are sustained by LSCs. Recently, an epigenetic signature associated with hematopoietic stem cell commitment was identified. These authors developed an epigenetic score, and AML patients with “low stem cell commitment”-associated epigenetic signature scores had significantly longer overall survival than patients with higher epigenetic scores (Bartholdy et al., Citation2014). Genome-wide screening in AML revealed no relevant differences in DNA methylation between stem cells, progenitor cells and/or mature cell populations. In contrast, thousands of genes changed their histone methylation (H3K4me3 and/or H3K27me3) status, with the most plasticity being observed in LSCs. Progressive loss of activating H3K4me3 status coincided with gene silencing during differentiation (Yamazaki et al., Citation2013). These data indicate that histone modifications, rather than promoter DNA methylation status, are involved in the switches from progenitor cells to LSCs in AML.
Various somatic alterations in genes encoding proteins that regulate DNA methylation and post-transcriptional histone modifications, have been identified (Dolnik et al., Citation2012; Nikoloski et al., Citation2012). These epigenetic modifiers account for a novel class of mutant disease alleles shown to have strong biological, clinical and potentially prognostic and/or therapeutic relevance in MDS and AML. Histone functions, variants, modifications, the enzymes that mediate these changes, their correlation with transcriptional status and what is known about their role in MDS and AML disease pathogenesis are discussed in detail in the “Targeting components of transcriptional regulation with relevance for MDS/AML” section.
Micro-RNA in gene regulation in MDS/AML
The human genome encodes for more than 1800 micro-RNAs (miR). These are small non-coding RNA molecules that regulate gene expression at the post-translational level by inhibiting protein translation and/or destabilizing target transcripts. Micro-RNAs are recognized as one of the major pillars that regulate gene expression and have been associated with most cellular functions, as well as with normal and malignant hematopoiesis (Vasilatou et al., Citation2010). Micro-RNAs may target key molecules of epigenetic reactions and their expression is another means of dynamically fine-tuning epigenetic regulation (Amiel et al., Citation2012). Micro-RNAs that control the epigenetic machinery have been termed “epi-micro-RNAs” (Vasilatou et al., Citation2013), and their own transcription may be subject to epigenetic regulation. Micro-RNAs may be down-regulated by aberrant epigenetic silencing, or by impaired synthesis by MDS mesenchymal stromal cells due to lack of ribonucleases necessary for micro-RNA maturation (Santamaria et al., Citation2012).
In recent years, several studies have investigated the role of micro-RNAs in MDS and AML. Altered micro-RNA expression is frequently observed (Jongen-Lavrencic et al., Citation2008; Vasilatou et al., Citation2013), and deregulation of certain micro-RNAs has been implicated in disease pathogenesis (Raynal et al., Citation2012; Zardo et al., Citation2012). Whereas global micro-RNA downregulation has been reported in these diseases, overexpression of single micro-RNAs has also been documented. In AML, e.g. overexpression of miR-29b was shown to induce downregulation of DNMT3, resulting in hypomethylation and re-expression of tumor suppressor genes (Garzon et al., Citation2009). In MDS patients, miR-125 was found to be significantly overexpressed, and this correlated with inhibition of erythroid differentiation as well as inferior overall survival (Ganan-Gomez et al., Citation2014). In contrast, miR-125 was found to be methylated (and thus expressed at lower levels) in AML patients, and reduced miR-125 expression status correlated with worse overall survival in this patient cohort (Dickstein et al., Citation2010; Ufkin et al., Citation2014). Obviously, micro-RNA function in cancer in general, and in MDS and AML in particular, is currently far from being understood (Chung & Park, Citation2014).
Nevertheless, certain micro-RNAs have been proposed as diagnostic and prognostic micro-RNAs (Erdogan et al., Citation2011; Lin et al., Citation2014; Sokol et al., Citation2011), as biomarkers (Li et al., Citation2014) and/or as therapeutic targets in MDS/AML (Gong et al., Citation2014; Su et al., Citation2014). Changes in micro-RNA expression levels were observed at response to therapy (Merkerova et al., Citation2014), and DAC resulted in demethylation and significant upregulation of miR-124 and miR-125 in MDS patients responding to treatment (Ufkin et al., Citation2014; Vasilatou et al., Citation2013). Therapeutic reactivation of micro-RNAs silenced by aberrant methylation with HMAs, is an as yet fairly unexplored terrain.
Long range epigenetic gene silencing (LRES) in the pathogenesis of MDS/AML
Gene silencing can “spread” along a chromosome and is thought to be a dynamic process. Spreading of gene silencing may occur in the relatively short range (<10 kilobases). This process involves local diffusion of histone-modifying enzymes from source binding sites to low affinity sites nearby, where they reside long enough to modify histone tails. This short-range “oozing” model involves recurrent rounds of deacetylation of H4K16 histone tails by silent information regulator (SIR) protein complexes, which propagate gene silencing along the chromosome (Oppikofer et al., Citation2013). Several hypothesis about discontinuous long-range gene regulation, and how elements of transcriptional control overcome the sometimes extreme distances between themselves and their target genes, as well as how they specifically select and communicate with the latter (without interfering with other genes in the vicinity), have been reviewed by others (Talbert & Henikoff, Citation2006). The most accepted model is that direct physical interaction between promoters and their distant distal enhancers (highly enriched for the permissive H3K4me and H3K27ac marks) is brought about by chromatin “hopping” (Phair et al., Citation2004) or “looping” (Doyle et al., Citation2014). The exact mechanisms underlying this looping are only beginning to be understood, but seem to involve the looping proteins cohesin and CCCTC-binding factor (CTCF). Binding sites for these proteins are located at the loop bases (Tark-Dame et al., Citation2014; Xu et al., Citation2014). Interestingly, both of these proteins seem to be relevantly involved in the pathogenesis of MDS and AML, and mutually exclusive mutations in components of the cohesion complex or its partner CTCF have been reported in 8% of MDS patients (Kon et al., Citation2013).
CTCF binding sites can act as nucleosome repositioning anchors, and thus represent a powerful means to regulate gene transcription (Fu et al., Citation2008). CTCF is a transcription factor and candidate tumor suppressor involved in the control of erythroid and myeloid growth and differentiation, and knock-down results in differentiation blocks (Torrano et al., Citation2005). CTCF Hypermethylation (and thus downregulation) of CTCF has also been linked to the pathogenesis of acute promyelocytic leukemia (Manodoro et al., Citation2014).
Cohesin is a multi-subunit protein complex, comprising four major subunits: structural maintenance of chromosomes 1A and 3 (SMC1A, SMC3), STAG1/2 and RAD21 (Leeke et al., Citation2014). The cohesion-complex is involved in chromosome pairing, DNA repair, and re-establishment of transcriptional regulation after cell division via “bookmarking” of transcription factor binding sites (Yan et al., Citation2013). Cohesin has been implicated to play a higher-order role in orchestration of hematopoiesis (Panigrahi & Pati, Citation2012), and impaired cohesin function has been linked with the development of dysplasia and disease progression in AML. As such, the cohesion-complex has recently been recognized as a novel genetic pathway associated with the development of MDS and AML. Mutations within all individual protein components of the cohesin-complex have been described, are mutually exclusive, and occur in 4–21% of MDS and AML patients (Leeke et al., Citation2014; Malcovati et al., Citation2014; Thol et al., Citation2014). A strong association between mutations affecting the cohesion-complex and nucleophosmin 1 (NPM1) was found, but no correlation with either response or survival could be demonstrated (Thol et al., Citation2014).
Long-range epigenetic silencing (LRES) involves polycomb responsive elements which recruit the histone methyltransferase enhancer of zeste homolog 2 (EZH2). EZH2 is then actively released or “hops” to a nearby low-affinity site to suppress transcription by methylation of adjacent H3K27. Epigenetic features of LRES are discussed in detail elsewhere (Harmston & Lenhard, Citation2013). Mutations in polycomb genes that affect histone function, such as EZH2 and additional sex combs like transcription regulator 1 (ASXL1), have recently been detected in MDS and AML, and are described in more detail below in “HMTs in MDS/AML” (Boultwood et al., Citation2010; El-Sharkawi et al., Citation2014; Nikoloski et al., Citation2010; Thol et al., Citation2011b; Wang et al., Citation2013b).
In general, it is thought that methylation is responsible for creating a long-lasting state of transcriptional repression, which is preceded by repressive histone modifications such as H3-acetylation, H3K27-trimethylation or demethylation of permissive H3K4me3 (Ng et al., Citation2007). LRES is a mechanism of gene inactivation that affects multiple adjoining CgP-islands observed in several human malignancies (Dallosso et al., Citation2012). DNA hypermethylation can thus span larger chromosomal domains and lead to the silencing of flanking unmethylated genes. In murine cancer models, molecular features involved in LRES include (i) DNA hypermethylation of specific CgP islands, and (ii) acquisition of inactive histone marks, both of which result in downregulation of gene expression (Forn et al., Citation2013). Genes embedded in LRES regions seem to be plastically regulated during physiological cell differentiation, but are constrained to coordinated repressive regulation in malignantly transformed cells (Forn et al., Citation2013). LRES was found to be correlated with the above mentioned CpG-island hypermethylator phenotype (CIMP) in colorectal cancer (Karpinski et al., Citation2008).
Targeting components of epigenetic transcriptional regulation with relevance for MDS/AML
In contrast to the irreversible nature of altered gene expression resulting from specific mutations, epigenetic modifications can be reversed and targeted for therapeutic interventions, which forms the basis of epigenetic cancer therapy (Goldberg et al., Citation2007). The most important components in the human epigenetic machinery that result in condensed chromatin formation and gene silencing are DNMTs and MBPs, which recruit transcriptional co-repressor complexes including HMTs and HDACs (Esteller, Citation2007). In contrast, other essential members of the epigenetic arsenal result in de-repression and transcriptional activation. These include methyl-cytosine dioxygenases, which represent a family of ten-eleven translocation (TET1-3) proteins, isocitrate dehydrogenases (IDH1-2), histone demethylases (HDMs), histone acetyltransferases (HATs) and bromodomain extra-terminal (BET) proteins. Mutations in epigenetic modulators are common in MDS and AML, and have been implicated in the development of these diseases (Fathi & Abdel-Wahab, Citation2012; Shih et al., Citation2012). These components are described in detail below, with a special focus on what is known about the role of mutant and wild-type forms in the development of MDS and AML, as well as on evolving therapeutic strategies to selectively target these enzymes. This review aims to highlight the clinical relevance and significance of the only two currently approved drugs for treatment of elderly patients with MDS and AML. We will therefore discuss how these enzymes and their functions are modulated by HMAs in the “Azanucleosides in MDS/AML” section.
Targeting of epigenetic modulators mediating transcriptional repression
DNMTs in MDS/AML
DNMTs enzymatically add a methyl group to cytosine in CpG dinucleotides in DNA (). Mammalian cells contain five DNMT isoforms, but only DNMT1, 3A and 3B methylate DNA (Bourc’his et al., Citation2001; Goll et al., Citation2006). DNMT2 methylates tRNAASP and DNMT3L is catalytically inactive, but formation of a DNMT3L–DNMT3A-complex results in enhancement and recruitment of DNMT3A activity to histone H3 tails, when H3K4 is not methylated (Ooi et al., Citation2007) (). DNMT3A and 3B exhibit predominant de-novo methyltransferase activity, initiating DNA methylation marks on unmethylated DNA (Okano et al., Citation1999) (), whereas DNMT1 is exclusively involved in the methylation of hemimethylated DNA, and is the main enzyme responsible for the maintenance or copying of DNA methylation patterns during DNA replication (Spada et al., Citation2007). DNMTs also attract HDACs to the methylated CgP-island, thus further stabilizing the silencing of the target gene (Herman & Baylin, Citation2003) ().
Figure 1. Biochemical pathways of DNA methylation and demethylation. (A) Physiologic state. (I) DNMTs add a methyl group to cytosines of CpG-islands. (II) TET2 catalyzes the conversion of 5-mC to 5-hmC. (III) AID mediates degradation of 5-hmC to 5-hmU. (IV) 5-hmU activates the BER-pathway, in which TDG or SMUG1 enable further degradation to unmethylated cytosine. (V) TET2 can also convert 5-mC to 5-fC and 5-caC. (VI) Both 5-fC and 5-caC are directly recognized and repaired by TDG-mediated BER. (VII) IDH1/2 converts isocitrate to α-KG which is an essential cofactor for TET2-mediated conversion of 5-mC to 5-hmC. (B) Effect of commonly occurring mutations in enzymes involved in DNA methylation and demethylation. (I) Mutant TET2 is unable to convert 5-mC to 5-hmC, which results in decreased 5-hmC levels, and thus inhibits demethylation. (II) IDH1/2 gain-of-function mutations result in a neomorphic enzyme activity that converts α-KG to 2-HG, thus inhibiting α-KG-dependent functions of TET2. (III) 2-HG also inhibits the JMJC-family of HDMs, and thus inhibits demethylation of histones. Both TET2 loss-of-function and IDH1/2 gain-of-function mutations result in reduced 5-hmC levels and result in global promoter hypermethylation.
The green field demarks methylation, whereas the red field demarks occurring demethylation. The yellow lightning flashes denote molecules that are being evaluated as therapeutic targets in MDS/AML.
Abbreviations: 2-HG, 2-hydroxyglutarate; 5-caC, 5-carboxyl-cytosine; 5-fC, 5-formyl-cytosine; 5-hmC, 5-hydroxy-methyl-cytosine; 5-hmU, 5-hydroxy-methyl-uridine; 5-mC, 5-methyl-cytosine; AID, activation-induced cytidine deaminase; α-KG, α-ketoglutarate; AML, acute myeloid leukemia; BER, base excision repair pathway; DNMT, DNA methyltransferase; HDM, histone demethylase; IDH, isocitrate dehydrogenase; JMJC, jumonji-domain-containing; MDS, myelodysplastic syndrome; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; SMUG, single-strand selective monofunctional uracil DNA glycosilase; TDG, thymine DNA glycosilase; TET, ten-eleven translocation.
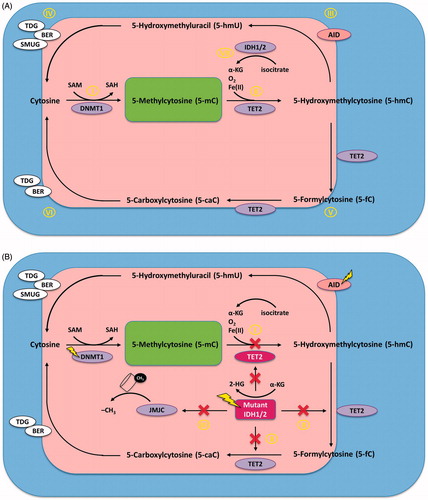
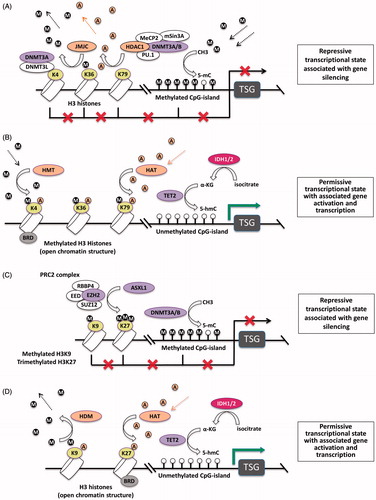
Figure 2. Enzyme network of epigenetic regulation of gene expression. The most relevant enzymatic systems known to modify DNA and/or histones are depicted. (A) DNMT3A and DNMT3B are responsible for de novo DNA methlyation. They catalize the addition of a methyl (CH3) group (denoted as “M”) at the 5-carbon atom of cytosine to form 5-mC in the context of CgP-islands in the promoter regions of genes. This blocks the access of transcription factors to the DNA and results in gene silencing. After binding methylated CpG, MeCP2 recruits and forms the MeCP2/DNMT3A-B/Sin3A/PU.1/HDAC1 co-repressor complex to silence transcription via histone deacetylation (denoted as “A”), which is mediated by HDAC1. JMJC is an HDM, which demethylates lysines 9 and 36 of histone H3. DNMT3L is catalytically inactive, but forms a complex with DNMT3A and recruits and enhances its activity to histone 3 when K4 is not methylated. (B) HMTs confer methylation of histone H3 at lysines 4, 36, and 79 (H3K4, H3K36 and H3K79) resulting in an open chromatin structure, which represents a permissive transcriptional state associated with gene activation. IDH2 enzymes hydroxylate isocitrate to α-KG, which is in turn utilized by TET2 to convert 5-mC to 5-hmC, ultimately resulting in DNA hypomethylation. HATs confer the acetylation of lysine residues on the N-terminus of histones, which is generally associated with active gene transcription. BRD proteins bind acetylated histones of target genes and activate transcription. (C) EZH2 is an H3K27 methyltransferase which requires formation of PRC2 to catalyze the addition of methyl groups to arginine and lysine residues on the N-terminal tail of histones. Methylation of histone H3 at lysines 9 and 27 (H3K9 and H3K27Me3) by the PRC2 complex represents a repressive transcriptional state associated with gene silencing. ASXL1 recruits and stabilizes the PRC2 complex at target locations in the genome. (D) HDMs demethylate H3K9 and H3K27 resulting in an open chromatin structure, and IDH2 and TET function along the same pathway to induce demethylaton of CgP-islands, ultimately resulting in a permissive transcriptional state. Most mutations of epigenetic modifiers in MDS and AML affect post-translational modifications on the N-terminal tail of histone 3 or at cytosines of DNA.
Purple enzymes are associated with loss-of-function mutations (DNMT3A, TET2, EZH2, ASXL1), whereas pink enzymes are associated with gain-of-function mutations of their respective genes (IDH1/2).
Abbreviations: 5-hmC, 5-hydroxy-methyl-cytosine; 5-mC, 5-methyl-cytosine; α-KG, α-ketoglutarate; AML, acute myeloid leukemia; ASXL1, additional sex combs like 1; BRD, bromodomain; DNMT, DNA methyltransferase; EZH2, enhancer of zeste homolog 2; HAT, histone acetyltransferase; HDAC1, histone deacatylase 1; HDM, histone demethylase; HMT, histone methyltransferase; IDH, isocitrate dehydrogenase; JMJC, jumonji-domain-containing; MDS, myelodysplastic syndrome; MeCP2, methyl CpG binding protein 2; PRC2, polycomb repressor binding complex 2; TET, ten-eleven translocation; TSG, tumor necrosis factor-stimulated genes.
Downregulation of DNMT1 coinciding with reduced global DNA methylation has been associated with “healthy aging”, and has been implicated to play a role in the development of early MDS (Karlic et al., Citation2014). In contrast, an upregulation of DNMT1 seems to be a transition step to more advanced MDS and AML (Langer et al., Citation2005). Overexpression, amplification and mutations in all three catalytically active human DNMT isoforms have been described in numerous cancers (Miremadi et al., Citation2007). Strong up-regulation of DNMT1 and 3A has been demonstrated in the vast majority of MDS bone marrow biopsies (Langer et al., Citation2005). DNMT overexpression is thought to be a consequence of enhanced cell division and cannot explain why certain CgP-islands become hypermethylated while others remaining unaffected. DNMT3A and 3B expression has been negatively correlated with MDS disease risk, being more pronounced in high-risk MDS (Hopfer et al., Citation2009).
DNMT mutations have mainly been described in DNMT3A so far (Dolnik et al., Citation2012). They occur in 8% of MDS patients and in 23% of newly diagnosed AML patients, and have been implicated in disease pathogenesis, and as prognostic parameters for worse overall survival (Hopfer et al., Citation2009; Ley et al., Citation2010; Ribeiro et al., Citation2012; Thol et al., Citation2011a; Walter et al., Citation2011; Yan et al., Citation2011). In the context of the DNMT inhibitors AZA or DAC, DNMT3A mutations may be predictive factors of better response and progression-free survival (Traina et al., Citation2014). DNMT3A mutations have been associated with decreased catalytic activity and DNA binding affinity, and are generally thought to enhance hypomethylation (Gowher et al., Citation2006). Decreased methylation of a select number of genomic loci was observed in AML patients with mutant versus wild-type DNMT3A (Ley et al., Citation2010), but the exact methylated loci that are altered and contribute to leukemogenesis are yet to be delineated. Homebox (HOX) genes have been identified, and very recently confirmed, as one such target (Qu et al., Citation2014). DNMT3A mutations in AML patients have been associated with a pattern of global hypomethylation that specifically targets HOX genes, resulting in gene expression changes (Qu et al., Citation2014). The HOX locus encodes a highly conserved family of transcription factors that specify cell lineage differentiation in hematopoiesis. The dysregulation of HOX genes has recently been implicated in leukemogenesis, where they have been shown to support the immortalization of leukemic cells, both as chimeric partners in fusion genes, and also when overexpressed in their wild-type form (Alharbi et al., Citation2013; Shah & Sukumar, Citation2010). Patients exhibiting these mutations have residual wild-type expression, and do not display global methylation changes or increased genomic instability (Ley et al., Citation2010), leaving the oncogenic mechanism of DNMT3A mutations unclear. It has been suggested that epigenetically regulated gene expression in MDS does not solely rely on DNMT activity, which supports the hypothesis of oncoprotein-driven site-specific methylation. However, the oncoproteins involved in such targeted methylation in MDS remain largely unknown (Hopfer et al., Citation2009; Quesnel, Citation2009).
AZA and DAC are first generation DNMT inhibitors, and the only two drugs approved for the treatment of elderly patients with MDS and AML. According to their clinical relevance and significance, these HMAs are discussed in considerable detail below in the “Effects of HMAs on transcriptional regulation in MDS/AML” section.
Several second generation DNMT inhibitors are currently being developed and include 5,6-dihydro-5-azacitidine (DHAC, NSC264880), which was found to be less toxic and more stable than DAC at doses that induce comparable DNA hypomethylation and gene reactivation (Matousova et al., Citation2011). DHAC has been evaluated in clinical trials as early as 1985 (Curt et al., Citation1985; Kratzke et al., Citation2008). Currently, there are no clinical trials listed for this substance; reasons remain obscure. On the other hand, another derivative, 5-fluoro-2′-deocycytidine (FdCyd) is the subject of ongoing clinical initiatives (Beumer et al., Citation2008; Guo et al., Citation2010; Zhao et al., Citation2012) ().
Table 1. Novel substances targeting the methylation machinery currently being evaluated in clinical trialsa.
Interestingly, several natural compounds have been found to modulate the expression of DNMTs and their associated proteins. The powerful antioxidant resveratrol which is extracted from grape skins, and present in both whole grapes and red wine, has recently been discovered to reduce DNMT1 and/or DNMT3B levels in breast cancer cell lines (Mirza et al., Citation2013), and also in tumor tissue in murine models of breast cancer (Qin et al., Citation2014). Resveratrol also influenced expression of HDAC1 and methyl CpG binding protein 2 (MeCP2). Similar results were obtained with the natural compounds curcumin, disulfiram, genistein and green tea extract (epigallocatechin gallate [EGCG]; Mirza et al., Citation2013). Clinical trials testing these substances in humans with malignancies including MDS and AML are underway ().
MBPs in MDS/AML
MBPs bind to methylated CgP-islands and mediate transcriptional repression of affected genes (Parry & Clarke, Citation2011). To date, 15 MBPs with largely redundant function have been identified, but little is known about the involvement of MBPs in leukemogenesis. Currently, only MeCP2 and the ubiquitin protein ligase UHRF1 (ubiquitin-like with PHD and RING finger domains 1) have been implicated in hematologic malignancies and leukemia (Parry & Clarke, Citation2011). UHRF1 is an oncogene that acts as a transcriptional repressor, drives DNA hypomethylation, and plays an essential role in cell proliferation and carcinogenesis/leukemogenesis (Guan et al., Citation2013; Unoki et al., Citation2009). This MBP plays a major role in the inheritance of several epigenetic marks during mitosis, is an epigenetic reader and acts as a “hub protein” that is involved in epigenetic information integration via its capability to sense the presence of methylated cytosines on both DNA and histones (Bronner et al., Citation2013). UHRF1 has an affinity for hemimethylated CpG DNA and recruits DNMT1 to ensure that the sequence becomes completely methylated following replication in order to maintain the epigenetic inheritance of DNA methylation (Bostick et al., Citation2007; Sharif et al., Citation2007) (). This role also applies to the maintenance of histone marks, specifically H3, by UHRF1-mediated recruitement of HMTs (Bronner et al., Citation2013; Rottach et al., Citation2010). UHRF1 also interacts with HDAC1 and recruits it to methlyated tumor suppressor genes (Jeanblanc et al., Citation2005) ().
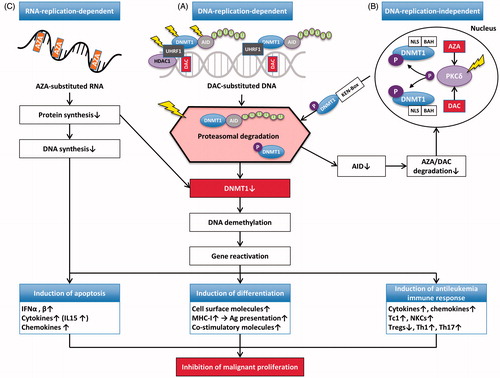
Figure 3. Mechanisms of action of AZA and DAC. (A) DAC is incorporated into DNA as a substitute for cytosine. This results in covalent trapping and depletion of DNMT1 (and AID), as well as in loss of DNA methylation marks. This eventually results in DNA demethylation and (re-)activation of gene expression. Depending on which genes are re-induced, this may lead to: (i) induction of apoptosis, (ii) induction of differentiation, and/or (iii) induction of an effective host anti-leukemia immune response, all of which ultimately result in inhibition of malignant proliferation. (B) DNMT1 is localized in the nucleus when NLS and BAH are present. HMAs induce hyperphosphorylation of DNMT1 via PKCδ. Phosphorylated DNMT1 is then targeted to the ubiquitination machinery, a process which requires KEN-Box. This leads to proteasomal degradation of both DNMT1 and AID. Lower levels of AID result in elevated levels of AZA/DAC, which can then further reduce DNMT1 levels via all the mechanisms described in this figure. (C) About 80–90% of AZA is incorporated into RNA resulting in mRNA and protein metabolism disruption, both of which ultimately inhibit malignant proliferation.
The yellow lightning flashes denote molecules that are being evaluated as therapeutic targets in MDS/AML.
Abbreviations: AID, activation induced cytidine deaminase; AML, acute myeloid leukemia; AZA, 5-azacitidine; BAH, bromo-adjacent homology; DAC, 5-aza-2′-deoxycytidine; DNMT1, DNA methyltransferase 1; HDAC1, histone deacatylase 1; HMAs, hypomethylating agents; IFNα, interferon alpha; IL15, interleukin 15; MDS, myelodysplastic syndrome; MHC, major histocompatibility complex; NKCs, natural keller cells; NLS, nuclear localization signal; P, phosphate; PKCδ, protein kinase C delta; Tc1, Type 1 CD8+ T cells; Th1, Type 1 T helper cell; Th17, Type 17 T helper cell; Tregs, regulatory T cells; U, uridine; UHRF1, ubiquitin-like with PHD and ring finger domains 1.
UHRF1 has been proposed both as a biomarker and as a therapeutic target (Bronner et al., Citation2007). The natural compound curcumin is thought to inhibit melanoma cell proliferation via regulation of UHRF1 and DNMT1 (Abusnina et al., Citation2011), and is currently being tested in various clinical trials ().
MeCP2, like UHRF1, acts as a global transcriptional repressor. MeCP2 is capable of long-range transcriptional repression, which is dependent on the local density of methylation and functional methyl-binding domains (Baubec et al., Citation2013; Nan et al., Citation1997). After binding to methylated CpG, MeCP2 recruits and forms the MeCP2/Sin3A/HDAC1 co-repressor complex to silence transcription via histone deacetylation (Nan et al., Citation1997) (). Additional methyl-CpG independent binding modes to chromatin have recently been described for MeCP2, but not for other MBPs (Baubec et al., Citation2013). MeCP2 has also been shown to link DNA methylation to histone methylation, and thus acts as a bridge between these two global epigenetics modifications. As mentioned above for UHRF1, this is mediated by recruitment of HMT activity by MeCP2, and results in particular results in increased H3K9 methylation (Fuks et al., Citation2003). MeCP2 also acts as a co-repressor of the transcription factor PU.1, which was originally characterized as a transcriptional activator essential for hematologic differentiation and myeloid development (Tenen et al., Citation1997). PU.1 deficiency is directly involved in the development of AML in mice (Basova et al., Citation2013), and humans (Bonadies et al., Citation2010). However, PU.1 can also act as a transcriptional repressor by binding directly to MeCP2 in the above mentioned complex (Hwang et al., Citation2004; Kihara-Negishi et al., Citation2001; Suzuki et al., Citation2003) (). In this sense, PU.1 has been shown to directly interact with DNMT3A and 3B in an MeCP2-dependent manner, resulting in the formation of the MeCP2/Sin3A/HDAC1/PU.1/DNMT3A/B co-repressor complex. The latter methylates CgP-islands in promoter regions with PU.1 binding sites (e.g. of p16INK4A (Kihara-Negishi et al., Citation2001; Suzuki et al., Citation2006) ()).
Recently, DAC was shown to reverse PU.1 mediated transcriptional repression (Aoyama et al., Citation2012). Similarly, AZA treatment significantly demethylated the PU.1 upstream regulatory element in AML cell lines in vitro (Curik et al., Citation2012). This led to up-regulation of PU.1, followed by de-repression of its transcriptional targets and onset of myeloid differentiation.
HMTs in MDS/AML
HMTs methylate arginine and lysine residues of histones, and are involved in the regulation of a wide range of processes including gene activity, chromatin structure and epigenetic memory (Martin & Zhang, Citation2005). All HMTs known to be involved in MDS/AML act as transcriptional repressors. As such, HMTs are major silencers of HOX genes, a group of transcription factors important for hematopoietic differentiation (Copur & Muller, Citation2013; Daser & Rabbitts, Citation2005; Eklund, Citation2011). Chromosomal aberrations involving loss of HMT function result in HOX protein overexpression, which leads to myeloproliferation and block of differentiation in AML cells in vitro (Argiropoulos & Humphries, Citation2007; Eklund, Citation2011).
The mixed lineage leukemia (MLL) gene, located on chromosome 11q23, is a recurrent locus of chromosomal translocation involved in leukemogenesis (Marschalek, Citation2011). MLL1 serves as an H3K4 methyltransferase. Eighty-seven different MLL rearrangements have been identified in addition to partial tandem duplications, all of which seem to confer worse prognosis with shorter survival (Basecke et al., Citation2006; Daser & Rabbitts, Citation2005). The four most frequent MLL translocations result in recruitment of DOT1-like histone H3 methyltransferase (DOT1L) to the fusion protein. DOT1L is an HMT that specifically methylates H3K79. Hypermethylation of H3K79 is a hallmark of MLL-rearranged AML, and critical for leukemogenesis induced by MLL-fusion proteins (Deshpande et al., Citation2013; Krivtsov et al., Citation2008; Okada et al., Citation2006).
HMTs have been identified as promising new targets in leukemia treatment (Flemming, Citation2012). As such, DOT1L inhibitors have been developed and shown to selectively kill cells bearing the MLL gene translocation in vitro, and to prolong survival in mouse and rat MLL xenograft models of AML (Daigle et al., Citation2011, Citation2013). Furthermore, DOT1L inhibition has recently been reported to sensitize MLL-rearranged AML to chemotherapy (Liu et al., Citation2014). The DOT1L inhibitor EPZ-5676 is the first to enter clinical trials ().
Table 2. DNA-replication-independent mechanisms of DNMT1 depletion.
Genome-wide profiling recently demonstrated ∼4600 gene promoters with increased H3K27Me3 in MDS patient samples, when compared with normal controls (Cheng et al., Citation2013). H3K27-hypermethylation has been linked to epigenetic inactivation of PU.1, and H3K27Me3 inhibitors were shown to upregulate both the expression of PU.1, as well as of its downstream genes, resulting in differentiation induction (Cheng et al., Citation2013). In this respect, another prominent member of the HMT group of enzymes is the H3K27 methyltransferase EZH2. EZH2 is the enzymatic member of the polycomb repressor complex 2 (PRC2), which initiates and maintains transcriptional silencing through post-translational histone modifications. EZH2 is only active when it is associated with the other PRC2 core components: embryonic ectoderm development (EED), suppressor of zeste 12 homolog (SUZ12) and retinoblastoma binding protein 4 (RBBP4); (Kogure et al., Citation2013; Margueron & Reinberg, Citation2011) (). EZH2 expression is essential for cancer survival (Varambally et al., Citation2008). Recent evidence supports an oncogenic role for EZH2, which enhances leukemogenicity and reinforces differentiation blockage in AML (Tanaka et al., Citation2012).
Whereas activating mutations have been found in diffuse large B-cell lymphomas (Morin et al., Citation2010), and wild-type EZH2 is commonly overexpressed in a variety of cancers (Bracken et al., Citation2003), only one report of EZH2 overexpression in MDS exists (Xu et al., Citation2011). Rather, a diverse range of loss-of-function mutations are seen in patients with MDS (10%), CMML (5%) and only rarely in AML (Abdel-Wahab et al., Citation2011; Grossmann et al., Citation2011; Xu & Li, Citation2012). Mutations in other PRC2 components are rare in myeloid malignancies, but have recently been reported for SUZ12 and EED in MDS (Puda et al., Citation2012; Score et al., Citation2012). EZH2 mutations, located at 7q36.1, have been associated with impaired survival in MDS (Bejar et al., Citation2011) and CMML (Grossmann et al., Citation2011). While loss-of-function mutations of EZH2 promoted the development of MDS, they attenuated transformation to AML in murine models (Sashida et al., Citation2014), or resulted in induction of differentiation in leukemic cells, and converted AML into an MDS/myeloproliferative neoplasm-like disease (Tanaka et al., Citation2012). This explains the rare occurrence of EZH2 mutations in AML. In contrast, ectopic expression of EZH2 seems to result in myeloid transformation (Herrera-Merchan et al., Citation2012).
Several small molecule inhibitors of EZH2 have been developed, and shown to induce tumor regression in hematologic tumor models (Knutson et al., Citation2012; McCabe et al., Citation2012; Qi et al., Citation2012). In vitro data suggest synergism between the EZH2 inhibitor 3-deazaneplanocin-A (DZNep) and DAC and/or HDAC inhibitors in targeting AML cells (Fiskus et al., Citation2009; Momparler et al., Citation2012, Citation2014; Zhou et al., Citation2011). Two EZH2 inhibitors have recently entered phase I clinical trials ().
ASXL1, located on 20q11, recruits and stabilizes the PRC2 complex at target locations in the genome, thus enabling EZH2 to methylate H3K27 (). Similar to the effect of aforementioned EZH2 mutations, ASXL1-loss results in compromised function of PCR2, with ensuing loss of the transcriptionally repressive H3K27 trimethylation, leading to up-regulation of HOXA genes (Abdel-Wahab et al., Citation2012). ASXL1 mutations result in severe myelodysplasia and MDS-like disease in mice (Inoue et al., Citation2013; Wang et al., Citation2014), occur in 49% of CMML, 5% of MDS and 14% of AML patients, and have been associated with poor prognosis (Abdel-Wahab & Levine, Citation2013; Abdel-Wahab et al., Citation2013; Bejar et al., Citation2011; Inoue et al., Citation2013; Itzykson et al., Citation2013; Metzeler et al., Citation2011a; Patnaik et al., Citation2013, Citation2014).
Other HMTs known to be involved in MDS and AML include the PR domain zinc finger (PRDM) proteins PRDM3 (EVI1), which maps to 3q26, and PRDM16 (MEL1), which maps to 1q36 (Duhoux et al., Citation2012; Mochizuki et al., Citation2000; Trubia et al., Citation2006). PRDM factors act as HMTs, and function in developmental contexts, in which they drive and maintain cell state transitions (Hohenauer & Moore, Citation2012), and have also been implicated in methylation of non-histone targets (Fog et al., Citation2012).
Targeting of epigenetic modulators mediating transcriptional activation
TET proteins in MDS/AML
Methyl-cytosine dioxygenases represent a family of three ten-eleven translocation (TET1–3) proteins that catalyze the conversion of 5-methylcytosine (5-mC) to 5-hydroxy-methylcytosine (5-hmC) (). TET proteins can also drive the 5-mC oxidation process further to produce 5-formylcytosine (5-fC) and 5-carboxylcytosine (5-caC) as reaction products (Pfeifer et al., Citation2014; Tahiliani et al., Citation2009) (, , and ). TET-mediated oxidation of 5-mC is thought to serve as a methylation repair pathway, that safeguards CgP-islands from occasional misdirected CpG-methylation, and thus has tumor suppressor function (Pfeifer et al., Citation2014). As mentioned briefly above, 5-hmC levels and patterns are also profoundly altered, and show a dramatic loss of 5-hmC in human cancers, including MDS/AML (Jin et al., Citation2011). 5-hmC depletion seems to be a universal occurrence in human malignancy, not limited to the presence of mutations in TET or IDH. As ascorbic acid is essential to optimal function of TET enzymes, it has been hypothesized that lack of vitamin C in cancer cells may be the cause for impaired function of TET proteins, resulting in the strongly decreased 5-hmC levels universally observed in malignancy (Pfeifer et al., Citation2014). TET-induced DNA demethylation is thought to operate gene-specifically, albeit the mechanism through which specificity is mediated remains obscure at this time-point. 5-hmC seems to be a fairly stable DNA base and not merely an intermediate in DNA demethylation. So far, no excision repair activity has been described that would effectively remove 5-hmC from DNA (Pfeifer et al., Citation2014).
Very recently, TET1 was shown to be a maintenance DNA demethylase that does not induce untargeted hypomethylation. Rather, TET1 mediates accumulation of 5-hmC at the edges of hypomethylated CpG-islands, and thus specifically prevents methylation spreading from methylated CpG-island edges into hypomethylated CpG-islands (Jin et al., Citation2014). As proof of principle, TET1 knockdown induced methlyation spreading (Jin et al., Citation2014).
Mutations in TET1 and TET2 have been observed in MDS, CMML and AML (Dolnik et al., Citation2012; Lorsbach et al., Citation2003). The mutant TET2 protein is unable to convert 5-mC to 5-hmC, resulting in decreased 5-hmC levels (; Madzo et al., Citation2013). The production of 5-hmC results in DNA demethylation and increased gene expression via three mechanisms: (i) active demethylation through the activation of AID (), which results in degradation of 5-hmC to 5-hm-uracil (5-hmU), which in turn activates the base excision repair (BER) pathway for further degradation to unmethylated cytosine (); (ii) passive DNA demethylation due to the inability of DNMT1 to recognize 5-hmC, resulting in loss of methylation marks in succeeding DNA-replication cycles (Pastor et al., Citation2011; Valinluck et al., Citation2004); (iii) 5-hmC blocks the binding of MBPs, in particular MeCP2 or MBD3, which usually confer transcriptional silencing (Ko et al., Citation2010; Pastor et al., Citation2011; Valinluck et al., Citation2004; Yildirim et al., Citation2011).
DNMT1 is incapable of methylating an unmethylated CpG-site on the strand opposite to a hydroxymethylated CpG-site (5-hmC-CpG). Moreover, the maintenance methylation cofactor UHFR1 (and most other MPBs that act as transcriptional repressors), is unable to interact with 5-hmC, with MeCP2 being the exception (Otani et al., Citation2013). As such, 5-hmC levels correlate positively with reduced methylation, reduced recruitment of repressive proteins, and elevated levels of transcription of 5-hmC-marked genes (Jin et al., Citation2011; Song et al., Citation2011). Measurement of 5-hmC levels in myeloid malignancies may thus prove a valuable diagnostic and prognostic tool to individually tailor therapies and assess therapeutic response (Ko et al., Citation2010).
Loss of TET2 leads to increased self-renewal capacity of hematopoietic stem cells, expansion of myeloid precursors and myeloid transformation in mice (Moran-Crusio et al., Citation2011). TET2 knockout mice develop a disease with MDS-like features (Li et al., Citation2011). TET2 mutations occur in 20–25% of MDS, 7–23% of AML and up to 53% of CMML patients (Kosmider et al., Citation2009b; Shih et al., Citation2012; Yamazaki et al., Citation2012). In line with the above, TET2-mutant AML has recently been shown to have the expected hypermethylation phenotype (Madzo et al., Citation2013). Accordingly, total 5-mC levels have been reported to be significantly higher in patients harboring TET2 loss-of function mutations, as compared to TET2 wild-type cases (Yamazaki et al., Citation2012). In humans, the prognostic relevance of these mutations in MDS, CMML and AML remains unclear, and may be disease-specific, as currently existing data are inconsistent (Abdel-Wahab et al., Citation2009; Kosmider et al., Citation2009a, Citationb; Liu et al., Citation2013; Metzeler et al., Citation2011b; Pollyea et al., Citation2011; Wang et al., Citation2013a). However, the presence of mutated TET2 has been associated with response to AZA and DAC in MDS and CMML by several (Braun et al., Citation2011; Itzykson et al., Citation2011; Traina et al., Citation2014; Voso et al., Citation2011), but not all groups (Pollyea et al., Citation2011).
Although TET2 has been discovered only recently, it is recognized as a key player in myeloid malignancies, and the race for the development of a TET2 inhibitor is ongoing. No clinical trials testing TET2 inhibitors were listed at ClinicalTrials.gov at the time of writing (November 2014).
IDH in MDS/AML
IDH enzymes convert isocitrate to α-ketoglutarate (α-KG; ), which is required for the conversion of 5-mC to 5-hmC by TET2 ( and ). IDH1 is present in the cytosol, whereas IDH2 is found in mitochondria.
Mutant IDH1 greatly accelerated the onset of myeloproliferative AML in mice in co-operation with the transcription factor HOXA9, which is involved in stem cell maintenance and hematopoietic differentiation, thus proving oncogenic potential (Argiropoulos & Humphries, Citation2007; Chaturvedi et al., Citation2013; Nakamura et al., Citation1996). IDH mutations occur in 3–4% of MDS and 15–33% of AML patients, with mutations in IDH2 being slightly more common than in IDH1 in cytogenetically normal AML (19% versus 14%) (Marcucci et al., Citation2010; Mardis et al., Citation2009). Results on the prognostic effects of IDH mutations in these diseases are divergent (Green et al., Citation2010, Citation2011; Patnaik et al., Citation2012). IDH1/2 mutations confer a gain-of-function with neomorphic enzyme activity, that converts α-KG to the “oncometabolite” 2-hydroxyglutarate (2-HG). Sequestering of α-KG, which is an essential cosubstrate for TET2, results in the inhibition of α-KG-dependent actions of TET2 (). Altered 2-HG/α-KG levels observed in cells with mutant IDH1/2 additionally result in the inhibition of the jumonji-domain-containing (JMJC) family of HDMs, another group of α-KG-dependent enzymes, which demethylate H3K9 and H3K26 (Chowdhury et al., Citation2011) ( and ). IDH gain-of-function mutations and TET2 loss-of-function mutations (which are mutually exclusive in AML) affect the same pathway, and ultimately induce the same global promoter hypermethylation phenotype (Dang et al., Citation2009; Madzo et al., Citation2013; Ward et al., Citation2010).
Inhibition of both groups of wild-type enzymes (IDH and TET2) results in hypermethylation. Therapeutic targeting of IDH1 seems promising (Levis, Citation2013). The first inhibitors of mutant IDH1 are being extensively characterized in vitro (Davis et al., Citation2014), and several clinical trials with IDH inhibitors are currently recruiting ().
Mutations in other enzymes involved in the citric acid cycle, such as fumarate hydratase (FH) and succinate dehydrogenase (SDH), have been observed in solid tumors, and result in accumulation of fumarate and succinate, both of which are competitive inhibitors of TET proteins (Xiao et al., Citation2012). Mutations in FH and SDH have been linked with lower levels of 5-hmC, thus coincide with alterations of genome-wide histone and DNA methylation, and are thought to contribute to tumorigenesis.
HDMs in MDS/AML
HDMs can reverse histone modifications by an oxidative demethylation reaction (Klose et al., Citation2006) (). As mentioned above, methylation of various histone lysine residues by HMTs can have diametral effects on gene expression. Thus, HDMs may also exert diverging effects, depending on which lysine residue is demethylated. Lysine-specific demethylase 1 (LSD1) demethylates mono- and di-methylated H3K4. High LSD1 expression blocks differentiation and confers a poor prognosis in AML. LSD1 inhibition resulted in increased methylation of H3K4, resulting in gene activation (Fiskus et al., Citation2014c). LSD1 is currently being explored as a therapeutic target in phase I clinical trials in AML and small cell lung cancer ().
In contrast to LSD1, JHDMs are capable of demethylating all three methylated states (mono-, di- and tri-methylated lysine), and have been shown to demethylate H3K36, H3K9 and H3K9/27 (). The link between this group of enzymes and cancer is still evolving. Recently, JHDM1B was reported to be highly expressed in primary AML samples and is possibly required for leukemia maintenance (Nakamura et al., Citation2013). In particular, JHDM1B promoted the proliferation of leukemic progenitor cells in murine models of AML via suppression of p15INK4b, and depletion of JHDM1B resulted in reduced colony formation in vitro (Nakamura et al., Citation2013; Sroczynska et al., Citation2014). Selective small molecule JHDM inhibitors are currently being developed (Luo et al., Citation2011; Xu et al., Citation2013), but have not yet reached clinical trials.
HATs in MDS/AML
HATs confer the acetylation of lysine residues on the N-terminus of histones, which is generally associated with active gene transcription (Yang, Citation2004) ( and ). Most HATs are present as components of large protein complexes and act as transcriptional co-activators. Many of them have also been shown to acetylate proteins other than histones (Yang, Citation2004). In AML (and more rarely in MDS and CMML), several HATs (EP300, CREBBP, MYST3 and MYST4) are commonly involved in chromosomal translocations, which are involved in leukemogenesis through aberrant acetylation of all core histones, as well as on various non-histone proteins (Camos et al., Citation2006; Murati et al., Citation2004; Pattabiraman et al., Citation2014; Shigeno et al., Citation2004). HATs have not been designated as therapeutic targets in hematology thus far.
BET proteins in MDS/AML
A bromodomain (BRD) is a structural motif that recognizes monoacetylated lysine residues such as those on the N-terminal tails of histones. BET proteins preferentially bind to acetylated histones, i.e. to regions where multiple acetylation sites exist in proximity. They play a role in chromatin remodeling and result in transcriptional activation of BET-target genes, such as the well-known oncogenes cMYC and Bcl2 (). Dysfunction of BET-proteins has been linked to cancer (Filippakopoulos et al., Citation2012), and members of the BET family have recently been identified as targets for modulation of chromatin dynamics.
BET inhibitors attenuate cell growth and survival in primary AML patient samples and murine models of acute leukemia, partially through the down-regulation of the critical oncogenes, MYC and Bcl2 (Chen et al., Citation2014; Dawson et al., Citation2014; Delmore et al., Citation2011; Fiskus et al., Citation2014b; Mertz et al., Citation2011; Valent & Zuber, Citation2014; Zuber et al., Citation2011). Of interest, nucleophosmin 1 (NPM1) has recently been shown to inhibit BRD4, and NPM1 mutations abrogate this function. Treatment of primary human AML cells with the BET-inhibitor I-BET151 could counteract the effect of mutated NPM1 (Dawson et al., Citation2014). Furthermore, the BRD4 inhibitor JQ1 was shown to be highly active in AML cells bearing both NPM1 and DNMT3A mutations, reported to be associated with poor prognosis (Stewart et al., Citation2013). JQ1 combined with the pan-HDAC inhibitor panabinostat prolonged survival of NOD/SCID mice engrafted with human NPM1 and FLT3-ITD mutated AML cell lines. Just weeks ago, synergism of JQ1 with FLT3 inhibitors in human AML cell lines was demonstrated (Fiskus et al., Citation2014a). Bromodomain inhibitors that have entered clinical trials are shown in .
HDACs in MDS/AML
HDACs promote gene repression through removal of acetyl groups from lysine residues in histone tails (). Hypoacetylation of histones by HDACs at the promoter region results in transcriptional repression of the target gene. At least 18 HDAC genes have been recognized in the human genome, and act mainly as part of large multi-protein complexes that function as transcriptional co-repressors (). HDACs are implicated in cancer through their aberrant recruitment and silencing of tumor suppressor genes.
Mutations in HDACs have not yet been described in MDS/AML, whereas a few reports of truncating mutations and expression changes in solid tumors have been published (Ozdag et al., Citation2006).
HDACs have been assessed as drug targets, and treatment with HDAC-inhibitors results in hyperacetylated histones and up-regulation of repressed genes. Until recently, DNA hypermethylation was thought of as a “molecular lock” resulting in gene silencing. However, serial in vitro testing with 24 different HDAC-inhibitors efficiently reactivated various genes silenced by DNA hypermethylation, without affecting the latter, but this effect was transient (2 weeks) (Raynal et al., Citation2012). Thus, induction of open chromatin formation via histone acetylation can result in transcription of genes whose promoter regions remain hypermethylated. The HDAC-inhibitors vorinostat and romidepsin have received FDA approval for cutaneous T-cell lymphoma, and the large number of clinical trials with these substances has been summarized by others (Copeland et al., Citation2009; Falkenberg & Johnstone, Citation2014; Johnstone, Citation2002). Many issues of HDAC inhibitors and their potential clinical utility in MDS remain only partially understood, and pose translational and clinical challenges (Stintzing et al., Citation2011). Combining DNMT inhibitors with HDAC inhibitors is an interesting field for clinical trials. Currently varying dosage and treatment schedules (sequential or concomitant application) need to be tested in order to evaluate whether the synergism of these substance classes that is observed in vitro, also occurs in vivo. Clinically relevant neurological toxicity is however dose limiting for most HDAC inhibitors.
Azanucleosides in MDS/AML
The most prominent example of clinical efficacy of epigenetic cancer therapy is DNA cytosine hypomethylation by HMAs. These drugs target the reversible process of promoter methylation, resulting in re-expression of previously silenced tumor suppressor genes. Below we will first go into detail regarding pharmacokinetics, pharmacodynamics, and molecular mechanisms of action of AZA and DAC, with a special focus on similarities, overlap and differences between these two structurally similar, but (partially) functionally divergent drugs. Next, we will focus on effects of HMAs on transcriptional regulation in MDS and AML.
Overview of azanucleoside pharmacokinetics and pharmacodynamics
The DNA double helix is composed of two polynucleotide strands. Nucleotides are the building blocks of nucleic acids and comprise a nucleoside, consisting of a nucleobase and a five-carbon sugar (ribose or deoxyribose), and at least one phosphate group. If the sugar is deoxyribose, the polymer is DNA. If the sugar is ribose, the polymer is RNA. DNA is composed of only four nucleobases: adenine (A), guanine (G), cytosine (C) and thymine (T). The DNA strands run in opposite directions to each other (antiparallel), using the following base-pairing rules: C-G and A-T. When DNA is transcribed to RNA, thymine is substituted for uridine (U).
AZA and DAC are structurally similar nucleoside analogue prodrugs that mimic physiological cytidine, but differ chemically in their ribose moiety. Both DAC and AZA are imported into cells by nucleoside transporters (Rius et al., Citation2009) (). Studies have shown a statistically significant correlation between the expression levels of nucleoside transporters (e.g. human equilibrative nucleoside transporter 1 [hENT1]) and the sensitivity of AML cells to nucleoside chemotherapeutics such as gemcitabine (Marce et al., Citation2006), fludarabine (Molina-Arcas et al., Citation2003) and cytarabine (Ara-C; Hubeek et al., Citation2005), as well as the DNMT inhibitors DAC and AZA, suggesting their potential role as predictive biomarkers for clinical response (Damaraju et al., Citation2012; Qin et al., Citation2009).
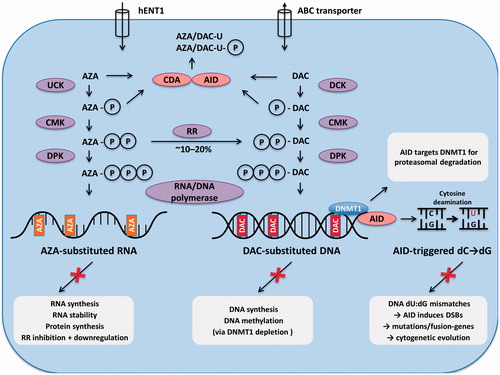
Figure 4. Membrane transporters and intracellular metabolism of AZA and DAC. AZA and DAC enter the cell via nucleoside transporters (e.g. hENT1). After triphosphorylation by the respective enzymes they are incorporated into RNA in the case of AZA, or into DNA in the case of DAC. Approximately 10–20% of AZA is reduced to DAC by RR, which is followed by incorporation into DNA. However, this step is self-limited and transient. Excess azanucleosides are rapidly deaminated to uracil-moieties by CDA. It is likely that AID can also perform this step. If the deamination process occurs on already DNA-incorporated DAC-cytidine residues, this will result in dU:dG mismatches on DNA, and may ultimately lead to DNA DSBs. AID-triggered DSBs can also be substrates for pro-oncogenic chromosomal translocations. AID may thus trigger leukemic evolution. In addition, AID targets DNMT1 for proteasomal degradation.
Abbreviations: ABC transporter, ATP-binding cassette transporter; AID, activation induced cytidine deaminase; AZA, 5-azacitidine; C, cytosine; CDA, cytidine deaminase; CMK, cytidine monophosphate kinase; DAC, 5-aza-2′-deoxycytidine; dC, deoxycytidine; DCK, deoxy-cytidine kinase; dG, deoxyguanine; DNMT1, DNA methyltransferase 1; DPK, diphosphate kinase; DSB, DNA double strand breaks; dU, deoxyuridine; G, guanine; hENT1, human equilibrative nucleoside transporter 1; P, phosphate; RR, ribonucleotide reductase; U, uridine; UCK, uridine-cytidine kinase.
After cellular uptake, azanucleosides need to be converted into active tri-phosphorylated nucleotides to become substrates for the DNA replication machinery, where they can substitute for cytosine. This reaction is catalyzed by different nucleoside metabolizing enzymes for AZA and DAC, respectively. DAC is tri-phosphorylated by deoxy-cytidine kinase (DCK), and can then be incorporated into newly synthesized DNA (where it pairs with guanine) with the help of DNA-polymerase (Momparler & Derse, Citation1979). In contrast, 80–90% of AZA is converted to ribonucleoside triphosphate by uridine–cytidine kinase (UCK), and is incorporated into RNA, leading to inhibition of protein synthesis (Li et al., Citation1970; Van Rompay et al., Citation2001) ().
DCK deficiency has been reported as a major mechanism of resistance to the nucleoside analogues DAC, Ara-C and gemcitabine (Galmarini et al., Citation2003; Qin et al., Citation2011; Saiki et al., Citation2012; Veuger et al., Citation2003). Furthermore, mutations in, and/or altered expression levels of nucleoside metabolizing enzymes, have been correlated with resistance to Ara-C and have been proposed as potential prognostic markers of response (Galmarini et al., Citation2001; Rathe & Largaespada, Citation2010; Yin et al., Citation2007). Silencing of the AZA metabolizing enzyme UCK1 reduced response to AZA in vitro. Very recently, MDS patients expressing lower levels of UCK1 were shown to be resistant to AZA, and had lower response rates and shorter overall survival (Valencia et al., Citation2014).
Ribonucleotide reductase (RR) reduces 10–20% of the diphosphate forms of AZA into deoxy-(DAC)-diphosphates, which can be incorporated into DNA after further phosphorylation to triphosphates (Kim et al., Citation1986) (). However, this process is transient and self-limited, which means that over time, the small fraction of AZA that is incorporated into DNA (initially 10–20%), is further reduced (Aimiuwu et al., Citation2012) ().
Data dating back to 1983 indicate rapid distribution of DAC between the extra- and intracellular compartments after i.v. injection. Penetration of the blood–brain barrier occurs, with DAC levels in the cerebrospinal fluid reaching 50% of the drugs plasma level (Chabot et al., Citation1983). Both drugs are rapidly cleared from systemic circulation: plasma half-life <1.5 to 1.8 h, as compared to a half-life of 7–21 h at 37 °C in neutral aqueous solutions in vitro (Liu et al., Citation2006; Yoo et al., Citation2007; Zhao et al., Citation2004). Systemic clearance exceeds the glomerular filtration rate and total renal blood flow, with urinary clearance of unchanged drug being <1% (van Groeningen et al., Citation1986). This suggests an important role for non-renal clearance (Chabot et al., Citation1983; Stresemann & Lyko, Citation2008).
In this context, cytidine deaminase (CDA) has been identified as the key enzyme catalyzing the deamination of cytidine, deoxycytidine and the cytidine analogues Ara-C, AZA and DAC, thereby destabilizing these drugs and rapidly reducing their half-life in vivo (). Retroviral overexpression of CDA caused significant resistance to DAC in vitro (Eliopoulos et al., Citation1998), and increased CDA expression/activity in human males in vivo has been linked with a reduced half-life of AZA and DAC, and possibly worse outcomes in MDS patients (Mahfouz et al., Citation2013). In rhesus monkeys, co-administration of CDA inhibitors with DAC improved pharmacokinetic profile and boosted plasma levels (Ferraris et al., Citation2014). Thus, a rational to combine inhibitors of CDA with HMA exists (Karahoca & Momparler, Citation2013). The CDA inhibitor tetrahydrouridine (THU) was assessed in humans in the 1990s and is being revived in clinical trials, with or without second generation DNMT inhibitors (Ferraris et al., Citation2014; Marsh et al., Citation1993) (). However, much care needs to be taken in titrating the dose of CDA inhibitors, as CDA downregulation was reported to be associated with toxic death in a patient exposed to the nucleoside analogue gemcitabine (Mercier et al., Citation2007).
Since both CDA and activation-induced cytidine deaminase (AID) catalyze cytidine deamination, AID is also likely to be involved in catabolism of cytidine analogues (). AID was originally described as a B-cell specific inducer of somatic hypermutation and class switch recombination in immunoglobulin genes, unique to activated germinal center B-cells. Specifically, AID deaminates the nucleoside cytidine and converts it to uridine, resulting in dU:dG mismatches on DNA. This causes DNA mutations and double strand breakage (Perez-Duran et al., Citation2007) (). Thus, in B-cells, AID is required for the generation of immunoglobulin diversity after V(D)J recombination (Honjo et al., Citation2004), an effect which is desired. However, via the same mechanism AID can also cause chromosomal translocations or mutations in proto-oncogenes, thus promoting tumor formation and/or leukemic evolution in a variety of hematologic malignancies (Kinoshita & Nonaka, Citation2006).
AID has also been shown to associate with and stabilize DNMT1. In a DNA-replication-dependent manner, AZA/DAC-substituted DNA binds the active site of AID and DNMT1. Indeed, AID is thought to target DNMT1 for ubiquitin-dependent proteasomal degradation, and thus represents an additional mechanism for DNMT1 depletion ( and ). As DNMT1 mRNA levels are not significantly affected, DNMT1 degradation is thought to be purely post-transcriptional (Tsai et al., Citation2014).
Therapeutic targeting of AID would theoretically result in: (i) higher levels of AZA/DAC due to reduced degradation to uracil-moieties; (ii) reduced genetic instability and thus reduced risk of cytogenetic evolution and disease progression; (iii) on the downside, DNMT1 depletion may be slightly hampered, although one might expect the multitude of collateral pathways for DNMT1 depletion to compensate for this. Interestingly, blockade of tumor necrosis factor α (TNFα) and/or interleukin 12 (IL12) has been shown to significantly suppress AID expression (Takai et al., Citation2012). The Anti-TNFα antibody infliximab and the TNFα-receptor fusion protein etanercept have been approved for clinical use in other entities (psoriasis, Crohn's disease, ulcerative colitis for infliximab, as well as rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis for etanercept), and have also shown clinical activity in MDS (Deeg et al., Citation2002; Raza et al., Citation2004; Scott et al., Citation2010a, Citationb; Stasi et al., Citation2005). The xanthin derivative pentoxifylline, approved for intermittent claudication from peripheral artery disease, also reduces the level of various cytokines, including TNFα; clinical activity in MDS and AML has been demonstrated (Erikci et al., Citation2008; Kim et al., Citation2006; Raza et al., Citation2000a, Citationb).
Molecular mechanisms of action of AZA and DAC
DNA-replication-dependent mechanism of DNMT1 depletion
AZA and DAC are inhibitors of DNMT1, which is completely depleted after HMA exposure, whereas DNMT3A is significantly less sensitive, and DNMT3B seems completely resistant to HMA-induced depletion (Ghoshal et al., Citation2005). DNMTs recognize C-G dinucleotides as their natural substrate. Formation of a reversible covalent bond between the DNA and DNMT is initiated by nucleophilic attack, which drives the transfer of a methyl group ( and ). The covalent complex is then usually resolved, resulting in release of the active DNMT enzyme.
The commonly accepted mechanism of action of HMAs is based on the initial report that incorporation of these structurally similar cytosine analogues into DNA inhibits the capacity of DNMTs to methylate DNA (Santi et al., Citation1984). Once DAC has been incorporated into DNA as a substitute for cytosine, DNMTs recognize DAC-G dinucleotides as their substrate. However, due to the slightly different chemical structure of the cytosine analogues AZA and DAC, as compared to naturally occurring cytosine (replacement of the cytosine C-5 by nitrogen), the covalent bond formation between DNMT1 and DAC-G becomes irreversible, which is in contrast to the reversible covalent bond that is formed between DNMT1 and C-G. Therefore, DNMT1 cannot be released from HMA-substituted DNA following methylation. This results in DNA-replication-dependent covalent trapping, depletion of functional DNMTs and loss of DNA methylation marks (Santi et al., Citation1984). In addition, the unavailability of DNMT1 prevents methylation of the newly replicated DNA strand. Thus, aberrant DNA methylation patterns are no longer reproduced in the daughter cells, leading to DNA demethylation and subsequent gene (re-)activation (Christman, Citation2002; Stresemann & Lyko, Citation2008). If re-expression of tumor suppressor genes occurs, these may subsequently inhibit malignant proliferation. Depending on which genes are re-induced, this may lead to an effective host anti-leukemia immune response via ameliorated antigen presentation through up-regulation of proteins involved in (tumor) antigen processing and re-induction of various cytokines and chemokines ().
DNA-replication-independent mechanisms of DNMT1 depletion
Several observations show that the formation of tight covalent complexes between DNMT1 and HMA-substituted DNA alone cannot explain many aspects of HMA mechanism of action, implicating additional DNA-replication-independent mechanisms of action (). In brief, DNMT1 activity decreases much faster than incorporation of HMAs into DNA (Creusot et al., Citation1982; Ghoshal et al., Citation2002, Citation2005), and DNMT1 depletion occurs even in the absence of (i) DNA replication and cell division (Datta et al., Citation2012; Easwaran et al., Citation2004; Ghoshal et al., Citation2005; Gius et al., Citation2004; Reither et al., Citation2003), and/or (ii) incorporation of AZA into DNA (Aimiuwu et al., Citation2012). This is, in part, thought to be due to the conserved KEN-box, which is a signature sequence for proteasomal degradation (Ghoshal et al., Citation2005). The data detailed in indicate that DNA-replication-dependent covalent bond formation between AZA/DAC-substituted DNA and DNMT1 is not always essential for enzyme degradation.
The following alternative DNA-replication and DNA-DNMT1 covalent bond formation independent pathway for HMA-mediated DNMT1 depletion has been proposed: HMAs induce degradation of DNMT1 in the nucleus via a proteasomal pathway that is dependent on HMA-induced hyperphosphorylation of DNMT1 by protein kinase C delta (PKCδ) (). Phosphorylated DNMT1 is then targeted to the ubiquitination machinery, with ensuing rapid proteasomal degradation. Multiple domains within DNMT1, including the KEN-box, nuclear localization signal (NLS) and bromo-adjacent homology (BAH) domains, are required for this process (Datta et al., Citation2012; Ghoshal et al., Citation2005). The importance of the conserved KEN-box is underlined by the observation that a DNMT1-KEN-box mutant is resistant to AZA/DAC-mediated degradation (Ghoshal et al., Citation2005). In this regard, both DNMT3A and 3B lack KEN-box, which explains why they are not targeted for proteasomal degradation by HMAs (Ghoshal et al., Citation2005). Both BAH and NLS are required for nuclear localization of DNMT1, which explains their necessity for HMA-induced degradation. Deletion of either of these domains results in cytoplasmic localization of DNMT1 (Ghoshal et al., Citation2005).
Although PKC mutations are very uncommon in AML (Redig & Platanias, Citation2008), the expression of various PKCs, including PKCδ, is often altered in both solid tumors and hematologic malignancies (Parker et al., Citation2014). PKCs are regarded as oncogenes and as participants in carcinogenesis and leukemogenesis (Koivunen et al., Citation2006; Steinberg, Citation2004). Elevated PKCδ expression has been described in several hematologic malignancies, including 45% of AML patients (Pearn et al., Citation2007).
PKCδ agonists would, in theory, enhance HMA-mediated DNMT1-depletion and possibly result in enhanced blast cell apoptosis via other mechanisms, as has been observed in solid tumors (Datta et al., Citation2012; Easwaran et al., Citation2004; Mochly-Rosen et al., Citation2012; Reither et al., Citation2003). PEP005, is an activator of various PKC isoenzymes, but its antileukemic effects on primary AML cells in vitro rely upon the activation of PKCδ (Kedei et al., Citation2004). These include: (i) reduction of the stem cell marker CXCR4, which is important for AML cell migration; (ii) induction of differentiation and increase in the expression of lineage-associated markers; (iii) induction of apoptosis in leukemic cells, but not in normal CD34+ hematopoietic cells; (iv) immunomodulatory effects such as increased T-cell recruitment, direct T-cell stimulatory effects, upregulation of T-cell co-stimulatory molecules CD80 and CD86 on dendritic cells, and induction of anticancer humoral immune responses with enhanced antibody dependent cellular cytotoxicity (Ersvaer et al., Citation2007, Citation2010; Hampson et al., Citation2005; Olsnes et al., Citation2009). Thus, PKCδ agonists such as PEP005 are a tantalizing target in AML. Although PKCδ modulators have been assessed in vitro and in murine models as a targeted cancer therapy, clinical trials have thus far only been performed with topical formulations in actinic keratosis, for which approval was gained. Systemic formulations have not yet reached clinical trials (Mochly-Rosen et al., Citation2012).
RNA-dependent inhibition of ribonucleotide reductase (RR) by AZA
As mentioned above, the majority (80–90%) of AZA is incorporated into RNA, resulting in mRNA and protein metabolism disruption, leading to apoptosis (; Aimiuwu et al., Citation2012; Hollenbach et al., Citation2010; Li et al., Citation1970). The first reports of AZA-mediated alteration in the processing of tRNA and rRNA, as well as inhibition of protein synthesis go as far back as 1973 (Cihak et al., Citation1974; Glover & Leyland-Jones, Citation1987; Lu & Randerath, Citation1980; Reichman & Penman, Citation1973).
Ribonucleotide reductase (RR) is the rate-limiting enzyme in DNA synthesis and consists of two subunits, RRM1 and RRM2. It generates the deoxyribonucleotides required for DNA synthesis and/or repair. AZA has recently been shown to be a potent and specific inhibitor of RRM2 in a murine AML model, AML cell lines, as well as in primary bone marrow cells from AML patients (Aimiuwu et al., Citation2012). Incorporation of AZA into RNA decreases RRM2 mRNA stability and inhibits RRM2 gene expression. Inhibition and/or down-regulation of RR causes a major perturbation of the deoxyribonucleotide pool, with a significant dose-dependent decrease of intracellular levels of all four deoxyribonucleotide levels by 60–90%. Furthermore, the conversion of AZA-diphosphate to DAC-diphosphate is terminated in a self-limited manner (Aimiuwu et al., Citation2012). These findings underline a profoundly different mode of action between AZA and DAC, which may explain the differences in observed clinical activities.
Interestingly, the RR inhibitor hydroxyurea was reported to block the ability of both AZA and DAC to induce DNA demethylation. This block was linked to cell cycle arrest in S-phase, thus inhibiting DNA-replication-dependent mechanisms of both drugs (Choi et al., Citation2007). This antagonism could be avoided with sequential treatment of hydroxyurea followed by AZA or DAC (Choi et al., Citation2007).
Similarities and differences in dose-dependent effects and mechanisms of action of AZA and DAC
AZA and DAC are usually viewed as mechanistically similar DNA HMAs. As mentioned above, it has only recently been (re-)acknowledged that AZA is mainly incorporated into RNA, whereas DAC is exclusively incorporated into DNA. Thus, the contention that AZA and DAC are interchangeable drugs (Coke versus Pepsi) needs to be rethought as it overlooks the additional mechanisms of action inherent to AZA (but not DAC). These are mediated via incorporation of AZA into newly synthesized RNA (including rRNAs, tRNAs, mRNAs and microRNAs), and result in the inhibition of protein synthesis.
Hypomethylating activity of AZA and DAC is only observed at low concentrations. In AML cell lines, DNMT1 levels are dramatically reduced with 100 mM DAC or 1 µM AZA. Furthermore, DNA hypomethylation occurs at even lower concentrations (30 nM DAC or 300 nM AZA) (Hollenbach et al., Citation2010). The difference in potency between AZA and DAC is reflected by the lower clinically used dosages (DAC:20 mg/m2 days 1–5; AZA: 75 mg/m2 days 1–7), and may be explained by the fact that ∼85% of AZA is incorporated into RNA and only ∼15% into DNA (Stresemann & Lyko, Citation2008). However, only minor substitution of 5-azacytosine for cytosine in DNA (approximately 0.3%) seems sufficient to inactivate more than 95% of DNMT1 (Creusot et al., Citation1982). The potent DNA hypomethylating activities of AZA and DAC are not observed at higher concentrations (∼3–10 µM), reflecting direct cytotoxicity, which is mediated by induction of apoptosis through DNA double strand breaks (Hollenbach et al., Citation2010).
Quantitative measurements of intracellular AZA-triphosphate and DAC-triphosphate levels are crucial, as they represent the amount of “activated” drug available for RNA- and DNA-incorporation, respectively. Furthermore, little is known about the quantitative pharmacokinetics of both drugs, and insight into this area may help improve dosing regimens. Several months ago, the first assay for quantitative detection of AZA-triphosphate in peripheral blood mononuclear cells was published (Derissen et al., Citation2014). Although sample number was very limited, AZA-triphosphate levels were 2.5–55 times higher than DAC-triphosphate concentrations previously published by the same group (Derissen et al., Citation2014; Jansen et al., Citation2012). This reflects the common difference between ribonucleotide (incorporated into RNA) and deoxyribonucleotide (incorporated into DNA) concentrations; endogenous ribonucleotide concentrations are typically several orders of magnitude higher than the corresponding deoxyribonucleotide concentrations (Cohen et al., Citation2009).
In vitro comparison of both drugs in human AML cell lines supports distinction of AZA and DAC as non-equivalent agents. Although shared mechanisms of action were revealed for DNA-incorporation-mediated markers of activity for AZA and DAC (e.g. DNMT1 depletion, DNA hypomethylation and DNA damage induction), distinctly different effects in their action on sensitivity/cell viability (AZA had a greater effect on reduction of cell viability), total protein synthesis (reduced only by AZA), and gene expression were reported (Hollenbach et al., Citation2010). HMA-modulated gene expression profiles were largely non-overlapping and drug-specific. Only 5–25% of the identified genes were regulated by both drugs. Generally, AZA regulated a greater number of genes than DAC (approximately twice as many after 48 h). AZA significantly down-regulated genes involved in the cell cycle, cell division and mitosis, whereas DAC preferentially up-regulated genes involved in cell differentiation (Flotho et al., Citation2009; Hollenbach et al., Citation2010). Mechanisms of resistance are also at least partly non-overlapping (Qin et al., Citation2009).
In summary, the differential effects of both drugs are very likely attributed to the incorporation of AZA into RNA. Importantly, AZA may have activity during all phases of the cell cycle via RNA incorporation, whereas DAC incorporation into DNA is restricted to S-phase, and may limit the number of affected cells at any given time (Hollenbach et al., Citation2010).
Initially, it was believed that DAC would be the more potent inhibitor of DNA hypomethylation (compared to AZA), as it can more readily be incorporated into DNA (100% DAC incorporation into DNA, as compared to 10–20% in the case of AZA) (Jones et al., Citation1983). However, survival data indirectly suggest clinical superiority of AZA (Fenaux et al., Citation2009; Gurion et al., Citation2010; Kantarjian et al., Citation2012; Kumar et al., Citation2010; Lee et al., Citation2013). It is thought that this may be precisely due to, and not despite, the fact that AZA is mainly incorporated into RNA (and not DNA) (Santini, Citation2009).
Effect of HMAs on transcriptional regulation
Effect of HMAs on DNA demethylation – correlation with response?
As previously discussed, cancer cells usually exhibit global hypomethylation and local hypermethylation of promoter regions. The events underlying genomic hypomethylation and localized hypermethylation of tumor suppressor genes in cancer cells, and why or how these types of genes are targeted, remain unclear. Interactions between regions of chromatin are pervasive and highly cell-type specific, and the mechanisms responsible for regulating the activity and kinetics of de novo methylation are poorly understood. In a model using colorectal cancer cell lines treated with DAC, it seems that feedback mediated regulation of DNMT activity remains intact in non-CIMP cells, but is defect in CIMP cells. In the latter, remethylation started without time-delay at a significantly slower rate, and continued at a constant low level (Wodarz et al., Citation2013). Thus, HMAs only achieve transient reactivation, with transcriptional silencing being restored approximately 2–4 weeks after drug exposure (Wodarz et al., Citation2013; Wong et al., Citation2013). This is why it is essential to adhere to the 4weekly treatment regimen.
Although aberrant DNA methylation is the most studied epigenetic alteration in MDS and AML, its role in prognosis and response to therapy is still unclear. Total genomic 5-mC levels decreased on average by 14% in AML patients treated with DAC (Klco et al., Citation2013; Yang et al., Citation2006), and global reductions in methylation were also observed in isolated AML blasts (Claus et al., Citation2013). Both oral and subcutaneous application of AZA significantly decreased both the number, as well as the methylation level, of highly methylated loci. Maximal effects were observed at day 15, but methylation levels returned to near-baseline level by the end of each cycle (day 28) (Garcia-Manero et al., Citation2011). Hypomethylation induced by HMAs results in significant variation in nuclear texture patterns, higher-order chromatin organization, distribution of heterochromatin regions, and ultimately in chromatin decondensation (Poplineau et al., Citation2013). Although aberrant DNA methylation has primarily been studied in the context of promoter CpG-islands in MDS and AML, it must be borne in mind, that CpG-islands represent only a small fraction of the methylome. In this regard, a very recent genome-wide methylation study demonstrated that differential methylation in AML was most pronounced in genomic areas far from CpG-islands, so-called “open sea regions” (Qu et al., Citation2014). In this context, DAC induces methylation changes with a preference for regions with higher baseline methylation. CpG-islands within gene bodies showed the same extent of DAC-induced hypomethylation as CpGs in promoter regions (Klco et al., Citation2013; Yan et al., Citation2012), but conflicting results exist (Wong et al., Citation2013).
Although some authors have observed significant correlation of HMA-induced hypomethylation with response and overall survival (Shen et al., Citation2010; Yang et al., Citation2006), uncertainty remains regarding the prognostic value of hypomethylation induction (Khan et al., Citation2013; Shen et al., Citation2010; Voso et al., Citation2014). Global methylation does not seem to differ between responders and non-responders to AZA or DAC (Daskalakis et al., Citation2002; Fandy et al., Citation2009). Hypomethylation occurred independently of the presence of significant disease and/or persistent cytogenetic aberrations (Yan et al., Citation2012). This failure to discriminate clinical responders from non-responders based on baseline methylation levels and/or changes in methylation treatment, has been attributed to the limitations of current techniques available to measure “global” DNA methylation (Voso et al., Citation2014). Measurement of global methylation levels cannot differentiate whether measured changes in methylation reflect: (i) therapeutic hypomethylation of the MDS/AML clone; (ii) cytoreduction resulting in decreased leukemia burden; or (iii) if they are a result of normal bone marrow recovery at the time of disease remission. It must further be considered that global methylation levels in blood DNA may vary in relation with age, gender, alcohol drinken, white blood cell count, as well as with the method used for methylation detection (Maegawa et al., Citation2014; Zhu et al., Citation2012). New methods, such as single-cell DNA-methylation analyses, may be more appropriate (Lorthongpanich et al., Citation2013).
Effect of HMAs on histone modifications – combinatorial therapeutic approaches
Genome-wide methylation studies have revealed that although HMAs induce global demethylation, only a limited number of genes are significantly reactivated, indicating the CpG demethylation is essential, but not sufficient to induce gene reactivation (Yang et al., Citation2012). As delineated above, methylation of DNA promoter elements and post-translational methylation/acetylation of histones, act in concert to epigenetically regulate gene expression. The movement of promoters and enhancers in and out of higher-order chromatin structures are associated with histone modifications. Again, the factors governing these changes are incompletely understood.
There is evidence of crosstalk between DNA methylation and histone post-translational modifications, whereby histone modifications are thought to provide labile transcriptional repression, and DNA methylation results in more permanent gene silencing (Cedar & Bergman, Citation2009; Ikegami et al., Citation2009; Raynal et al., Citation2012). Although detailed studies of histone modifications in MDS and AML remain to be described, they are thought to be critically involved in hematologic diseases, including leukemias (Issa, Citation2010). Several years ago, it was demonstrated that DAC induced gene expression is not necessarily dependent on DNA demethylation: DAC was shown to force expression from genes with persisting promoter CgP-island hypermethylation, which suggests that DAC is capable of influencing other factors involved in gene expression (Mossman et al., Citation2010). Recent data indicate that long-term transcriptional reactivation of epigenetically silenced genes with DAC cannot be adequately explained by DNA hypomethylation alone.
It is becoming increasingly clear, that HMA induced gene reactivation not only involves DNA methylation states, but also the balance between histone H3K4 and H3K27 methylation, as well as H3 acetylation. These histone modifications clearly define solid tumor cell lines with alternate responsiveness to HMA treatment (Mayor et al., Citation2011; Mossman et al., Citation2010). In a first step, DAC induces DNA demethylation, which is followed by rapid enrichment of (i) acetylated (ac)H3K9/14, (ii) trimethlyated H3K4(me3), and (iii) the histone variant H2A.Z, at demethylated promoter regions. This ultimately results in nucleosome depletion, which plays a key role in reconstructing chromatin architecture around demethylated promoters, leading to transcriptional activation (Yang et al., Citation2012). Of relevance, H2A.Z was shown to promote DAC induced gene re-expression, but had minimal effects on constitutively active genes (Yang et al., Citation2012). It has been demonstrated that long-term gene reactivation requires increased H3 acetylation, increased H3K4 trimethylation, hypomethylation of H3K27, in addition to DNA hypomethylation (Mossman et al., Citation2010). In contrast, hypermethylated genes that did not show increased H3 acetylation were only transiently re-expressed by DAC, before reverting to an inactive state (Mossman et al., Citation2010). This has been further underlined by intriguing observations that HDAC inhibitors can reactivate silenced genes without affecting their hypermethylated promoter state (Raynal et al., Citation2012).
All of the above findings provide a preclinical rational to combine DNMT inhibitors with drugs that inhibit histone methylation (inhibitors of HMTs) and/or facilitate histone acetylation (HDAC inhibitors). In vitro synergistic data provides theoretical promise of combining DNA-HMAs with HDAC-inhibitors (Zhu & Otterson, Citation2003) and/or inhibitors of histone methylation (Fiskus et al., Citation2009; Momparler et al., Citation2012, Citation2014; Zhou et al., Citation2011). However, clinically relevant results are yet to be demonstrated (Garcia-Manero, Citation2012; Quintas-Cardama et al., Citation2011; Stintzing et al., Citation2011).
Conclusions
Knowledge regarding the exact role of methylation and, in particular, oncoprotein-driven site-specific methylation is scarce. Measurable changes in methylation are likely markers of drug-bioavailability and adequate concentration, and appear necessary for response to HMA. However, demethylation of promoter regions by itself is insufficient for gene reactivation and response, and requires additional epigenetic changes in histones/nucleosomes.
A plethora of aberrations (e.g. loss-/gain-of-function mutations or up-/down-regulation) in components of the methylation machinery have been implicated in the development of MDS/AML. Moreover, as mere assessment of changes in global methylation status is unlikely to predict response or overall survival to commonly used HMAs in these patients, many of these components have become targets for predicting treatment outcomes. In particular, data have shown that mutational assessment of DNMT3A and TET2 may provide prognostic information for the likelihood of response and progression-free survival in patients treated with HMAs. Modulation of HOX genes and the transcription factor PU.1 may also be relevant for response prediction. Proteins involved in the intracellular transport and metabolism of HMAs are also under investigation as potential biomarkers for response or resistance to HMA therapy.
In addition to these potential predictors of treatment outcomes with HMAs, many components of the methylation machinery have been identified as novel therapeutic targets themselves. Notable examples include mutant IDH1 and UHRF1. It is clear from the data discussed in this review that the HMAs AZA and DAC have only partly overlapping mechanisms of action. This is likely due to AZA being incorporated into both DNA and RNA, while DAC is incorporated into DNA only. These differing biochemical interactions set the stage for us to speculate that there may be additive or synergistic effects when both drugs are combined.
In the last decade, much insight has been gained regarding the epigenetic machinery, as well as on basic molecular mechanisms of action of HMAs. However, despite this new wealth in knowledge, almost nothing is known about the precise factors that translate above discussed modes of action into clinical efficacy. Our current understanding remains rudimentary regarding how, why, and in whom, HMAs result in clinical efficacy significantly prolonging overall survival in approximately half of the patients, while being inefficacious in the other half. Further genome-wide analyses of the methylome is expected to bring more clarity into the complexity of epigenetic dysregulation in MDS and AML. Hopefully, methylation signatures will be reveiled, that can be used for prognostication, therapeutic decision making, and/or monitoring of therapeutic response.
Acknowledgements
The authors would like to acknowledge and thank Lucinda Huxley from FireKite, part of the KnowledgePoint360 Group, an Ashfield company, for proofreading as an English native speaker, as well as editing of the references and figures to the required journal format.
Declaration of interest
Consultant or advisory role: Lisa Pleyer, Janssen-Cilag, Celgene, Bristol-Myers Squibb, Novartis; Richard Greil, Bristol-Myers-Squibb, Cephalon, Celgene. Honoraria: Lisa Pleyer, Celgene, Bristol-Myers Squibb, Novartis, AOP Orphan Pharmaceuticals; Richard Greil, Amgen, Eisai, Mundipharma, Merck, Janssen-Cilag, Genentech, Novartis, Astra-Zeneca, Cephalon, Boehringer-Ingelheim, Pfizer, Roche, Bristol-Myers Squibb, Sanofi Aventis. Research funding: Richard Greil: Janssen-Cilag, GSK, Amgen, Genentech, Ratiopharm, Celgene, Pfizer, Mundipharma, Cephalon.
References
- Abdel-Wahab O, Adli M, LaFave LM, et al. (2012). ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 22:180–193
- Abdel-Wahab O, Gao J, Adli M, et al. (2013). Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J Exp Med 210:2641–2659
- Abdel-Wahab O, Levine RL. (2013). Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood 121:3563–3572
- Abdel-Wahab O, Mullally A, Hedvat C, et al. (2009). Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 114:144–147
- Abdel-Wahab O, Pardanani A, Patel J, et al. (2011). Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia 25:1200–1202
- Abusnina A, Keravis T, Yougbare I, et al. (2011). Anti-proliferative effect of curcumin on melanoma cells is mediated by PDE1A inhibition that regulates the epigenetic integrator UHRF1. Mol Nutr Food Res 55:1677–1689
- Aimiuwu J, Wang H, Chen P, et al. (2012). RNA-dependent inhibition of ribonucleotide reductase is a major pathway for 5-azacytidine activity in acute myeloid leukemia. Blood 119:5229–5238
- Alharbi RA, Pettengell R, Pandha HS, Morgan R. (2013). The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia 27:1000–1008
- Amiel J, de PL, Henrion-Caude A. (2012). miRNA, development and disease. Adv Genet 80:1–36
- Anand R, Marmorstein R. (2007). Structure and mechanism of lysine-specific demethylase enzymes. J Biol Chem 282:35425–35429
- Aoyama S, Nakano H, Danbara M, et al. (2012). The differentiating and apoptotic effects of 2-aza-5′-deoxycytidine are dependent on the PU.1 expression level in PU.1-transgenic K562 cells. Biochem Biophys Res Commun 420:775–781
- Argiropoulos B, Humphries RK. (2007). Hox genes in hematopoiesis and leukemogenesis. Oncogene 26:6766–6776
- Bartholdy B, Christopeit M, Will B, et al. (2014). HSC commitment-associated epigenetic signature is prognostic in acute myeloid leukemia. J Clin Invest 124:1158–1167
- Basecke J, Whelan JT, Griesinger F, Bertrand FE. (2006). The MLL partial tandem duplication in acute myeloid leukaemia. Br J Haematol 135:438–449
- Basova P, Pospisil V, Savvulidi F, et al. (2013). Aggressive acute myeloid leukemia in PU.1/p53 double-mutant mice. Oncogene 33:4735–4745
- Baubec T, Ivanek R, Lienert F, Schubeler D. (2013). Methylation-dependent and -independent genomic targeting principles of the MBD protein family. Cell 153:480–492
- Bejar R, Stevenson K, Abdel-Wahab O, et al. (2011). Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 364:2496–2506
- Beumer JH, Parise RA, Newman EM, et al. (2008). Concentrations of the DNA methyltransferase inhibitor 5-fluoro-2′-deoxycytidine (FdCyd) and its cytotoxic metabolites in plasma of patients treated with FdCyd and tetrahydrouridine (THU). Cancer Chemother Pharmacol 62:363–368
- Bird A. (2007). Perceptions of epigenetics. Nature 447:396–398
- Bonadies N, Pabst T, Mueller BU. (2010). Heterozygous deletion of the PU.1 locus in human AML. Blood 115:331–334
- Bostick M, Kim JK, Esteve PO, et al. (2007). UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317:1760–1764
- Boultwood J, Perry J, Pellagatti A, et al. (2010). Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia 24:1062–1065
- Bourc’his D, Xu GL, Lin CS, et al. (2001). Dnmt3L and the establishment of maternal genomic imprints. Science 294:2536–2539
- Bracken AP, Pasini D, Capra M, et al. (2003). EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J 22:5323–5335
- Braun T, Itzykson R, Renneville A, et al. (2011). Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: A phase 2 trial. Blood 118:3824–3831
- Bronner C, Achour M, Arima Y, et al. (2007). The UHRF family: Oncogenes that are drugable targets for cancer therapy in the near future? Pharmacol Ther 115:419–434
- Bronner C, Krifa M, Mousli M. (2013). Increasing role of UHRF1 in the reading and inheritance of the epigenetic code as well as in tumorogenesis. Biochem Pharmacol 86:1643–1649
- Camos M, Esteve J, Jares P, et al. (2006). Gene expression profiling of acute myeloid leukemia with translocation t(8;16)(p11;p13) and MYST3-CREBBP rearrangement reveals a distinctive signature with a specific pattern of HOX gene expression. Cancer Res 66:6947–6954
- Cedar H, Bergman Y. (2009). Linking DNA methylation and histone modification: Patterns and paradigms. Nat Rev Genet 10:295–304
- Cervantes F. (2014). How I treat myelofibrosis. Blood 124:2635–2642
- Chabot GG, Rivard GE, Momparler RL. (1983). Plasma and cerebrospinal fluid pharmacokinetics of 5-Aza-2′-deoxycytidine in rabbits and dogs. Cancer Res 43:592–597
- Chaturvedi A, Araujo Cruz MM, Jyotsana N, et al. (2013). Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 122:2877–2887
- Chen C, Liu Y, Rappaport AR, et al. (2014). MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 25:652–665
- Cheng JX, Anastasi J, Watanabe K, et al. (2013). Genome-wide profiling reveals epigenetic inactivation of the PU.1 pathway by histone H3 lysine 27 trimethylation in cytogenetically normal myelodysplastic syndrome. Leukemia 27:1291–1300
- Choi SH, Byun HM, Kwan JM, et al. (2007). Hydroxycarbamide in combination with azacitidine or decitabine is antagonistic on DNA methylation inhibition. Br J Haematol 138:616–623
- Chowdhury R, Yeoh KK, Tian YM, et al. (2011). The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 12:463–469
- Christiansen DH, Andersen MK, Pedersen-Bjergaard J. (2003). Methylation of p15INK4B is common, is associated with deletion of genes on chromosome arm 7q and predicts a poor prognosis in therapy-related myelodysplasia and acute myeloid leukemia. Leukemia 17:1813–1819
- Christman JK. (2002). 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 21:5483–5495
- Chung SS, Park CY. (2014). MicroRNA dysregulation in the myelodysplastic syndromes. Microrna 2:174–186
- Cihak A, Weiss JW, Pitot HC. (1974). Characterization of polyribosomes and maturation of ribosomal RNA in hepatoma cells treated with 5-azacytidine. Cancer Res 34:3003–3009
- Claus R, Pfeifer D, Almstedt M, et al. (2013). Decitabine induces very early in vivo DNA methylation changes in blasts from patients with acute myeloid leukemia. Leuk Res 37:190–196
- Cohen S, Megherbi M, Jordheim LP, et al. (2009). Simultaneous analysis of eight nucleoside triphosphates in cell lines by liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 877:3831–3840
- Copeland RA, Solomon ME, Richon VM. (2009). Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov 8:724–732
- Copur O, Muller J. (2013). The histone H3-K27 demethylase Utx regulates HOX gene expression in Drosophila in a temporally restricted manner. Development 140:3478–3485
- Costello JF, Fruhwald MC, Smiraglia DJ, et al. (2000). Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet 24:132–138
- Creusot F, Acs G, Christman JK. (1982). Inhibition of DNA methyltransferase and induction of Friend erythroleukemia cell differentiation by 5-azacytidine and 5-aza-2′-deoxycytidine. J Biol Chem 257:2041–2048
- Curik N, Burda P, Vargova K, et al. (2012). 5-Azacitidine in aggressive myelodysplastic syndromes regulates chromatin structure at PU.1 gene and cell differentiation capacity. Leukemia 26:1804–1811
- Curt GA, Kelley JA, Fine RL, et al. (1985). A phase I and pharmacokinetic study of dihydro-5-azacytidine (NSC 264880). Cancer Res 45:3359–3363
- Daigle SR, Olhava EJ, Therkelsen CA, et al. (2011). Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20:53–65
- Daigle SR, Olhava EJ, Therkelsen CA, et al. (2013). Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 122:1017–1025
- Dallosso AR, Oster B, Greenhough A, et al. (2012). Long-range epigenetic silencing of chromosome 5q31 protocadherins is involved in early and late stages of colorectal tumorigenesis through modulation of oncogenic pathways. Oncogene 31:4409–4419
- Dalmasso MC, Onyango DO, Naguleswaran A, et al. (2009). Toxoplasma H2A variants reveal novel insights into nucleosome composition and functions for this histone family. J Mol Biol 392:33–47
- Damaraju VL, Mowles D, Yao S, et al. (2012). Role of human nucleoside transporters in the uptake and cytotoxicity of azacitidine and decitabine. Nucleosides Nucleotides Nucleic Acids 31:236–255
- Dang L, White DW, Gross S, et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462:739–744
- Daser A, Rabbitts TH. (2005). The versatile mixed lineage leukaemia gene MLL and its many associations in leukaemogenesis. Semin Cancer Biol 15:175–188
- Daskalakis M, Nguyen TT, Nguyen C, et al. (2002). Demethylation of a hypermethylated P15/INK4B gene in patients with myelodysplastic syndrome by 5-Aza-2′-deoxycytidine (decitabine) treatment. Blood 100:2957–2964
- Datta J, Ghoshal K, Motiwala T, Jacob ST. (2012). Novel insights into the molecular mechanism of action of DNA hypomethylating agents: Role of protein kinase C delta in decitabine-induced degradation of DNA methyltransferase 1. Genes Cancer 3:71–81
- Davis MI, Gross S, Shen M, et al. (2014). Biochemical, cellular and biophysical characterization of a potent inhibitor of mutant isocitrate dehydrogenase IDH1. J Biol Chem 289:13717–13725
- Dawson MA, Gudgin EJ, Horton SJ, et al. (2014). Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia 28:311–320
- de la Cruz X, Lois S, Sanchez-Molina S, Martinez-Balbas MA. (2005). Do protein motifs read the histone code? Bioessays 27:164–175
- de Lavallade H, Khoder A, Hart M, et al. (2013). Tyrosine kinase inhibitors impair B-cell immune responses in CML through off-target inhibition of kinases important for cell signaling. Blood 122:227–238
- Deeg HJ, Gotlib J, Beckham C, et al. (2002). Soluble TNF receptor fusion protein (etanercept) for the treatment of myelodysplastic syndrome: A pilot study. Leukemia 16:162–164
- Delmore JE, Issa GC, Lemieux ME, et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146:904–917
- Derissen EJ, Hillebrand MJ, Rosing H, et al. (2014). Quantitative determination of azacitidine triphosphate in peripheral blood mononuclear cells using liquid chromatography coupled with high-resolution mass spectrometry. J Pharm Biomed Anal 90:7–14
- Deshpande AJ, Chen L, Fazio M, et al. (2013). Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood 121:2533–2541
- Dickstein J, Senyuk V, Premanand K, et al. (2010). Methylation and silencing of miRNA-124 by EVI1 and self-renewal exhaustion of hematopoietic stem cells in murine myelodysplastic syndrome. Proc Natl Acad Sci USA 107:9783–9788
- Dobbelstein M, Moll U. (2014). Targeting tumour-supportive cellular machineries in anticancer drug development. Nat Rev Drug Discov 13:179–196
- Dolnik A, Engelmann JC, Scharfenberger-Schmeer M, et al. (2012). Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood 120:e83–e92
- Doyle B, Fudenberg G, Imakaev M, Mirny LA. (2014). Chromatin loops as allosteric modulators of enhancer-promoter interactions. PLoS Comput Biol 10:e1003867
- Duhoux FP, Ameye G, Montano-Almendras CP, et al. (2012). PRDM16 (1p36) translocations define a distinct entity of myeloid malignancies with poor prognosis but may also occur in lymphoid malignancies. Br J Haematol 156:76–88
- Easwaran HP, Schermelleh L, Leonhardt H, Cardoso MC. (2004). Replication-independent chromatin loading of Dnmt1 during G2 and M phases. EMBO Rep 5:1181–1186
- Eklund E. (2011). The role of Hox proteins in leukemogenesis: Insights into key regulatory events in hematopoiesis. Crit Rev Oncog 16:65–76
- Eliopoulos N, Cournoyer D, Momparler RL. (1998). Drug resistance to 5-aza-2′-deoxycytidine, 2′,2′-difluorodeoxycytidine, and cytosine arabinoside conferred by retroviral-mediated transfer of human cytidine deaminase cDNA into murine cells. Cancer Chemother Pharmacol 42:373–378
- El-Sharkawi D, Ali A, Evans CM, et al. (2014). ASXL1 mutations are infrequent in young patients with primary acute myeloid leukemia and their detection has a limited role in therapeutic risk stratification. Leuk Lymphoma 55:1326–1331
- Erdogan B, Facey C, Qualtieri J, et al. (2011). Diagnostic microRNAs in myelodysplastic syndrome. Exp Hematol 39:915–926
- Erikci AA, Ozturk A, Karagoz B, et al. (2008). Results of combination therapy with amifostine, pentoxifylline, ciprofloxacin and dexamethasone in patients with myelodysplastic syndrome and acute myeloid leukemia. Hematology 13:289–292
- Ersvaer E, Hampson P, Hatfield K, et al. (2007). T cells remaining after intensive chemotherapy for acute myelogenous leukemia show a broad cytokine release profile including high levels of interferon-gamma that can be further increased by a novel protein kinase C agonist PEP005. Cancer Immunol Immunother 56:913–925
- Ersvaer E, Kittang AO, Hampson P, et al. (2010). The protein kinase C agonist PEP005 (ingenol 3-angelate) in the treatment of human cancer: A balance between efficacy and toxicity. Toxins (Basel) 2:174–194
- Esteller M. (2007). Epigenetic gene silencing in cancer: The DNA hypermethylome. Hum Mol Genet 16 Spec No 1:R50–R59
- Falkenberg KJ, Johnstone RW. (2014). Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov 13:673–691
- Fandy TE, Herman JG, Kerns P, et al. (2009). Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood 114:2764–2773
- Fathi AT, Abdel-Wahab O. (2012). Mutations in epigenetic modifiers in myeloid malignancies and the prospect of novel epigenetic-targeted therapy. Adv Hematol 2012: Article ID 469592, 12 pages, doi: 10.1155/2012/469592
- Feinberg AP, Tycko B. (2004). The history of cancer epigenetics. Nat Rev Cancer 4:143–153
- Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. (2009). Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol 10:223–232
- Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. (2010). Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol 28:562–569
- Ferraris D, Duvall B, Delahanty G, et al. (2014). Design, synthesis, and pharmacological evaluation of fluorinated tetrahydrouridine derivatives as inhibitors of cytidine deaminase. J Med Chem 57:2582–2588
- Ferreira BV, Harrison C. (2014). How many JAK inhibitors in myelofibrosis? Best Pract Res Clin Haematol 27:187–195
- Figueroa ME, Lugthart S, Li Y, et al. (2010). DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 17:13–27
- Figueroa ME, Skrabanek L, Li Y, et al. (2009). MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood 114:3448–3458
- Filippakopoulos P, Picaud S, Mangos M, et al. (2012). Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149:214–231
- Fiskus W, Sharma S, Qi J, et al. (2014a). BET protein antagonist JQ1 is synergistically lethal with FLT3 tyrosine kinase inhibitor (TKI) and overcomes resistance to FLT3-TKI in AML cells expressing FLT-ITD. Mol Cancer Ther 13:2315–2327
- Fiskus W, Sharma S, Qi J, et al. (2014b). Highly active combination of BRD4 antagonist and histone deacetylase inhibitor against human acute myelogenous leukemia cells. Mol Cancer Ther 13:1142–1154
- Fiskus W, Sharma S, Shah B, et al. (2014c). Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia 28:2155–2164
- Fiskus W, Wang Y, Sreekumar A, et al. (2009). Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood 114:2733–2743
- Flemming A. (2012). Deal watch: Epizyme-Celgene deal signals interest in new class of epigenetic drugs. Nat Rev Drug Discov 11:508
- Flotho C, Claus R, Batz C, et al. (2009). The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia 23:1019–1028
- Fog CK, Galli GG, Lund AH. (2012). PRDM proteins: Important players in differentiation and disease. Bioessays 34:50–60
- Forn M, Munoz M, Tauriello DV, et al. (2013). Long range epigenetic silencing is a trans-species mechanism that results in cancer specific deregulation by overriding the chromatin domains of normal cells. Mol Oncol 7:1129–1141
- Fu Y, Sinha M, Peterson CL, Weng Z. (2008). The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet 4:e1000138
- Fuks F, Hurd PJ, Wolf D, et al. (2003). The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem 278:4035–4040
- Galmarini CM, Graham K, Thomas X, et al. (2001). Expression of high Km 5′-nucleotidase in leukemic blasts is an independent prognostic factor in adults with acute myeloid leukemia. Blood 98:1922–1926
- Galmarini CM, Thomas X, Graham K, et al. (2003). Deoxycytidine kinase and cN-II nucleotidase expression in blast cells predict survival in acute myeloid leukaemia patients treated with cytarabine. Br J Haematol 122:53–60
- Ganan-Gomez I, Wei Y, Yang H, et al. (2014). Overexpression of miR-125a in myelodysplastic syndrome CD34+ cells modulates NF-kappaB activation and enhances erythroid differentiation arrest. PLoS One 9:e93404
- Garcia-Manero G, Gore SD, Cogle C, et al. (2011). Phase I study of oral azacitidine in myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia. J Clin Oncol 29:2521–2527
- Garcia-Manero G. (2012). Can we improve outcomes in patients with acute myelogenous leukemia? Incorporating HDAC inhibitors into front-line therapy. Best Pract Res Clin Haematol 25:427–435
- Garzon R, Liu S, Fabbri M, et al. (2009). MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 113:6411–6418
- Ghoshal K, Datta J, Majumder S, et al. (2002). Inhibitors of histone deacetylase and DNA methyltransferase synergistically activate the methylated metallothionein I promoter by activating the transcription factor MTF-1 and forming an open chromatin structure. Mol Cell Biol 22:8302–8319
- Ghoshal K, Datta J, Majumder S, et al. (2005). 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol 25:4727–4741
- Gius D, Cui H, Bradbury CM, et al. (2004). Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach. Cancer Cell 6:361–371
- Glover AB, Leyland-Jones B. (1987). Biochemistry of azacitidine: A review. Cancer Treat Rep 71:959–964
- Goldberg AD, Allis CD, Bernstein E. (2007). Epigenetics: A landscape takes shape. Cell 128:635–638
- Goll MG, Kirpekar F, Maggert KA, et al. (2006). Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311:395–398
- Gong JN, Yu J, Lin HS, et al. (2014). The role, mechanism and potentially therapeutic application of microRNA-29 family in acute myeloid leukemia. Cell Death Differ 21:100–112
- Gowher H, Loutchanwoot P, Vorobjeva O, et al. (2006). Mutational analysis of the catalytic domain of the murine Dnmt3a DNA-(cytosine C5)-methyltransferase. J Mol Biol 357:928–941
- Green CL, Evans CM, Hills RK, et al. (2010). The prognostic significance of IDH1 mutations in younger adult patients with acute myeloid leukemia is dependent on FLT3/ITD status. Blood 116:2779–2782
- Green CL, Evans CM, Zhao L, et al. (2011). The prognostic significance of IDH2 mutations in AML depends on the location of the mutation. Blood 118:409–412
- Gronbaek K, Muller-Tidow C, Perini G, et al. (2012). A critical appraisal of tools available for monitoring epigenetic changes in clinical samples from patients with myeloid malignancies. Haematologica 97:1380–1388
- Grossmann V, Kohlmann A, Eder C, et al. (2011). Molecular profiling of chronic myelomonocytic leukemia reveals diverse mutations in >80% of patients with TET2 and EZH2 being of high prognostic relevance. Leukemia 25:877–879
- Guan D, Factor D, Liu Y, et al. (2013). The epigenetic regulator UHRF1 promotes ubiquitination-mediated degradation of the tumor-suppressor protein promyelocytic leukemia protein. Oncogene 32:3819–3828
- Guo D, Myrdal PB, Karlage KL, et al. (2010). Stability of 5-fluoro-2′-deoxycytidine and tetrahydrouridine in combination. AAPS PharmSciTech 11:247–252
- Gurion R, Vidal L, Gafter-Gvili A, et al. (2010). 5-Azacitidine prolongs overall survival in patients with myelodysplastic syndrome – a systematic review and meta-analysis. Haematologica 95:303–310
- Hampson P, Chahal H, Khanim F, et al. (2005). PEP005, a selective small-molecule activator of protein kinase C, has potent antileukemic activity mediated via the delta isoform of PKC. Blood 106:1362–1368
- Harmston N, Lenhard B. (2013). Chromatin and epigenetic features of long-range gene regulation. Nucleic Acids Res 41:7185–7199
- Herman JG, Baylin SB. (2003). Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 349:2042–2054
- Herrera-Merchan A, Arranz L, Ligos JM, et al. (2012). Ectopic expression of the histone methyltransferase Ezh2 in haematopoietic stem cells causes myeloproliferative disease. Nat Commun 3:623. doi: 10.1038/ncomms1623
- Hohenauer T, Moore AW. (2012). The Prdm family: Expanding roles in stem cells and development. Development 139:2267–2282
- Hollenbach PW, Nguyen AN, Brady H, et al. (2010). A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS One 5:e9001
- Honjo T, Muramatsu M, Fagarasan S. (2004). AID: How does it aid antibody diversity? Immunity 20:659–668
- Hopfer O, Komor M, Koehler IS, et al. (2009). Aberrant promotor methylation in MDS hematopoietic cells during in vitro lineage specific differentiation is differently associated with DNMT isoforms. Leuk Res 33:434–442
- Hubeek I, Stam RW, Peters GJ, et al. (2005). The human equilibrative nucleoside transporter 1 mediates in vitro cytarabine sensitivity in childhood acute myeloid leukaemia. Br J Cancer 93:1388–1394
- Hwang CK, Kim CS, Choi HS, et al. (2004). Transcriptional regulation of mouse mu opioid receptor gene by PU.1. J Biol Chem 279:19764–19774
- Ikegami K, Ohgane J, Tanaka S, et al. (2009). Interplay between DNA methylation, histone modification and chromatin remodeling in stem cells and during development. Int J Dev Biol 53:203–214
- Inoue D, Kitaura J, Togami K, et al. (2013). Myelodysplastic syndromes are induced by histone methylation-altering ASXL1 mutations. J Clin Invest 123:4627–4640
- Issa JP. (2010). Epigenetic changes in the myelodysplastic syndrome. Hematol Oncol Clin North Am 24:317–330
- Itzykson R, Fenaux P. (2014). Epigenetics of myelodysplastic syndromes. Leukemia 28:497–506
- Itzykson R, Kosmider O, Cluzeau T, et al. (2011). Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia 25:1147–1152
- Itzykson R, Kosmider O, Renneville A, et al. (2013). Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol 31:2428–2436
- Jaenisch R, Bird A. (2003). Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat Genet 33:245–254
- Jansen RS, Rosing H, Wijermans PW, et al. (2012). Decitabine triphosphate levels in peripheral blood mononuclear cells from patients receiving prolonged low-dose decitabine administration: A pilot study. Cancer Chemother Pharmacol 69:1457–1466
- Jeanblanc M, Mousli M, Hopfner R, et al. (2005). The retinoblastoma gene and its product are targeted by ICBP90: A key mechanism in the G1/S transition during the cell cycle. Oncogene 24:7337–7345
- Jiang Y, Dunbar A, Gondek LP, et al. (2009). Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood 113:1315–1325
- Jin C, Lu Y, Jelinek J, et al. (2014). TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res 42:6956–6971
- Jin SG, Jiang Y, Qiu R, et al. (2011). 5-Hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations. Cancer Res 71:7360–7365
- Johnstone RW. (2002). Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat Rev Drug Discov 1:287–299
- Jones PA, Taylor SM, Wilson VL. (1983). Inhibition of DNA methylation by 5-azacytidine. Recent Results Cancer Res 84:202–211
- Jones PA, Taylor SM. (1980). Cellular differentiation, cytidine analogs and DNA methylation. Cell 20:85–93
- Jongen-Lavrencic M, Sun SM, Dijkstra MK, et al. (2008). MicroRNA expression profiling in relation to the genetic heterogeneity of acute myeloid leukemia. Blood 111:5078–5085
- Kantarjian HM, Thomas XG, Dmoszynska A, et al. (2012). Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 30:2670–2677
- Karahoca M, Momparler RL. (2013). Pharmacokinetic and pharmacodynamic analysis of 5-aza-2′-deoxycytidine (decitabine) in the design of its dose-schedule for cancer therapy. Clin Epigenetics 5:3. doi: 10.1186/1868-7083-5-3
- Karlic H, Herrmann H, Varga F, et al. (2014). The role of epigenetics in the regulation of apoptosis in myelodysplastic syndromes and acute myeloid leukemia. Crit Rev Oncol Hematol 90:1–16
- Karpinski P, Ramsey D, Grzebieniak Z, et al. (2008). The CpG island methylator phenotype correlates with long-range epigenetic silencing in colorectal cancer. Mol Cancer Res 6:585–591
- Kedei N, Lundberg DJ, Toth A, et al. (2004). Characterization of the interaction of ingenol 3-angelate with protein kinase C. Cancer Res 64:3243–3255
- Khan H, Vale C, Bhagat T, Verma A. (2013). Role of DNA methylation in the pathogenesis and treatment of myelodysplastic syndromes. Semin Hematol 50:16–37
- Kihara-Negishi F, Yamamoto H, Suzuki M, et al. (2001). In vivo complex formation of PU.1 with HDAC1 associated with PU.1-mediated transcriptional repression. Oncogene 20:6039–6047
- Kim CH, Marquez VE, Mao DT, et al. (1986). Synthesis of pyrimidin-2-one nucleosides as acid-stable inhibitors of cytidine deaminase. J Med Chem 29:1374–1380
- Kim MK, Lee JL, Cho HS, et al. (2006). The hematologic response to anti-apoptotic cytokine therapy: Results of pentoxifylline, ciprofloxacin, and dexamethasone treatment for patients with myelodysplastic syndrome. J Korean Med Sci 21:40–45
- Kinoshita K, Nonaka T. (2006). The dark side of activation-induced cytidine deaminase: Relationship with leukemia and beyond. Int J Hematol 83:201–207
- Klco JM, Spencer DH, Lamprecht TL, et al. (2013). Genomic impact of transient low-dose decitabine treatment on primary AML cells. Blood 121:1633–1643
- Klose RJ, Kallin EM, Zhang Y. (2006). JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet 7:715–727
- Knutson SK, Wigle TJ, Warholic NM, et al. (2012). A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol 8:890–896
- Ko M, Huang Y, Jankowska AM, et al. (2010). Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 468:839–843
- Kogure M, Takawa M, Saloura V, et al. (2013). The oncogenic polycomb histone methyltransferase EZH2 methylates lysine 120 on histone H2B and competes ubiquitination. Neoplasia 15:1251–1261
- Koivunen J, Aaltonen V, Peltonen J. (2006). Protein kinase C (PKC) family in cancer progression. Cancer Lett 235:1–10
- Kon A, Shih LY, Minamino M, et al. (2013). Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet 45:1232–1237
- Kosmider O, Gelsi-Boyer V, Cheok M, et al. (2009a). TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs). Blood 114:3285–3291
- Kosmider O, Gelsi-Boyer V, Ciudad M, et al. (2009b). TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica 94:1676–1681
- Kratzke RA, Wang X, Wong L, et al. (2008). Response to the methylation inhibitor dihydro-5-azacytidine in mesothelioma is not associated with methylation of p16INK4a: Results of cancer and leukemia group B 159904. J Thorac Oncol 3:417–421
- Krivtsov AV, Feng Z, Lemieux ME, et al. (2008). H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 14:355–368
- Kroeger H, Jelinek J, Estecio MR, et al. (2008). Aberrant CpG island methylation in acute myeloid leukemia is accentuated at relapse. Blood 112:1366–1373
- Kumar A, List AF, Hozo I, et al. (2010). Decitabine versus 5-azacitidine for the treatment of myelodysplastic syndrome: Adjusted indirect meta-analysis. Haematologica 95:340–342
- Langer F, Dingemann J, Kreipe H, Lehmann U. (2005). Up-regulation of DNA methyltransferases DNMT1, 3A, and 3B in myelodysplastic syndrome. Leuk Res 29:325–329
- Lee JH, Choi Y, Kim SD, et al. (2013). Comparison of 7-day azacitidine and 5-day decitabine for treating myelodysplastic syndrome. Ann Hematol 92:889–897
- Leeke B, Marsman J, O’Sullivan JM, Horsfield JA. (2014). Cohesin mutations in myeloid malignancies: Underlying mechanisms. Exp Hematol Oncol 3:13. doi: 10.1186/2162-3619-3-13
- Levis M. (2013). Targeting IDH: The next big thing in AML. Blood 122:2770–2771
- Ley TJ, Ding L, Walter MJ, et al. (2010). DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363:2424–2433
- Li LH, Olin EJ, Buskirk HH, Reineke LM. (1970). Cytotoxicity and mode of action of 5-azacytidine on L1210 leukemia. Cancer Res 30:2760–2769
- Li Q, Liu L, Li W. (2014). Identification of circulating microRNAs as biomarkers in diagnosis of hematologic cancers: A meta-analysis. Tumour Biol 35:10467–10478
- Li Z, Cai X, Cai CL, et al. (2011). Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 118:4509–4518
- Lin X, Wang Z, Zhang R, Feng W. (2014). High serum microRNA-335 level predicts aggressive tumor progression and unfavorable prognosis in pediatric acute myeloid leukemia. Clin Transl Oncol 2014 Oct 10. [Epub ahead of print]
- Liu W, Deng L, Song Y, Redell M. (2014). DOT1L inhibition sensitizes MLL-rearranged AML to chemotherapy. PLoS One 9:e98270
- Liu X, Zhang G, Yi Y, et al. (2013). Decreased 5-hydroxymethylcytosine levels are associated with TET2 mutation and unfavorable overall survival in myelodysplastic syndromes. Leuk Lymphoma 54:2466–2473
- Liu Z, Marcucci G, Byrd JC, et al. (2006). Characterization of decomposition products and preclinical and low dose clinical pharmacokinetics of decitabine (5-aza-2′-deoxycytidine) by a new liquid chromatography/tandem mass spectrometry quantification method. Rapid Commun Mass Spectrom 20:1117–1126
- Lopez-Serra L, Esteller M. (2008). Proteins that bind methylated DNA and human cancer: Reading the wrong words. Br J Cancer 98:1881–1885
- Lorsbach RB, Moore J, Mathew S, et al. (2003). TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 17:637–641
- Lorthongpanich C, Cheow LF, Balu S, et al. (2013). Single-cell DNA-methylation analysis reveals epigenetic chimerism in preimplantation embryos. Science 341:1110–1112
- Lu LJ, Randerath K. (1980). Mechanism of 5-azacytidine-induced transfer RNA cytosine-5-methyltransferase deficiency. Cancer Res 40:2701–2705
- Luger K, Mader AW, Richmond RK, et al. (1997). Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389:251–260
- Luo X, Liu Y, Kubicek S, et al. (2011). A selective inhibitor and probe of the cellular functions of Jumonji C domain-containing histone demethylases. J Am Chem Soc 133:9451–9456
- Madzo J, Vasanthakumar A, Godley LA. (2013). Perturbations of 5-hydroxymethylcytosine patterning in hematologic malignancies. Semin Hematol 50:61–69
- Maegawa S, Gough SM, Watanabe-Okochi N, et al. (2014). Age-related epigenetic drift in the pathogenesis of MDS and AML. Genome Res 24:580–591
- Mahfouz RZ, Jankowska A, Ebrahem Q, et al. (2013). Increased CDA expression/activity in males contributes to decreased cytidine analog half-life and likely contributes to worse outcomes with 5-azacytidine or decitabine therapy. Clin Cancer Res 19:938–948
- Malcovati L, Rumi E, Cazzola M. (2014). Somatic mutations of calreticulin in myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms. Haematologica 99:1650–1652
- Manodoro F, Marzec J, Chaplin T, et al. (2014). Loss of imprinting at the 14q32 domain is associated with microRNA overexpression in acute promyelocytic leukemia. Blood 123:2066–2074
- Marce S, Molina-Arcas M, Villamor N, et al. (2006). Expression of human equilibrative nucleoside transporter 1 (hENT1) and its correlation with gemcitabine uptake and cytotoxicity in mantle cell lymphoma. Haematologica 91:895–902
- Marcucci G, Maharry K, Wu YZ, et al. (2010). IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: A Cancer and Leukemia Group B study. J Clin Oncol 28:2348–2355
- Mardis ER, Ding L, Dooling DJ, et al. (2009). Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361:1058–1066
- Margueron R, Reinberg D. (2011). The polycomb complex PRC2 and its mark in life. Nature 469:343–349
- Marschalek R. (2011). Mechanisms of leukemogenesis by MLL fusion proteins. Br J Haematol 152:141–154
- Marsh JH, Kreis W, Barile B, et al. (1993). Therapy of refractory/relapsed acute myeloid leukemia and blast crisis of chronic myeloid leukemia with the combination of cytosine arabinoside, tetrahydrouridine, and carboplatin. Cancer Chemother Pharmacol 31:481–484
- Martin C, Zhang Y. (2005). The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol 6:838–849
- Matousova M, Votruba I, Otmar M, et al. (2011). 2-Deoxy-5,6-dihydro-5-azacytidine – A less toxic alternative of 2-deoxy-5-azacytidine: A comparative study of hypomethylating potential. Epigenetics 6:769–776
- Mayor R, Munoz M, Coolen MW, et al. (2011). Dynamics of bivalent chromatin domains upon drug induced reactivation and resilencing in cancer cells. Epigenetics 6:1138–1148
- McCabe MT, Brandes JC, Vertino PM. (2009). Cancer DNA methylation: Molecular mechanisms and clinical implications. Clin Cancer Res 15:3927–3937
- McCabe MT, Ott HM, Ganji G, et al. (2012). EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492:108–112
- Mercier C, Raynal C, Dahan L, et al. (2007). Toxic death case in a patient undergoing gemcitabine-based chemotherapy in relation with cytidine deaminase downregulation. Pharmacogenet Genomics 17:841–844
- Merkerova MD, Krejcik Z, Belickova M, et al. (2014). Genome-wide miRNA profiling in myelodysplastic syndrome with del(5q) treated with lenalidomide. Eur J Haematol 2014 Oct 7. [Epub ahead of print]. doi: 10.1111/ejh.12458
- Mertz JA, Conery AR, Bryant BM, et al. (2011). Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci USA 108:16669–16674
- Metzeler KH, Becker H, Maharry K, et al. (2011a). ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood 118:6920–6929
- Metzeler KH, Maharry K, Radmacher MD, et al. (2011b). TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: A Cancer and Leukemia Group B study. J Clin Oncol 29:1373–1381
- Miremadi A, Oestergaard MZ, Pharoah PD, Caldas C. (2007). Cancer genetics of epigenetic genes. Hum Mol Genet 16 Spec No 1:R28–R49
- Mirza S, Sharma G, Parshad R, et al. (2013). Expression of DNA methyltransferases in breast cancer patients and to analyze the effect of natural compounds on DNA methyltransferases and associated proteins. J Breast Cancer 16:23–31
- Mochizuki N, Shimizu S, Nagasawa T, et al. (2000). A novel gene, MEL1, mapped to 1p36.3 is highly homologous to the MDS1/EVI1 gene and is transcriptionally activated in t(1;3)(p36;q21)-positive leukemia cells. Blood 96:3209–3214
- Mochly-Rosen D, Das K, Grimes KV. (2012). Protein kinase C, an elusive therapeutic target? Nat Rev Drug Discov 11:937–957
- Molina-Arcas M, Bellosillo B, Casado FJ, et al. (2003). Fludarabine uptake mechanisms in B-cell chronic lymphocytic leukemia. Blood 101:2328–2334
- Momparler RL, Cote S, Momparler LF, Idaghdour Y. (2014). Epigenetic therapy of acute myeloid leukemia using 5-aza-2′-deoxycytidine (decitabine) in combination with inhibitors of histone methylation and deacetylation. Clin Epigenetics 6:19. doi: 10.1186/1868-7083-6-19
- Momparler RL, Derse D. (1979). Kinetics of phosphorylation of 5-aza-2′-deoxyycytidine by deoxycytidine kinase. Biochem Pharmacol 28:1443–1444
- Momparler RL, Idaghdour Y, Marquez VE, Momparler LF. (2012). Synergistic antileukemic action of a combination of inhibitors of DNA methylation and histone methylation. Leuk Res 36:1049–1054
- Moran-Crusio K, Reavie L, Shih A, et al. (2011). Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20:11–24
- Morin RD, Johnson NA, Severson TM, et al. (2010). Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 42:181–185
- Mossman D, Kim KT, Scott RJ. (2010). Demethylation by 5-aza-2′-deoxycytidine in colorectal cancer cells targets genomic DNA whilst promoter CpG island methylation persists. BMC Cancer 10:366
- Muller-Tidow C, Klein HU, Hascher A, et al. (2010). Profiling of histone H3 lysine 9 trimethylation levels predicts transcription factor activity and survival in acute myeloid leukemia. Blood 116:3564–3571
- Murati A, Adelaide J, Mozziconacci MJ, et al. (2004). Variant MYST4-CBP gene fusion in a t(10;16) acute myeloid leukaemia. Br J Haematol 125:601–604
- Nakamura S, Tan L, Nagata Y, et al. (2013). JmjC-domain containing histone demethylase 1B-mediated p15(Ink4b) suppression promotes the proliferation of leukemic progenitor cells through modulation of cell cycle progression in acute myeloid leukemia. Mol Carcinog 52:57–69
- Nakamura T, Largaespada DA, Lee MP, et al. (1996). Fusion of the nucleoporin gene NUP98 to HOXA9 by the chromosome translocation t(7;11)(p15;p15) in human myeloid leukaemia. Nat Genet 12:154–158
- Nan X, Campoy FJ, Bird A. (1997). MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88:471–481
- Ng K, Pullirsch D, Leeb M, Wutz A. (2007). Xist and the order of silencing. EMBO Rep 8:34–39
- Nikoloski G, Langemeijer SM, Kuiper RP, et al. (2010). Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet 42:665–667
- Nikoloski G, van der Reijden BA, Jansen JH. (2012). Mutations in epigenetic regulators in myelodysplastic syndromes. Int J Hematol 95:8–16
- Okada Y, Jiang Q, Lemieux M, et al. (2006). Leukaemic transformation by CALM-AF10 involves upregulation of Hoxa5 by hDOT1L. Nat Cell Biol 8:1017–1024
- Okano M, Bell DW, Haber DA, Li E. (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99:247–257
- Olsnes AM, Ersvaer E, Ryningen A, et al. (2009). The protein kinase C agonist PEP005 increases NF-kappaB expression, induces differentiation and increases constitutive chemokine release by primary acute myeloid leukaemia cells. Br J Haematol 145:761–774
- Ooi SK, Qiu C, Bernstein E, et al. (2007). DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448:714–717
- Oppikofer M, Kueng S, Gasser SM. (2013). SIR-nucleosome interactions: Structure–function relationships in yeast silent chromatin. Gene 527:10–25
- Otani J, Kimura H, Sharif J, et al. (2013). Cell cycle-dependent turnover of 5-hydroxymethyl cytosine in mouse embryonic stem cells. PLoS One 8:e82961
- Ozdag H, Teschendorff AE, Ahmed AA, et al. (2006). Differential expression of selected histone modifier genes in human solid cancers. BMC Genomics 7:90
- Panigrahi AK, Pati D. (2012). Higher-order orchestration of hematopoiesis: Is cohesin a new player? Exp Hematol 40:967–973
- Parker PJ, Justilien V, Riou P, et al. (2014). Atypical protein kinase Ciota as a human oncogene and therapeutic target. Biochem Pharmacol 88:1–11
- Parry L, Clarke AR. (2011). The roles of the methyl-CpG binding proteins in cancer. Genes Cancer 2:618–630
- Pastor WA, Pape UJ, Huang Y, et al. (2011). Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature 473:394–397
- Patnaik MM, Hanson CA, Hodnefield JM, et al. (2012). Differential prognostic effect of IDH1 versus IDH2 mutations in myelodysplastic syndromes: A Mayo Clinic study of 277 patients. Leukemia 26:101–105
- Patnaik MM, Itzykson R, Lasho TL, et al. (2014). ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: A two-center study of 466 patients. Leukemia 28:2206–2212
- Patnaik MM, Padron E, LaBorde RR, et al. (2013). Mayo prognostic model for WHO-defined chronic myelomonocytic leukemia: ASXL1 and spliceosome component mutations and outcomes. Leukemia 27:1504–1510
- Pattabiraman DR, McGirr C, Shakhbazov K, et al. (2014). Interaction of c-Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood 123:2682–2690
- Pearn L, Fisher J, Burnett AK, Darley RL. (2007). The role of PKC and PDK1 in monocyte lineage specification by Ras. Blood 109:4461–4469
- Pennings S, Muyldermans S, Meersseman G, Wyns L. (1989). Formation, stability and core histone positioning of nucleosomes reassembled on bent and other nucleosome-derived DNA. J Mol Biol 207:183–192
- Perez-Duran P, de Yebenes V, Ramiro AR. (2007). Oncogenic events triggered by AID, the adverse effect of antibody diversification. Carcinogenesis 28:2427–2433
- Pfeifer GP, Xiong W, Hahn MA, Jin SG. (2014). The role of 5-hydroxymethylcytosine in human cancer. Cell Tissue Res 356:631–641
- Phair RD, Scaffidi P, Elbi C, et al. (2004). Global nature of dynamic protein-chromatin interactions in vivo: Three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol Cell Biol 24:6393–6402
- Pleyer L, Burgstaller S, Girschikofsky M, et al. (2014a). Azacitidine in 302 patients with WHO-defined acute myeloid leukemia: Results from the Austrian Azacitidine Registry of the AGMT-Study Group. Ann Hematol 93:1825–1838
- Pleyer L, Germing U, Sperr WR, et al. (2014b). Azacitidine in CMML: Matched-pair analyses of daily-life patients reveal modest effects on clinical course and survival. Leuk Res 38:475–483
- Pleyer L, Stauder R, Burgstaller S, et al. (2013). Azacitidine in patients with WHO-defined AML – Results of 155 patients from the Austrian Azacitidine Registry of the AGMT-Study Group. J Hematol Oncol 6:32. doi: 10.1186/1756-8722-6-32
- Pollyea DA, Raval A, Kusler B, et al. (2011). Impact of TET2 mutations on mRNA expression and clinical outcomes in MDS patients treated with DNA methyltransferase inhibitors. Hematol Oncol 29:157–160
- Poplineau M, Doliwa C, Schnekenburger M, et al. (2013). Epigenetically induced changes in nuclear textural patterns and gelatinase expression in human fibrosarcoma cells. Cell Prolif 46:127–136
- Puda A, Milosevic JD, Berg T, et al. (2012). Frequent deletions of JARID2 in leukemic transformation of chronic myeloid malignancies. Am J Hematol 87:245–250
- Qi W, Chan H, Teng L, et al. (2012). Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci USA 109:21360–21365
- Qin T, Castoro R, El Ahdab S, et al. (2011). Mechanisms of resistance to decitabine in the myelodysplastic syndrome. PLoS One 6:e23372
- Qin T, Jelinek J, Si J, et al. (2009). Mechanisms of resistance to 5-aza-2′-deoxycytidine in human cancer cell lines. Blood 113:659–667
- Qin W, Zhang K, Clarke K, et al. (2014). Methylation and miRNA effects of resveratrol on mammary tumors vs. normal tissue. Nutr Cancer 66:270–277
- Qu Y, Lennartsson A, Gaidzik VI, et al. (2014). Differential methylation in CN-AML preferentially targets non-CGI regions and is dictated by DNMT3A mutational status and associated with predominant hypomethylation of HOX genes. Epigenetics 9:1108–1119
- Quesnel B. (2009). Methyltransferases in myelodysplastic syndromes: Guilty or not guilty? Leuk Res 33:601–602
- Quintas-Cardama A, Santos FP, Garcia-Manero G. (2011). Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia 25:226–235
- Rathe SK, Largaespada DA. (2010). Deoxycytidine kinase is downregulated in Ara-C-resistant acute myeloid leukemia murine cell lines. Leukemia 24:1513–1515
- Raynal NJ, Si J, Taby RF, et al. (2012). DNA methylation does not stably lock gene expression but instead serves as a molecular mark for gene silencing memory. Cancer Res 72:1170–1181
- Raza A, Candoni A, Khan U, et al. (2004). Remicade as TNF suppressor in patients with myelodysplastic syndromes. Leuk Lymphoma 45:2099–2104
- Raza A, Qawi H, Andric T, et al. (2000a). Pentoxifylline, ciprofloxacin and dexamethasone improve the ineffective hematopoiesis in myelodysplastic syndrome patients; malignancy. Hematology 5:275–284
- Raza A, Qawi H, Lisak L, et al. (2000b). Patients with myelodysplastic syndromes benefit from palliative therapy with amifostine, pentoxifylline, and ciprofloxacin with or without dexamethasone. Blood 95:1580–1587
- Redig AJ, Platanias LC. (2008). Protein kinase C signalling in leukemia. Leuk Lymphoma 49:1255–1262
- Reichman M, Penman S. (1973). The mechanism of inhibition of protein synthesis by 5-azacytidine in HeLa cells. Biochim Biophys Acta 324:282–289
- Reither S, Li F, Gowher H, Jeltsch A. (2003). Catalytic mechanism of DNA-(cytosine-C5)-methyltransferases revisited: Covalent intermediate formation is not essential for methyl group transfer by the murine Dnmt3a enzyme. J Mol Biol 329:675–684
- Ribeiro AF, Pratcorona M, Erpelinck-Verschueren C, et al. (2012). Mutant DNMT3A: A marker of poor prognosis in acute myeloid leukemia. Blood 119:5824–5831
- Rius M, Stresemann C, Keller D, et al. (2009). Human concentrative nucleoside transporter 1-mediated uptake of 5-azacytidine enhances DNA demethylation. Mol Cancer Ther 8:225–231
- Romermann D, Hasemeier B, Metzig K, et al. (2008). Global increase in DNA methylation in patients with myelodysplastic syndrome. Leukemia 22:1954–1956
- Rosenthal A, Mesa RA. (2014). Janus kinase inhibitors for the treatment of myeloproliferative neoplasms. Expert Opin Pharmacother 15:1265–1276
- Rottach A, Frauer C, Pichler G, et al. (2010). The multi-domain protein Np95 connects DNA methylation and histone modification. Nucleic Acids Res 38:1796–1804
- Saied MH, Marzec J, Khalid S, et al. (2012). Genome wide analysis of acute myeloid leukemia reveal leukemia specific methylome and subtype specific hypomethylation of repeats. PLoS One 7:e33213
- Saiki Y, Yoshino Y, Fujimura H, et al. (2012). DCK is frequently inactivated in acquired gemcitabine-resistant human cancer cells. Biochem Biophys Res Commun 421:98–104
- Santamaria C, Muntion S, Roson B, et al. (2012). Impaired expression of DICER, DROSHA, SBDS and some microRNAs in mesenchymal stromal cells from myelodysplastic syndrome patients. Haematologica 97:1218–1224
- Santi DV, Norment A, Garrett CE. (1984). Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc Natl Acad Sci USA 81:6993–6997
- Santini V, Melnick A, Maciejewski JP, et al. (2013). Epigenetics in focus: Pathogenesis of myelodysplastic syndromes and the role of hypomethylating agents. Crit Rev Oncol Hematol 88:231–245
- Santini V. (2009). Azacitidine: Activity and efficacy as an epigenetic treatment of myelodysplastic syndromes. Expert Rev Hematol 2:121–127
- Sashida G, Harada H, Matsui H, et al. (2014). Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat Commun 5:4177. doi:10.1038/ncomms5177
- Score J, Hidalgo-Curtis C, Jones AV, et al. (2012). Inactivation of polycomb repressive complex 2 components in myeloproliferative and myelodysplastic/myeloproliferative neoplasms. Blood 119:1208–1213
- Scott BL, Ramakrishnan A, Fosdal M, et al. (2010a). Anti-thymocyte globulin plus etanercept as therapy for myelodysplastic syndromes (MDS): A phase II study. Br J Haematol 149:706–710
- Scott BL, Ramakrishnan A, Storer B, et al. (2010b). Prolonged responses in patients with MDS and CMML treated with azacitidine and etanercept. Br J Haematol 148:944–947
- Shah N, Sukumar S. (2010). The Hox genes and their roles in oncogenesis. Nat Rev Cancer 10:361–371
- Sharif J, Muto M, Takebayashi S, et al. (2007). The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450:908–912
- Shen L, Kantarjian H, Guo Y, et al. (2010). DNA methylation predicts survival and response to therapy in patients with myelodysplastic syndromes. J Clin Oncol 28:605–613
- Shigeno K, Yoshida H, Pan L, et al. (2004). Disease-related potential of mutations in transcriptional cofactors CREB-binding protein and p300 in leukemias. Cancer Lett 213:11–20
- Shih AH, Abdel-Wahab O, Patel JP, Levine RL. (2012). The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer 12:599–612
- Sokol L, Caceres G, Volinia S, et al. (2011). Identification of a risk dependent microRNA expression signature in myelodysplastic syndromes. Br J Haematol 153:24–32
- Song CX, Szulwach KE, Fu Y, et al. (2011). Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol 29:68–72
- Sorm F, Piskala A, Cihak A, Vesely J. (1964). 5-Azacytidine, a new, highly effective cancerostatic. Experientia 20:202–203
- Spada F, Haemmer A, Kuch D, et al. (2007). DNMT1 but not its interaction with the replication machinery is required for maintenance of DNA methylation in human cells. J Cell Biol 176:565–571
- Sroczynska P, Cruickshank VA, Bukowski JP, et al. (2014). shRNA screening identifies JMJD1C as being required for leukemia maintenance. Blood 123:1870–1882
- Stasi R, Amadori S, Newland AC, Provan D. (2005). Infliximab chimeric antitumor necrosis factor-a monoclonal antibody as potential treatment for myelodysplastic syndromes. Leuk Lymphoma 46:509–516
- Steinberg SF. (2004). Distinctive activation mechanisms and functions for protein kinase Cdelta. Biochem J 384:449–459
- Stewart HJ, Horne GA, Bastow S, Chevassut TJ. (2013). BRD4 associates with p53 in DNMT3A-mutated leukemia cells and is implicated in apoptosis by the bromodomain inhibitor JQ1. Cancer Med 2:826–835
- Stintzing S, Kemmerling R, Kiesslich T, et al. (2011). Myelodysplastic syndrome and histone deacetylase inhibitors: “to be or not to be acetylated”? J Biomed Biotechnol 2011:214143. doi: 10.1155/2011/214143
- Stresemann C, Lyko F. (2008). Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer 123:8–13
- Su R, Lin HS, Zhang XH, et al. (2014). MiR-181 family: Regulators of myeloid differentiation and acute myeloid leukemia as well as potential therapeutic targets. Oncogene 2014 Sep 1. [Epub ahead of print]. doi: 10.1038/onc.2014.274
- Suzuki M, Yamada T, Kihara-Negishi F, et al. (2003). Direct association between PU.1 and MeCP2 that recruits mSin3A-HDAC complex for PU.1-mediated transcriptional repression. Oncogene 22:8688–8698
- Suzuki M, Yamada T, Kihara-Negishi F, et al. (2006). Site-specific DNA methylation by a complex of PU.1 and Dnmt3a/b. Oncogene 25:2477–2488
- Tahiliani M, Koh KP, Shen Y, et al. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324:930–935
- Takai A, Marusawa H, Minaki Y, et al. (2012). Targeting activation-induced cytidine deaminase prevents colon cancer development despite persistent colonic inflammation. Oncogene 31:1733–1742
- Talbert PB, Henikoff S. (2006). Spreading of silent chromatin: Inaction at a distance. Nat Rev Genet 7:793–803
- Tanaka S, Miyagi S, Sashida G, et al. (2012). Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood 120:1107–1117
- Tark-Dame M, Jerabek H, Manders EM, et al. (2014). Depletion of the chromatin looping proteins CTCF and cohesin causes chromatin compaction: Insight into chromatin folding by polymer modelling. PLoS Comput Biol 10:e1003877
- Tenen DG, Hromas R, Licht JD, Zhang DE. (1997). Transcription factors, normal myeloid development, and leukemia. Blood 90:489–519
- Thol F, Bollin R, Gehlhaar M, et al. (2014). Mutations in the cohesin complex in acute myeloid leukemia: Clinical and prognostic implications. Blood 123:914–920
- Thol F, Damm F, Ludeking A, et al. (2011a). Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol 29:2889–2896
- Thol F, Friesen I, Damm F, et al. (2011b). Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J Clin Oncol 29:2499–2506
- Tien HF, Tang JH, Tsay W, et al. (2001). Methylation of the p15(INK4B) gene in myelodysplastic syndrome: It can be detected early at diagnosis or during disease progression and is highly associated with leukaemic transformation. Br J Haematol 112:148–154
- Timp W, Feinberg AP. (2013). Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer 13:497–510
- Torrano V, Chernukhin I, Docquier F, et al. (2005). CTCF regulates growth and erythroid differentiation of human myeloid leukemia cells. J Biol Chem 280:28152–28161
- Toyota M, Ahuja N, Ohe-Toyota M, et al. (1999). CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 96:8681–8686
- Traina F, Visconte V, Elson P, et al. (2014). Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia 28:78–87
- Trubia M, Albano F, Cavazzini F, et al. (2006). Characterization of a recurrent translocation t(2;3)(p15-22;q26) occurring in acute myeloid leukaemia. Leukemia 20:48–54
- Tsai CT, Yang PM, Chern TR, et al. (2014). AID downregulation is a novel function of the DNMT inhibitor 5-aza-deoxycytidine. Oncotarget 5:211–223
- Ufkin ML, Peterson S, Yang X, et al. (2014). miR-125a regulates cell cycle, proliferation, and apoptosis by targeting the ErbB pathway in acute myeloid leukemia. Leuk Res 38:402–410
- Unoki M, Brunet J, Mousli M. (2009). Drug discovery targeting epigenetic codes: The great potential of UHRF1, which links DNA methylation and histone modifications, as a drug target in cancers and toxoplasmosis. Biochem Pharmacol 78:1279–1288
- Valencia A, Masala E, Rossi A, et al. (2014). Expression of nucleoside-metabolizing enzymes in myelodysplastic syndromes and modulation of response to azacitidine. Leukemia 28:621–628
- Valent P, Zuber J. (2014). BRD4: A BET(ter) target for the treatment of AML? Cell Cycle 13:689–690
- Valinluck V, Tsai HH, Rogstad DK, et al. (2004). Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res 32:4100–4108
- van Groeningen CJ, Leyva A, O’Brien AM, et al. (1986). Phase I and pharmacokinetic study of 5-aza-2′-deoxycytidine (NSC 127716) in cancer patients. Cancer Res 46:4831–4836
- Van Rompay AR, Norda A, Linden K, et al. (2001). Phosphorylation of uridine and cytidine nucleoside analogs by two human uridine-cytidine kinases. Mol Pharmacol 59:1181–1186
- Varambally S, Cao Q, Mani RS, et al. (2008). Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 322:1695–1699
- Vasilatou D, Papageorgiou S, Pappa V, et al. (2010). The role of microRNAs in normal and malignant hematopoiesis. Eur J Haematol 84:1–16
- Vasilatou D, Papageorgiou SG, Dimitriadis G, Pappa V. (2013). Epigenetic alterations and microRNAs: New players in the pathogenesis of myelodysplastic syndromes. Epigenetics 8:561–570
- Veuger MJ, Honders MW, Spoelder HE, et al. (2003). Inactivation of deoxycytidine kinase and overexpression of P-glycoprotein in AraC and daunorubicin double resistant leukemic cell lines. Leuk Res 27:445–453
- Voso MT, Fabiani E, Piciocchi A, et al. (2011). Role of BCL2L10 methylation and TET2 mutations in higher risk myelodysplastic syndromes treated with 5-azacytidine. Leukemia 25:1910–1913
- Voso MT, Santini V, Fabiani E, et al. (2014). Why methylation is not a marker predictive of response to hypomethylating agents. Haematologica 99:613–619
- Walter MJ, Ding L, Shen D, et al. (2011). Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 25:1153–1158
- Wang J, Ai X, Gale RP, et al. (2013a). TET2, ASXL1 and EZH2 mutations in Chinese with myelodysplastic syndromes. Leuk Res 37:305–311
- Wang J, Li Z, He Y, et al. (2014). Loss of Asxl1 leads to myelodysplastic syndrome-like disease in mice. Blood 123:541–553
- Wang X, Dai H, Wang Q, et al. (2013b). EZH2 mutations are related to low blast percentage in bone marrow and -7/del(7q) in de novo acute myeloid leukemia. PLoS One 8:e61341
- Ward PS, Patel J, Wise DR, et al. (2010). The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17:225–234
- Wodarz D, Boland CR, Goel A, Komarova NL. (2013). Methylation kinetics and CpG-island methylator phenotype status in colorectal cancer cell lines. Biol Direct 8:14. doi: 10.1186/1745-6150-8-14
- Wong YF, Jakt LM, Nishikawa S. (2013). Prolonged treatment with DNMT inhibitors induces distinct effects in promoters and gene-bodies. PLoS One 8:e71099
- Xiao M, Yang H, Xu W, et al. (2012). Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 26:1326–1338
- Xu F, Li X, Wu L, et al. (2011). Overexpression of the EZH2, RING1 and BMI1 genes is common in myelodysplastic syndromes: Relation to adverse epigenetic alteration and poor prognostic scoring. Ann Hematol 90:643–653
- Xu F, Li X. (2012). The role of histone methyltransferase EZH2 in myelodysplastic syndromes. Expert Rev Hematol 5:177–185
- Xu M, Zhao GN, Lv X, et al. (2014). CTCF controls HOXA cluster silencing and mediates PRC2-repressive higher-order chromatin structure in NT2/D1 cells. Mol Cell Biol 34:3867–3879
- Xu W, Podoll JD, Dong X, et al. (2013). Quantitative analysis of histone demethylase probes using fluorescence polarization. J Med Chem 56:5198–5202
- Yamazaki J, Estecio MR, Lu Y, et al. (2013). The epigenome of AML stem and progenitor cells. Epigenetics 8:92–104
- Yamazaki J, Taby R, Vasanthakumar A, et al. (2012). Effects of TET2 mutations on DNA methylation in chronic myelomonocytic leukemia. Epigenetics 7:201–207
- Yan J, Enge M, Whitington T, et al. (2013). Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell 154:801–813
- Yan P, Frankhouser D, Murphy M, et al. (2012). Genome-wide methylation profiling in decitabine-treated patients with acute myeloid leukemia. Blood 120:2466–2474
- Yan XJ, Xu J, Gu ZH, et al. (2011). Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet 43:309–315
- Yang AS, Doshi KD, Choi SW, et al. (2006). DNA methylation changes after 5-aza-2′-deoxycytidine therapy in patients with leukemia. Cancer Res 66:5495–5503
- Yang X, Noushmehr H, Han H, et al. (2012). Gene reactivation by 5-aza-2′-deoxycytidine-induced demethylation requires SRCAP-mediated H2A.Z insertion to establish nucleosome depleted regions. PLoS Genet 8:e1002604
- Yang XJ. (2004). The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res 32:959–976
- Yildirim O, Li R, Hung JH, et al. (2011). Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell 147:1498–1510
- Yin B, Tsai ML, Hasz DE, et al. (2007). A microarray study of altered gene expression after cytarabine resistance in acute myeloid leukemia. Leukemia 21:1093–1097
- Yoo CB, Jeong S, Egger G, et al. (2007). Delivery of 5-aza-2′-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res 67:6400–6408
- Zardo G, Ciolfi A, Vian L, et al. (2012). Polycombs and microRNA-223 regulate human granulopoiesis by transcriptional control of target gene expression. Blood 119:4034–4046
- Zhao M, Rudek MA, He P, et al. (2004). Quantification of 5-azacytidine in plasma by electrospray tandem mass spectrometry coupled with high-performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci 813:81–88
- Zhao Q, Fan J, Hong W, et al. (2012). Inhibition of cancer cell proliferation by 5-fluoro-2′-deoxycytidine, a DNA methylation inhibitor, through activation of DNA damage response pathway. Springerplus 1:65. doi: 10.1186/2193-1801-1-65
- Zhao X, Yang F, Li S, et al. (2014). CpG island methylator phenotype of myelodysplastic syndrome identified through genome-wide profiling of DNA methylation and gene expression. Br J Haematol 165:649–658
- Zhou J, Bi C, Cheong LL, et al. (2011). The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood 118:2830–2839
- Zhu WG, Otterson GA. (2003). The interaction of histone deacetylase inhibitors and DNA methyltransferase inhibitors in the treatment of human cancer cells. Curr Med Chem Anticancer Agents 3:187–199
- Zhu ZZ, Hou L, Bollati V, et al. (2012). Predictors of global methylation levels in blood DNA of healthy subjects: A combined analysis. Int J Epidemiol 41:126–139
- Zuber J, Shi J, Wang E, et al. (2011). RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478:524–528