Abstract
In a previous issue of Annals of Medicine, we presented evidence in support of the concept that an abnormally increased production of reactive oxygen species plays a central role in the genesis and progression of cardiovascular disease. While a number of preclinical lines of evidence support this concept, and despite the results of many studies suggesting a beneficial impact of antioxidant drugs on endothelial function, large clinical trials have failed to demonstrate a benefit of antioxidants on cardiovascular outcomes. Studies exploring the possibility that classical antioxidants such as vitamin C, vitamin E, selenium, or folic acid may improve the prognosis of patients with cardiac disease have substantially reported neutral—and occasionally negative—results. In contrast, medications such as statins, ACE inhibitors, certain β-blockers, or angiotensin I receptor blockers, which possess indirect ‘ancillary’ antioxidant properties, have been associated with beneficial effects in both preclinical studies and large clinical trials. The reasons for the failure of the ‘direct’ approach to antioxidant therapy, and for the success of the therapy with these drugs, are discussed in the present review.
| Abbreviations | ||
| eNOS | = | endothelial nitric oxide synthase |
| NO | = | nitric oxide |
| ROS | = | reactive oxygen species |
Key messages
Despite a sound biological background, clinical trials testing the impact of antioxidant therapy on cardiovascular outcomes have reported neutral—and occasionally negative—results.
Similarly, exogenous administration of nitric oxide with organic nitrates does not seem to improve cardiovascular outcome.
In contrast, a more complex approach with substances such as statins, ACE inhibitors, certain β-blockers, or angiotensin I receptor blockers, which possess antioxidant properties and stimulate nitric oxide production, has been associated with beneficial effects in both preclinical studies and large clinical trials.
Introduction
A number of lines of evidence support the importance of the vascular endothelium in preventing the genesis and the progression of cardiovascular disease. Functions of the endothelium include the control of vascular tone (a role that is critical, for instance, in unstable coronary syndromes, vasomotor angina, and microvascular angina), in the modulation of inflammation, and in the promotion or inhibition of vascular growth and of platelet aggregation and coagulation (all critically important in the setting of acute coronary syndromes). Abnormalities in these delicate processes, a condition termed ‘endothelial dysfunction’, have been shown in patients with coronary atherosclerosis and have been associated with worse outcome (higher incidence of cardiovascular events, progression of disease, in-stent restenosis, as well as higher all-cause mortality) (Citation1–7). These lines of evidence have prompted the investigation of the mechanisms of endothelial dysfunction in the hope that their correction would lead, through a ‘resetting’ of endothelial biochemistry to its normal status, to a prognostic improvement. Mirroring the complexity of endothelial function, however, the investigation of the mechanisms underlying an impairment in the biochemical homeostasis of this tissue has proven to be a challenging field. Although a number of factors have been shown to come into play, a growing body of evidence emphasizes the role of an abnormally increased production of reactive oxygen species (ROS). The mechanisms through which such an increased bioavailability of ROS may lead to endothelial dysfunction and vascular pathology have been discussed in detail in a review published by our group in a previous issue of Annals of Medicine (Citation8); the present review will summarize the current concepts concerning old and new pharmaceutical approaches for fighting increased oxidative stress in the vascular tissue.
The balance between oxidative stress and NO as an important determinant of cardiovascular disease
The numerous properties of NO, which have a central importance in the endogenous mechanisms of defense and repair that prevent or reduce the damage of acute coronary syndromes, have been described in our previous review article (Citation8). In all conditions where an absolute or relative NO deficit is present, i.e. when NO is insufficiently produced or too rapidly scavenged, atherogenetic processes are initiated or accelerated (Citation9,Citation10).
As described in our previous paper (Citation8), ROS and NO should not be considered two entirely separate entities and biochemical pathways. Indeed, ROS modify both the bioavailability and the effects of NO through several mechanisms. First, the ROS superoxide may bind to NO to form the highly reactive intermediate peroxynitrite (ONOO−). The speed of this reaction is about ten times faster than the dismutation of superoxide by the superoxide dismutase (Citation11,Citation12), and, when produced in concentrations that exceed the endogenous scavenging capacities, superoxide may substantially limit NO bioavailability and its effects. Second, although the product of superoxide dismutation, hydrogen peroxide, acutely stimulates NO production, chronic exposure to this ROS derivate increases superoxide production by NADPH oxidases, thus causing oxidative stress (Citation13). Third, recent studies indicate that ONOO− may influence the synthesis of the other endothelial mediators. Research from our group has indeed shown that an increased bioavailability of ONOO− in the setting of prolonged therapy with organic nitrates is associated with damage and impaired function of the enzyme prostacyclin synthase and therefore leads to a reduced production of vasodilator, antiaggregant, and anti-inflammatory prostaglandins (Citation14). Similarly, increased hydroxyl radical, superoxide, and ONOO− may impair the function of the endothelial NO synthase (eNOS) by reducing the bioavailability of its co-enzyme tetrahydrobiopterin (BH4), a phenomenon known as ‘eNOS uncoupling’ (Citation8). As well, an increased production of ROS is associated with that of asymmetric dimethylarginine (ADMA), a derivate of arginine which, unlike its molecular analog L-arginine, cannot serve as substrate for the eNOS, but still may compete for the active site of eNOS and of the L-arginine transporters (Citation15). The result of the competition between L-arginine and ADMA would thus be an inefficient NO production and an inefficient cellular uptake of L-arginine, resulting in impaired NO production and further increased ROS bioavailability. Interestingly, several lines of evidence demonstrate that the bioavailability of ADMA is linked with that of ROS: activation of membrane oxidases results in increased ADMA levels (Citation16), and the activity of the enzymes responsible for the synthesis and the catabolism of ADMA are redox-sensitive (for review, see (Citation15)).
Two conclusions can be gathered from these preliminary considerations. The first is that activation of any of the vascular superoxide sources described in our previous paper will promote ONOO− formation. The second is that the bioavailability of ROS is in a tight equilibrium with that of other endothelial mediators: in case of increased ROS production, a number of positive feedback mechanisms are activated that lead to further ROS production and to inhibition of other endothelium-derived factors.
In line with this concept, and the general idea that correction of the abnormally elevated ROS bioavailability might retard or prevent cardiovascular disease, a number of interventions have been proposed to restore the redox status in the vascular milieu. In general, three possible strategies can be followed to correct the balance towards an increased vascular NO synthesis (): the first and simplest one consists of the administration of classical antioxidants such as vitamin E and/or C that are directly aimed at reducing vascular ROS bioavailability; the second strategy is based on the administration of NO donors such as organic nitrates, in the hope that they will serve to increase vascular NO bioavailability. Finally, the most sophisticated approach is aimed at the stimulation of vascular NO production and the simultaneous inhibition of vascular ROS production. A number of lines of evidence show that only the last-mentioned of these strategies may lead to clinical benefits and to a better prognosis.
Figure 1. In general, increased oxidative stress in vascular tissue can be addressed by three different approaches: Treatment with compounds that possess direct antioxidant properties, exogenous administration of NO, or administration of compounds that decrease ROS bioavailability while increasing that of NO.
Direct antioxidants: ‘small’ successes, large failures
‘Traditional’ antioxidants have a number of properties that, at least in theory, may help restore vascular homeostasis in the setting of increased ROS production (). Beyond being able to react directly with most ROS, thus preventing—at least in theory—the interaction of these molecules with cellular structures and with NO, antioxidants may interfere with the damaging effects of ROS (Citation17). For instance, vitamin C or folic acid increases the recycling of trihydrobiopterin radical to BH4 (the functioning co-enzyme of eNOS), thus preventing or reversing ONOO−-induced eNOS uncoupling (Citation18). Folic acid, according to some authors, may even compensate for the absolute absence of BH4 in the active site of the eNOS (Citation19), which is compatible with the evidence that this B vitamin restores eNOS function even in the absence of BH4 (Citation20).
Figure 2. Scheme of the key players in ROS (reactive oxygen species)-induced damage and possible interventions. Gray lines mean inhibition or scavenging. Antioxidants such as vitamin C and E ‘only’ act by scavenging free radicals after they are formed. Organic nitrates increase the bioavailability of nitric oxide (NO), but at the same time increase that of ROS and inhibit the nitric oxide synthase. Angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARB), and statins act on the sources of ROS while at the same time stimulating endogenous NO production.
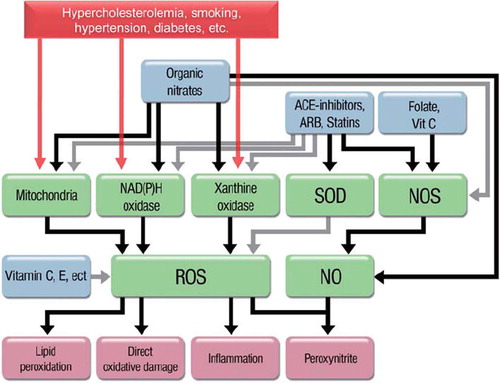
In line with these concepts, a number of studies have demonstrated the beneficial role of ‘traditional’ antioxidants on endothelial function. Vitamin C, vitamin E, co-enzyme Q10, folic acid, and polyphenols (just to mention some of the numerous antioxidant compounds available) improve endothelial responses when administered acutely in a number of conditions associated with cardiovascular pathology (). Even more interestingly, the degree of improvement of endothelial dysfunction in response to vitamin C (as an indirect measure of the oxidative burden) can be used as a prognostic indicator: patients with established coronary artery disease who show an improvement in endothelial function in response to intra-arterial infusions of vitamin C, i.e. those patients in whom oxidative stress can be demonstrated, have indeed a worse prognosis as compared to those who show no response to vitamin C (Citation1).
Table I. Studies demonstrating an effect of direct antioxidants on endothelial function in the presence of different conditions.
Results of large observational trials lend support to these preclinical findings, demonstrating that higher intake of antioxidants is associated with decreased cardiovascular and all-cause morbidity and mortality. For instance (only to quote some of the larger reports), a meta-analysis of cohort studies including more than 374,000 subjects (Citation21) with a follow-up of up to 15 years reported that high dietary vitamin E and vitamin C intake was associated with a lower incidence of coronary heart disease. Similarly, trials have demonstrated a reduced mortality from coronary heart disease in patients with high dietary flavonoid intake (Citation22,Citation23), an observation that was not modified after adjustment for traditional cardiovascular risk factors and for the intake of other antioxidant vitamins. As well, the ATBC study showed that higher serum vitamin E portends a lower mortality for cancer and cardiovascular disease (Citation24), and the NHANES-II study showed that low serum vitamin C levels are associated with increased mortality for the same causes (Citation25).
These mechanistic and observational studies obviously raised the expectations based on the hypothesis that supplementation with antioxidants could interfere with the pathophysiology of cardiovascular disease and improve patients’ prognosis. Given the low cost of most of these interventions, this hypothesis has led in the last 10 years to a number of interventional trials testing whether administration of antioxidant drugs could reduce the incidence of events and improve the life expectancy of patients with or at risk for cardiovascular disease. Frustrating the expectations of a rising industry, however, the clinical trials that reported positive results are a net minority (among others, one reporting a benefit of vitamin E supplementation in secondary prevention of cardiovascular disease (Citation26), another one demonstrating that vitamin E plus C reduces the progression of carotid intima-media thickening in hypercholesterolemic patients (Citation27), and a third one demonstrating reduced all-cause mortality after supplementation with a mix of selenium, vitamin E, and beta-carotene (Citation28)). In contrast, the trials reporting a neutral—or even a negative (Citation29)—impact of antioxidant supplementation are the large majority: in the meta-analysis by Ye et al., while dietary intake of antioxidants was associated with decreased incidence of coronary artery disease, supplementation with vitamin C or E did not have additional effects (Citation21); similarly, another meta-analysis (Citation30) demonstrated a substantial lack of efficacy for different doses of β-carotene and vitamin E in diverse population groups.
A selection of these studies, including both mechanistic studies and large-scale trials, is presented in . As of folic acid (which, as discussed above, might help prevent or reverse eNOS uncoupling and endothelial dysfunction), a number of studies showed no prognostic impact. In one trial, the combination of low-dose folic acid (0.8 mg/day) together with vitamin B12 and vitamin B6 effectively lowered plasma homocysteine levels in patients with a history of acute myocardial infarction, but was associated with a trend towards increased cardiovascular risk () (Citation31). In line with this observation, some studies have even questioned the safety (rather than efficacy) of prescribing antioxidant vitamins: a meta-analysis from Miller et al. with 135,967 participants in 19 clinical trials suggested that high-dosage vitamin E (> 400 IU/day) paradoxically increases all-cause mortality () (Citation32). Similarly, the HOPE (Citation33) and HOPE TOO (Citation34) trials reported an increased incidence of chronic congestive heart failure and acute left heart decompensation in the vitamin E-treated group (400 IU/day) during a follow-up of up to 7 years. Likewise, in a study enrolling 1,923 postmenopausal women with diabetes mellitus type II, supplemental vitamin C (> 300 mg/day) showed a positive association with mortality end-points (Citation35). As well, an increased overall mortality was in certain studies associated with supplementation of β-carotene, vitamin A, and vitamin E, possibly associated with increases in cancer mortality (Citation36). Based on these negative findings, it is clear that the use of antioxidant supplementation for prevention of cardiovascular disease should rather be discouraged. Future trials will have to test whether administration of one (or more) of the several compounds belonging to the family of polyphenols might provide better results. A number of mechanistic trials have demonstrated that these molecules possess antioxidant, anti-inflammatory, antiaggregant, preconditioning-mimetic, and endothelium-stimulating properties that have been associated with protection against a broad variety of diseases including ischemic heart disease, cancer, Alzheimer's disease, diabetes, inflammation, and infection in epidemiological studies (reviewed in (Citation37) and (Citation38)), and prospective, large-scale, randomized intervention trials are awaited.
Figure 3. Kaplan-Meier estimates of the probability of reaching the primary end-point (a composite of fatal and non-fatal myocardial infarction, fatal and non-fatal stroke, and sudden death attributed to coronary heart disease) in patients receiving placebo or three forms of vitamin B supplementation. Figure from (Citation31), with permission.
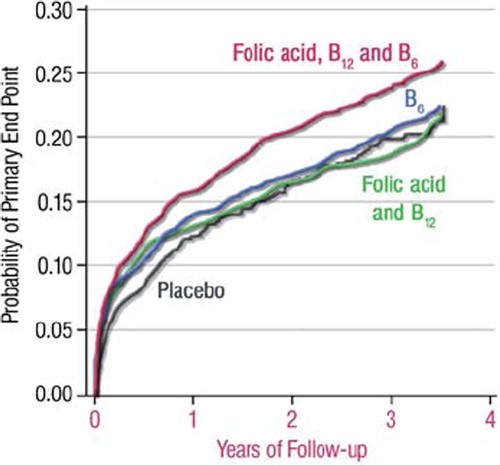
Figure 4. Evidence of a dose-response relationship between vitamin E supplementation and all-cause mortality in a meta-analysis of randomized, controlled trials. (ADCS = Alzheimer's Disease Cooperative Study; AREDS = Age-Related Eye Diseases Study; ATBC = Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group; CHAOS = Cambridge Heart Antioxidant Study; DATATOP = Deprenyl and Tocopherol Antioxidative Therapy of Parkinsonism; GISSI-Prevenzione = Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarcto Miocardio Prevenzione; HOPE = Heart Outcomes Prevention Evaluation; MIN.VIT. AOX = The Geriatrie/MINeraux, VITamines, et AntiOXydants Network; MRC/BHF HPS = Medical Research Council/British Heart Foundation Heart Protection Study; PPP = Primary Prevention Project; PPS = Polyp Prevention Study; REACT = Roche European American Cataract Trial; SPACE = Secondary Prevention with Antioxidants of Cardiovascular disease in Endstage renal disease; SU.VI.MAX = SUpplementation en VItamines et Mineraux AntioXydants; VECAT = Vitamin E, Cataracts, and Age-Related Maculopathy; WAVE = Women's Angiographic Vitamin and Estrogen.) (Adapted from (Citation32), with permission.)
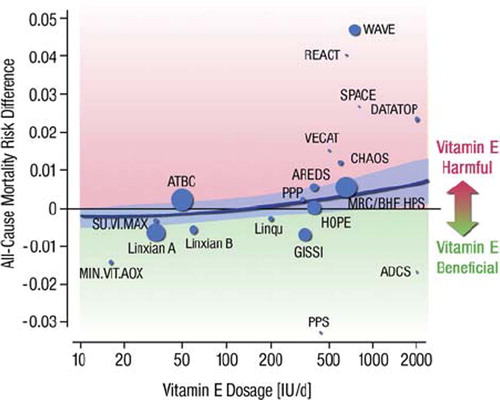
Table II. Large clinical trials and mechanistic studies investigating the impact of antioxidants on clinical end-point and the progression of atherosclerosis.
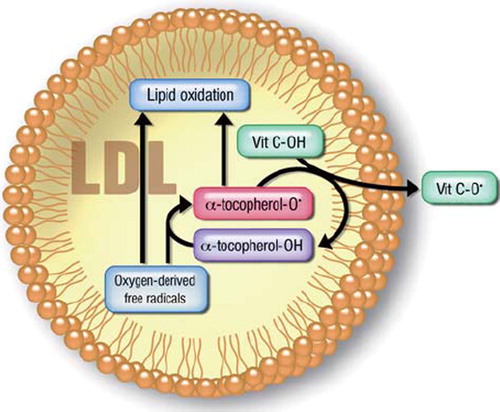
Several reasons have been advocated to motivate the discrepancy between preclinical observations and large clinical intervention trials with antioxidants, and the deceiving results of the latter do not necessarily rule out the role of oxidative stress in cardiovascular pathophysiology. Among these is the possibility that the molecules tested were inadequate in terms of potency: for instance, the rate constant of the reaction between vitamin C and superoxide is 105 lower than that between superoxide and NO, which makes this antioxidant an effective ROS scavenger only at concentrations such as those achieved by i.v. or intra-arterial administration (Citation39). Further, the possibility that their bioavailability could be insufficient after oral administration (Citation40) or that they might have negative effects that compensate for their protective ones (for instance, antioxidants have been shown to inhibit preconditioning (Citation41)) have to be considered. As well, it has been proposed that treatment with vitamin C and vitamin E might result in the formation of so-called vitamin E or C radicals, which possess paradoxical pro-oxidant properties (Citation42) () and might also promote ROS formation (for instance hydroxyl radical (Citation43)). In support of this hypothesis, experimental evidence from both animals and patients suggests that lipid peroxidation does proceed in the vascular wall even in the presence of vitamin E (Citation44).
Figure 5. Scheme of vitamin E-induced lipid peroxidation. Reduced vitamin E (α-tocopherol-OH) may react with oxygen free radicals, thus preventing direct free radical-induced lipid peroxidation. However, this reaction results in the production of a tocopherol radical (α-tocopherol-O˙), which may also cause lipid peroxidation. In the presence of vitamin C (or another reductant), this tocopherol radical may be scavenged. Thus, in the absence of other antioxidants, vitamin E may paradoxically act as an oxidant.
In the case of folic acid, the rationale proposed to justify the several negative findings is that high dosages (in the range of 10 mg/day) of this vitamin would be necessary to achieve cardiovascular effects (Citation20). However, the administration of such high dosages is not deprived of toxicity, and studies have shown that even in this case the cardiovascular benefit, if any (Citation45), is marginal. Further, of all the arguments brought forward to explain the failure of traditional antioxidants in primary and secondary prevention, probably the most potent one is based on the observation that the action of traditional antioxidants lacks site-specificity: vascular cells are extremely complex, and most of the phenomena that lead to the development of endothelial dysfunction and atherosclerosis occur in specific subcellular microenvironments. Even assuming that we had a highly bioavailable, highly potent antioxidant, non-targeted administration of this molecule would indiscriminately interfere with a number of processes, including some protective ones (for instance ischemic preconditioning, a protective phenomenon that is at least in part mediated by transient formation of ROS (Citation41)), and not necessarily, given the high velocity of any reaction involving ROS, with the deleterious ones.
Collectively, the above findings reject the role of ‘simple’ antioxidants as a possible therapeutic strategy. Another important issue is the lack of an accepted, clinically useful, parameter for oxidative stress as an individual marker of whether the patients being treated with an antioxidant have oxidative stress at all and whether the applied antioxidant is indeed working. As stated by Steinberg and Witztum, so far no clinical trials have attempted to assess the efficacy of the antioxidant regime used (Citation46). By analogy, it is as if a cholesterol-lowering drug was being tested for efficacy in preventing CAD events but without measurements of cholesterol as part of the protocol.
The second strategy: administration of exogenous NO
An alternative strategy, still aimed at restoring the cellular redox balance towards NO, consists of the exogenous supplementation of this free radical via administration of NO donors such as organic nitrates ( and ).
Since they release NO (or more likely a NO-derived compound (Citation47)) directly to the endothelium and the vascular media, organic nitrates are used clinically to compensate for the coronary artery constriction that is often associated with atherosclerosis and endothelial dysfunction. Expanding this concept, some authors have proposed that nitrates may act as substitutes of the endothelium-derived NO, an hypothesis based on studies reporting decreased progression, or even regression, of atherosclerosis in animals administered isosorbide or pentaerythrityl tetranitrate (Citation48,Citation49). Additionally, nitrates (like endothelial NO) have been proposed to exert antiaggregant effects (Citation50), a property that would obviously prevent (or, in clinical practice, benefit patients presenting with) acute coronary syndromes. In the last 10 years, however, the hypothesis that organic nitrates like nitroglycerin and isosorbide mono- or dinitrate could restore vascular homeostasis has been proved wrong. Research in animals and in humans has definitely shown that, rather than treating it, these drugs actually cause endothelial dysfunction and increase oxidative stress through (at least) three different mechanisms: uncoupling of the mitochondrial respiratory chain (Citation51) (resulting in inefficient ATP production), activation of ROS-producing enzymes (including eNOS uncoupling) (Citation51–53), and direct reaction of the nitrate-derived NO with vascular superoxide to form ONOO−. Recent papers from our group investigate all these phenomena (reviewed in (Citation41,Citation54)): by uncoupling the mitochondrial respiratory chain, nitroglycerin causes a burst of ROS that can be easily observed in isolated mitochondrial preparations. These ROS (or more properly RNS, reactive nitrogen species, for instance ONOO−) would then leave the mitochondrial matrix through the permeability transition pore (also opened by nitroglycerin), entering the cytoplasm where they produce a number of changes, including eNOS uncoupling, activation of further ROS production by membrane oxidases, and direct oxidative damage. Although brief oxidative bursts might have some beneficial effects, traditional nitrates such as nitroglycerin, isosorbide mono- and dinitrate most likely have a neutral impact or even accelerate, rather than inhibit, oxidative stress upon chronic administration. Notably, the existence of organic nitrates deprived of pro-oxidant effects, such as pentaerythrityl tetranitrate, suggests that some forms of external administration of NO might still be a possibility (Citation55). To date, these considerations remain at the preclinical level, and (despite the existence of preliminary evidence suggesting a negative impact of nitrates such as nitroglycerin or isosorbides on cardiovascular morbidity and mortality) a long-awaited large clinical trial testing the prognostic impact of nitrate therapy remains to be conducted.
Intermediate between the first and the second strategy, the administration of exogenous L-arginine has been proposed as a mechanism to recouple eNOS activity and increase NO bioavailability. By increasing cellular levels of L-arginine, one would favor the competition of this substrate with ADMA for the active site of eNOS, decreasing the impact of this endogenous inhibitor of NO synthesis. Although exogenous administration of L-arginine has been associated with an improved endothelial function in a variety of settings (), and with the prevention of nitrate tolerance (Citation56), its impact on cardiovascular prognosis remains to be established.
The third, and most effective, strategy
The efficacy of the first two strategies (removal of ROS via antioxidants or exogenous administration of NO) appears thus to be inadequate for a complex pathophysiology such as that of cardiovascular disease. In contrast, the third approach, i.e. simultaneous stimulation of NO production and inhibition of vascular ROS, appears to be more promising (). Interestingly, this hypothesis followed the reverse path: while the idea of administering antioxidants or NO donors stemmed from the understanding of the role of these mediators in the cardiovascular system, evidence of a prognostic benefit of several classes of drugs that could not entirely be explained by their ‘primary’ effects (cholesterol-lowering, antihypertensive, etc.) preceded mechanistic explanations. Along with statins, a number of other drugs, including angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor (AT)1 blockers, and beta-receptor blockers such as the NO-releasing nebivolol or the antioxidant carvedilol, have all been shown to possess such combined antioxidant properties.
The vascular ‘antioxidant’ action of ACE inhibitors, like their hemodynamic properties, is justified by the inhibition that these drugs exert on the renin-angiotensin axis. Not only does angiotensin II cause vasoconstriction via stimulation of smooth muscle AT1 receptor and by increasing the expression of preproendothelin within endothelial cells and smooth muscle cells (Citation57), but it also promotes recruitment of monocytes and macrophages into the vessel wall and stimulates smooth muscle cells mitogenesis and extracellular matrix formation (Citation58,Citation59).
Beyond their positive impact on all these effects of angiotensin II, ACE inhibitors have important implications in the limitation of angiotensin II-induced vascular oxidative stress. As described in our previous paper, all vascular cell types express membrane NADH and/or NADPH oxidases that are among the most important cellular sources of superoxide anion (Citation8). Importantly, the major stimulus for the activity of these enzymes is indeed angiotensin II, and the activation of the local and/or the systemic renin-angiotensin system is constantly associated with oxidative stress. Animal models of angiotensin II-induced hypertension and of accelerated atherosclerosis demonstrate a relationship between plaque formation, ROS formation, and increased vascular tone (Citation60), and ACE and NADPH oxidase activity is increased in atherosclerotic plaques and in patients with risk factors for coronary artery disease (Citation61).
Further strengthening the implications of ACE inhibition, the ACE also functions as an endothelial kininase II. Inhibition of kininase II leads to increased bioavailability of bradykinin, which, via B2 receptors, induces the release of vasodilator and antioxidant substances such as nitric oxide (NO), endothelium-derived hyperpolarizing factor, and prostacyclin (Citation62). Thus, ACE inhibitors possess effective antioxidant properties that are mediated, on one side, by the inhibition of NAD(P)H oxidases, and, on the other, by the stimulation of the release of NO. This combined strategy results in a much more effective and consistent antioxidant effect. Although it is clearly impossible to determine which of the (antioxidant, antiproliferative, hemodynamic, etc.) properties of ACE inhibition is responsible for their prognostic impact, it is well accepted that therapy with these drugs is associated with reduced mortality and morbidity in patients with cardiovascular risk factors, acute myocardial infarction, and chronic congestive heart failure.
Similar considerations apply to the group of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors, or statins. Statins are a group of lipid-lowering drugs that have been consistently shown to improve the prognosis in patients with coronary or peripheral arterial disease, heart failure, hypercholesterolemia, and several other conditions (Citation63–68). Throughout these studies, it has been proposed since the late 1990s that the benefit of statins extends beyond their cholesterol-lowering effects and includes improvement of endothelial function, stabilization of atherosclerotic plaques, inhibition of oxidative stress and inflammation, and inhibition of thrombogenic responses (for a review see (Citation69)): for instance, therapy with statins is associated with a lower risk for coronary artery disease as compared to patients with comparable serum cholesterol levels treated with placebo (Citation70) and with reduced events as compared to what would be expected based on cholesterol levels alone (Citation71). These data seem to be confirmed by more recent trials comparing statins with equipotent (in terms of cholesterol reduction) treatment with ezetimibe: as compared to statins, cholesterol-lowering with ezetimibe has a much lower impact on endothelial dysfunction (Citation72), and it does not provide additional benefit in terms of progression of intima-media thickness. The importance of the anti-inflammatory properties of statins is well exemplified by the results of several studies which showed a benefit, in terms of mortality and morbidity, in patients with normal cholesterol but evidence of an activated immune system (Citation73). Similarly, in studies like the CARE and the AFCAPS, changes in reactive C protein levels in response to statin therapy were associated with an improved prognosis independently of the effect on cholesterol levels (Citation74,Citation75). Notably, statins increase NO bioavailability by stabilizing eNOS mRNA, by increasing intracellular BH4 bioavailability, by stimulating eNOS activity, and possibly also by reducing ADMA levels (Citation76) (although negative data have also been reported (Citation77)); indeed, the role of NO in the above effects is confirmed by the fact that they can be blocked or prevented by eNOS inhibitors or eNOS knock-out. Further, statins reverse oxidative stress by decreasing the expression and the activity of NADPH oxidase (Citation78), an effect that overlaps with that described above for ACE inhibitors (for a review, see (Citation79)). Like for ACE inhibitors, it would be impossible to quantify the relative importance of these properties versus their lipid-lowering effects towards the observed reduction in mortality and morbidity in the clinical setting. However, the importance of the biological properties of these drugs is not only compatible with the pathophysiology of cardiovascular disease, but is also in line with the findings of a number of clinical trials showing decreased progression of intima-media thickness, improved endothelial function, and improved outcome after treatment with these drugs (partially listed in ; for reviews, see (Citation80–82)).
Table III. A short list of studies demonstrating a benefit of angiotensin inhibition or statin therapy on intima-media thickness (IMT) and of trials showing blood pressure or cholesterol-independent effects on cardiovascular outcomes. Notably, an effect on IMT has also been shown for calcium channel inhibitors (Citation142).
Conclusions
The observation that, despite preclinical evidence, the administration of ROS scavengers does not modify cardiovascular outcomes brings us to admit that our understanding of cardiovascular pathophysiology is still far from being complete. Different targets for treatment and different treatment modalities need to be developed, and clinical evidence of their efficacy needs to be provided before these interventions are implemented.
Based on the above considerations, therapy with direct antioxidant drugs or with NO donors might have been a naive attempt in a complex pathophysiology. Oxidative stress is an attractive target for novel therapies, as it represents the common pathway through which different risk factors exert their deleterious impact on the cardiovascular system. Several issues, however, still remain. Mechanistic studies are necessary to characterize better the sequence of events leading from ROS to vascular damage, and whether there are particular components of this sequence that can be specifically targeted pharmacologically. We are also still looking for the perfect marker for oxidative stress, helping us to identify people who are subjected to oxidative stress and who most likely benefit from treatment with antioxidants.
Drugs that are specifically aimed at these processes need to be developed in order to avoid a blind, untargeted (and ineffective) approach to ROS damage. As well, it is necessary to characterize, both in terms of clinical implications and mechanisms, the ‘pleiotropic’ effects of drugs such as ACE inhibitors and statins in order to exploit new classes of medications. For instance, a number of studies show that statins, even when administered in dosages that have no impact on lipids, have potent anti-ischemic properties. However seen, therapy with antioxidants is still in its infancy.
Declaration of interest: The authors state no conflict of interest and have received no payment in preparation of this manuscript.
References
- Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation. 2001;104:2673–8.
- Lippincott MF, Carlow A, Desai A, Blum A, Rodrigo M, Patibandla S, . Relation of endothelial function to cardiovascular risk in women with sedentary occupations and without known cardiovascular disease. Am J Cardiol. 2008; 102:348–52.
- Fichtlscherer S, Breuer S, Zeiher AM. Prognostic value of systemic endothelial dysfunction in patients with acute coronary syndromes: further evidence for the existence of the ‘vulnerable’ patient. Circulation. 2004;110:1926–32.
- Heitzer T, Baldus S, von Kodolitsch Y, Rudolph V, Meinertz T. Systemic endothelial dysfunction as an early predictor of adverse outcome in heart failure. Arterioscler Thromb Vasc Biol. 2005;25:1174–9.
- Akcakoyun M, Kargin R, Tanalp AC, Pala S, Ozveren O, Akcay M, . Predictive value of noninvasively determined endothelial dysfunction for long-term cardiovascular events and restenosis in patients undergoing coronary stent implantation: a prospective study. Coron Artery Dis. 2008;19:337–43.
- Thanyasiri P, Kathir K, Celermajer DS, Adams MR. Endothelial dysfunction and restenosis following percutaneous coronary intervention. Int J Cardiol. 2007;119:362–7.
- Kitta Y, Nakamura T, Kodama Y, Takano H, Umetani K, Fujioka D, . Endothelial vasomotor dysfunction in the brachial artery is associated with late in-stent coronary restenosis. J Am Coll Cardiol. 2005;46:648–55.
- Munzel T, Sinning C, Post F, Warnholtz A, Schulz E. Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction. Ann Med. 2008;40:180–96.
- Herrmann J, Lerman A. The endothelium—the cardiovascular health barometer. Herz. 2008;33:343–53.
- Vita JA, Keaney JF Jr. Endothelial function: a barometer for cardiovascular risk? Circulation. 2002;106:640–2.
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271(5 Pt 1):C1424–37.
- Munzel T, Heitzer T, Harrison DG. The physiology and pathophysiology of the nitric oxide/superoxide system. Herz. 1997;22:158–72.
- Boulden BM, Widder JD, Allen JC, Smith DA, Al-Baldawi RN, Harrison DG, . Early determinants of H2O2-induced endothelial dysfunction. Free Radic Biol Med. 2006;41:810–7.
- Hink U, Oelze M, Kolb P, Bachschmid M, Zou MH, Daiber A, . Role for peroxynitrite in the inhibition of prostacyclin synthase in nitrate tolerance. J Am Coll Cardiol. 2003;42:1826–34.
- Sydow K, Munzel T. ADMA and oxidative stress. Atheroscler Suppl. 2003;4:41–51.
- Luo Z, Teerlink T, Griendling K, Aslam S, Welch WJ, Wilcox CS. Angiotensin II and NADPH oxidase increase ADMA in vascular smooth muscle cells. Hypertension. 2010;56:498–504.
- Cooke MS, Evans MD, Podmore ID, Herbert KE, Mistry N, Mistry P, . Novel repair action of vitamin C upon in vivo oxidative DNA damage. FEBS Lett. 1998;439:363–7.
- Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J Biol Chem. 2003;278:22546–54.
- Hyndman ME, Verma S, Rosenfeld RJ, Anderson TJ, Parsons HG. Interaction of 5-methyltetrahydrofolate and tetrahydrobiopterin on endothelial function. Am J Physiol Heart Circ Physiol. 2002;282:H2167–72.
- Moat SJ, Clarke ZL, Madhavan AK, Lewis MJ, Lang D. Folic acid reverses endothelial dysfunction induced by inhibition of tetrahydrobiopterin biosynthesis. Eur J Pharmacol. 2006;530:250–8.
- Ye Z, Song H. Antioxidant vitamins intake and the risk of coronary heart disease: meta-analysis of cohort studies. Eur J Cardiovasc Prev Rehabil. 2008;15:26–34.
- Hertog MG, Feskens EJ, Hollman PC, Katan MB, Kromhout D. Dietary antioxidant flavonoids and risk of coronary heart disease: the Zutphen Elderly Study. Lancet. 1993;342:1007–11.
- Mink PJ, Scrafford CG, Barraj LM, Harnack L, Hong CP, Nettleton JA, . Flavonoid intake and cardiovascular disease mortality: a prospective study in postmenopausal women. Am J Clin Nutr. 2007;85:895–909.
- Wright ME, Lawson KA, Weinstein SJ, Pietinen P, Taylor PR, Virtamo J, . Higher baseline serum concentrations of vitamin E are associated with lower total and cause-specific mortality in the Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study. Am J Clin Nutr. 2006; 84:1200–7.
- Loria CM, Klag MJ, Caulfield LE, Whelton PK. Vitamin C status and mortality in US adults. Am J Clin Nutr. 2000; 72:139–45.
- Stephens NG, Parsons A, Schofield PM, Kelly F, Cheeseman K, Mitchinson MJ. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet. 1996;347:781–6.
- Salonen RM, Nyyssonen K, Kaikkonen J, Porkkala-Sarataho E, Voutilainen S, Rissanen TH, . Six-year effect of combined vitamin C and E supplementation on atherosclerotic progression: the Antioxidant Supplementation in Atherosclerosis Prevention (ASAP) Study. Circulation. 2003;107:947–53.
- Qiao YL, Dawsey SM, Kamangar F, Fan JH, Abnet CC, Sun XD, . Total and cancer mortality after supplementation with vitamins and minerals: follow-up of the Linxian General Population Nutrition Intervention Trial. J Natl Cancer Inst. 2009;101:507–18.
- Lee IM, Cook NR, Gaziano JM, Gordon D, Ridker PM, Manson JE, . Vitamin E in the primary prevention of cardiovascular disease and cancer: the Women's Health Study: a randomized controlled trial. JAMA. 2005;294: 56–65.
- Vivekananthan DP, Penn MS, Sapp SK, Hsu A, Topol EJ. Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. Lancet. 2003;361:2017–23.
- Bonaa KH, Njolstad I, Ueland PM, Schirmer H, Tverdal A, Steigen T, . Homocysteine lowering and cardiovascular events after acute myocardial infarction. N Engl J Med. 2006;354:1578–88.
- Miller ER 3rd, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med. 2005;142:37–46.
- Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145–53.
- Lonn E, Bosch J, Yusuf S, Sheridan P, Pogue J, Arnold JM, . Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA. 2005;293:1338–47.
- Lee DH, Folsom AR, Harnack L, Halliwell B, Jacobs DR Jr. Does supplemental vitamin C increase cardiovascular disease risk in women with diabetes? Am J Clin Nutr. 2004;80:1194–200.
- Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA. 2007;297:842–57.
- Michalska M, Gluba A, Mikhailidis DP, Nowak P, Bielecka-Dabrowa A, Rysz J, . The role of polyphenols in cardiovascular disease. Med Sci Monit. 2010;16: RA110–9.
- Hooper L, Kroon PA, Rimm EB, Cohn JS, Harvey I, Le Cornu KA, . Flavonoids, flavonoid-rich foods, and cardiovascular risk: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2008;88:38–50.
- Tomasian D, Keaney JF, Vita JA. Antioxidants and the bioactivity of endothelium-derived nitric oxide. Cardiovasc Res. 2000;47:426–35.
- Padayatty SJ, Sun H, Wang Y, Riordan HD, Hewitt SM, Katz A, . Vitamin C pharmacokinetics: implications for oral and intravenous use. Ann Intern Med. 2004;140: 533–7.
- Gori T, Parker JD. Nitrate-induced toxicity and preconditioning: a rationale for reconsidering the use of these drugs. J Am Coll Cardiol. 2008;52:251–4.
- Chen Q, Espey MG, Sun AY, Lee JH, Krishna MC, Shacter E, . Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc Natl Acad Sci U S A. 2007;104:8749–54.
- Hara S, Mizukami H, Mukai T, Kurosaki K, Kuriiwa F, Endo T. Involvement of extracellular ascorbate and iron in hydroxyl radical generation in rat striatum in carbon monoxide poisoning. Toxicology. 2009;264:69–73.
- Witztum JL, Steinberg D. Role of oxidized low density lipoprotein in atherogenesis. J Clin Invest. 1991;88:1785–92.
- Zoungas S, McGrath BP, Branley P, Kerr PG, Muske C, Wolfe R, . Cardiovascular morbidity and mortality in the Atherosclerosis and Folic Acid Supplementation Trial (ASFAST) in chronic renal failure: a multicenter, randomized, controlled trial. J Am Coll Cardiol. 2006;47: 1108–16.
- Witztum JL, Steinberg D. The oxidative modification hypothesis of atherosclerosis: does it hold for humans? Trends Cardiovasc Med. 2001;11:93–102.
- Kleschyov AL, Oelze M, Daiber A, Huang Y, Mollnau H, Schulz E, . Does nitric oxide mediate the vasodilator activity of nitroglycerin? Circ Res. 2003;93:e104–12.
- Kojda G, Noack E. Effects of pentaerythrityl-tetranitrate and isosorbide-5-mononitrate in experimental atherosclerosis. Agents Actions Suppl. 1995;45:201–6.
- Kojda G, Stein D, Kottenberg E, Schnaith EM, Noack E. In vivo effects of pentaerythrityl-tetranitrate and isosorbide-5-mononitrate on the development of atherosclerosis and endothelial dysfunction in cholesterol-fed rabbits. J Cardiovasc Pharmacol. 1995;25:763–73.
- Chirkov YY, Chirkova LP, Horowitz JD. Nitroglycerin tolerance at the platelet level in patients with angina pectoris. Am J Cardiol. 1997;80:128–31.
- Wenzel P, Mollnau H, Oelze M, Schulz E, Wickramanayake JM, Muller J, . First evidence for a crosstalk between mitochondrial and NADPH oxidase–derived reactive oxygen species in nitroglycerin-triggered vascular dysfunction. Antioxid Redox Signal. 2008;10:1435–47.
- Gori T, Burstein JM, Ahmed S, Miner SE, Al-Hesayen A, Kelly S, . Folic acid prevents nitroglycerin-induced nitric oxide synthase dysfunction and nitrate tolerance: a human in vivo study. Circulation. 2001;104:1119–23.
- Gori T, Mak SS, Kelly S, Parker JD. Evidence supporting abnormalities in nitric oxide synthase function induced by nitroglycerin in humans. J Am Coll Cardiol. 2001;38: 1096–101.
- Munzel T, Daiber A, Mulsch A. Explaining the phenomenon of nitrate tolerance. Circ Res. 2005;97:618–28.
- Gori T, Daiber A. Non-hemodynamic effects of organic nitrates and the distinctive characteristics of pentaerithrityl tetranitrate. Am J Cardiovasc Drugs. 2009;9:7–15.
- Parker JO, Parker JD, Caldwell RW, Farrell B, Kaesemeyer WH. The effect of supplemental L-arginine on tolerance development during continuous transdermal nitroglycerin therapy. J Am Coll Cardiol. 2002;39:1199–203.
- Wong PC, Hart SD, Duncia JV, Timmermans PB. Nonpeptide angiotensin II receptor antagonists. Studies with DuP 753 and EXP3174 in dogs. Eur J Pharmacol. 1991;202: 323–30.
- Tummala PE, Chen XL, Sundell CL, Laursen JB, Hammes CP, Alexander RW, . Angiotensin II induces vascular cell adhesion molecule-1 expression in rat vasculature: A potential link between the renin-angiotensin system and atherosclerosis. Circulation. 1999;100:1223–9.
- Chen XL, Tummala PE, Olbrych MT, Alexander RW, Medford RM. Angiotensin II induces monocyte chemoattractant protein-1 gene expression in rat vascular smooth muscle cells. Circ Res. 1998;83:952–9.
- Warnholtz A, Nickenig G, Schulz E, Macharzina R, Brasen JH, Skatchkov M, . Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: evidence for involvement of the renin-angiotensin system. Circulation. 1999;99:2027–33.
- Guzik TJ, West NE, Black E, McDonald D, Ratnatunga C, Pillai R, . Vascular superoxide production by NAD(P)H oxidase: association with endothelial dysfunction and clinical risk factors. Circ Res. 2000;86:E85–90.
- Mombouli JV, Vanhoutte PM. Kinins and the vascular actions of converting enzyme inhibitors. Curr Opin Nephrol Hypertens. 1994;3:481–4.
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. 1994;344:1383–9.
- Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, . The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335:1001–9.
- Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med. 1998;339:1349–57.
- West of Scotland Coronary Prevention Study: implications for clinical practice. The WOSCOPS Study Group. Eur Heart J. 1996;17:163–4.
- Downs JR, Clearfield M, Weis S, Whitney E, Shapiro DR, Beere PA, . Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279:1615–22.
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7–22.
- Liao JK. Clinical implications for statin pleiotropy. Curr Opin Lipidol. 2005;16:624–9.
- Weitz-Schmidt G, Welzenbach K, Brinkmann V, Kamata T, Kallen J, Bruns C, . Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat Med. 2001;7:687–92.
- Zhou Q, Liao JK. Pleiotropic effects of statins. Basic research and clinical perspectives. Circ J. 2010;74:818–26.
- Landmesser U, Bahlmann F, Mueller M, Spiekermann S, Kirchhoff N, Schulz S, . Simvastatin versus ezetimibe: pleiotropic and lipid-lowering effects on endothelial function in humans. Circulation. 2005;111:2356–63.
- Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, . Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–207.
- Ridker PM, Rifai N, Clearfield M, Downs JR, Weis SE, Miles JS, . Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med. 2001;344: 1959–65.
- Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation. 1999;100:230–5.
- Lu TM, Ding YA, Leu HB, Yin WH, Sheu WH, Chu KM. Effect of rosuvastatin on plasma levels of asymmetric dimethylarginine in patients with hypercholesterolemia. Am J Cardiol. 2004;94:157–61.
- Valkonen VP, Laakso J, Paiva H, Lehtimaki T, Lakka TA, Isomustajarvi M, . Asymmetrical dimethylarginine (ADMA) and risk of acute coronary events. Does statin treatment influence plasma ADMA levels? Atheroscler Suppl. 2003;4:19–22.
- Wassmann S, Laufs U, Muller K, Konkol C, Ahlbory K, Baumer AT, . Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2002;22:300–5.
- Zhou Q, Liao JK. Statins and cardiovascular diseases: from cholesterol lowering to pleiotropy. Curr Pharm Des. 2009;15:467–78.
- Riccioni G. Statins and carotid intima-media thickness reduction: an up-to-date review. Curr Med Chem. 2009;16: 1799–805.
- Mills EJ, Wu P, Chong G, Ghement I, Singh S, Akl EA, . Efficacy and safety of statin treatment for cardiovascular disease: a network meta-analysis of 170 255 patients from 76 randomized trials. QJM. 2010 Oct 7 (Epub ahead of print).
- Bakris G. Are there effects of renin-angiotensin system antagonists beyond blood pressure control? Am J Cardiol. 2010;105(1 Suppl):21A–29A.
- Cangemi R, Angelico F, Loffredo L, Del Ben M, Pignatelli P, Martini A, . Oxidative stress-mediated arterial dysfunction in patients with metabolic syndrome: Effect of ascorbic acid. Free Radic Biol Med. 2007;43:853–9.
- Plantinga Y, Ghiadoni L, Magagna A, Giannarelli C, Franzoni F, Taddei S, . Supplementation with vitamins C and E improves arterial stiffness and endothelial function in essential hypertensive patients. Am J Hypertens. 2007; 20:392–7.
- Teede HJ, Giannopoulos D, Dalais FS, Hodgson J, McGrath BP. Randomised, controlled, cross-over trial of soy protein with isoflavones on blood pressure and arterial function in hypertensive subjects. J Am Coll Nutr. 2006; 25:533–40.
- Mangoni AA, Sherwood RA, Swift CG, Jackson SH. Folic acid enhances endothelial function and reduces blood pressure in smokers: a randomized controlled trial. J Intern Med. 2002;252:497–503.
- Young JM, Shand BI, McGregor PM, Scott RS, Frampton CM. Comparative effects of enzogenol and vitamin C supplementation versus vitamin C alone on endothelial function and biochemical markers of oxidative stress and inflammation in chronic smokers. Free Radic Res. 2006;40: 85–94.
- Takase B, Etsuda H, Matsushima Y, Ayaori M, Kusano H, Hamabe A, . Effect of chronic oral supplementation with vitamins on the endothelial function in chronic smokers. Angiology. 2004;55:653–60.
- Neunteufl T, Priglinger U, Heher S, Zehetgruber M, Soregi G, Lehr S, . Effects of vitamin E on chronic and acute endothelial dysfunction in smokers. J Am Coll Cardiol. 2000;35:277–83.
- Heiss C, Kleinbongard P, Dejam A, Perre S, Schroeter H, Sies H, . Acute consumption of flavanol-rich cocoa and the reversal of endothelial dysfunction in smokers. J Am Coll Cardiol. 2005;46:1276–83.
- Mangoni AA, Arya R, Ford E, Asonganyi B, Sherwood RA, Ouldred E, . Effects of folic acid supplementation on inflammatory and thrombogenic markers in chronic smokers. A randomised controlled trial. Thromb Res. 2003; 110:13–17.
- Antoniades C, Tousoulis D, Tentolouris C, Toutouza M, Marinou K, Goumas G, . Effects of antioxidant vitamins C and E on endothelial function and thrombosis/fibrinolysis system in smokers. Thromb Haemost. 2003; 89:990–5.
- Thorne S, Mullen MJ, Clarkson P, Donald AE, Deanfield JE. Early endothelial dysfunction in adults at risk from atherosclerosis: different responses to L-arginine. J Am Coll Cardiol. 1998;32:110–6.
- Skyrme-Jones RA, O'Brien RC, Berry KL, Meredith IT. Vitamin E supplementation improves endothelial function in type I diabetes mellitus: a randomized, placebo-controlled study. J Am Coll Cardiol. 2000;36:94–102.
- Anderson RA, Evans LM, Ellis GR, Khan N, Morris K, Jackson SK, . Prolonged deterioration of endothelial dysfunction in response to postprandial lipaemia is attenuated by vitamin C in Type 2 diabetes. Diabet Med. 2006;23: 258–64.
- Economides PA, Khaodhiar L, Caselli A, Caballero AE, Keenan H, Bursell SE, . The effect of vitamin E on endothelial function of micro- and macrocirculation and left ventricular function in type 1 and type 2 diabetic patients. Diabetes. 2005;54:204–11.
- Pena AS, Wiltshire E, Gent R, Hirte C, Couper J. Folic acid improves endothelial function in children and adolescents with type 1 diabetes. J Pediatr. 2004;144:500–4.
- Mangoni AA, Sherwood RA, Asonganyi B, Swift CG, Thomas S, Jackson SH. Short-term oral folic acid supplementation enhances endothelial function in patients with type 2 diabetes. Am J Hypertens. 2005;18(2 Pt 1):220–6.
- van Etten RW, de Koning EJ, Verhaar MC, Gaillard CA, Rabelink TJ. Impaired NO-dependent vasodilation in patients with Type II (non-insulin-dependent) diabetes mellitus is restored by acute administration of folate. Diabetologia. 2002;45:1004–10.
- Hamilton SJ, Chew GT, Watts GF. Coenzyme Q10 improves endothelial dysfunction in statin-treated type 2 diabetic patients. Diabetes Care. 2009;32:810–2.
- Title LM, Ur E, Giddens K, McQueen MJ, Nassar BA. Folic acid improves endothelial dysfunction in type 2 diabetes—an effect independent of homocysteine-lowering. Vasc Med. 2006;11:101–9.
- Regensteiner JG, Popylisen S, Bauer TA, Lindenfeld J, Gill E, Smith S, . Oral L-arginine and vitamins E and C improve endothelial function in women with type 2 diabetes. Vasc Med. 2003;8:169–75.
- Engler MM, Engler MB, Malloy MJ, Chiu EY, Schloetter MC, Paul SM, . Antioxidant vitamins C and E improve endothelial function in children with hyperlipidemia: Endothelial Assessment of Risk from Lipids in Youth (EARLY) Trial. Circulation. 2003;108:1059–63.
- Neunteufl T, Kostner K, Katzenschlager R, Zehetgruber M, Maurer G, Weidinger F. Additional benefit of vitamin E supplementation to simvastatin therapy on vasoreactivity of the brachial artery of hypercholesterolemic men. J Am Coll Cardiol. 1998;32:711–6.
- Verhaar MC, Wever RM, Kastelein JJ, van Dam T, Koomans HA, Rabelink TJ. 5-methyltetrahydrofolate, the active form of folic acid, restores endothelial function in familial hypercholesterolemia. Circulation. 1998;97:237–41.
- Hamabe A, Takase B, Uehata A, Kurita A, Ohsuzu F, Tamai S. Impaired endothelium-dependent vasodilation in the brachial artery in variant angina pectoris and the effect of intravenous administration of vitamin C. Am J Cardiol. 2001;87:1154–9.
- Cangemi R, Loffredo L, Carnevale R, Perri L, Patrizi MP, Sanguigni V, . Early decrease of oxidative stress by atorvastatin in hypercholesterolaemic patients: effect on circulating vitamin E. Eur Heart J. 2008;29:54–62.
- Chan YH, Lau KK, Yiu KH, Li SW, Chan HT, Tam S, . Isoflavone intake in persons at high risk of cardiovascular events: implications for vascular endothelial function and the carotid atherosclerotic burden. Am J Clin Nutr. 2007;86:938–45.
- Title LM, Cummings PM, Giddens K, Genest JJ Jr, Nassar BA. Effect of folic acid and antioxidant vitamins on endothelial dysfunction in patients with coronary artery disease. J Am Coll Cardiol. 2000;36:758–65.
- Moens AL, Claeys MJ, Wuyts FL, Goovaerts I, Van Hertbruggen E, Wendelen LC, . Effect of folic acid on endothelial function following acute myocardial infarction. Am J Cardiol. 2007;99:476–81.
- Doshi S, McDowell I, Moat S, Lewis M, Goodfellow J. Folate improves endothelial function in patients with coronary heart disease. Clin Chem Lab Med. 2003;41: 1505–12.
- Doshi SN, McDowell IF, Moat SJ, Payne N, Durrant HJ, Lewis MJ, . Folic acid improves endothelial function in coronary artery disease via mechanisms largely independent of homocysteine lowering. Circulation. 2002;105:22–6.
- Stanger O, Semmelrock HJ, Wonisch W, Bos U, Pabst E, Wascher TC. Effects of folate treatment and homocysteine lowering on resistance vessel reactivity in atherosclerotic subjects. J Pharmacol Exp Ther. 2002;303:158–62.
- Heiss C, Jahn S, Taylor M, Real WM, Angeli FS, Wong ML, . Improvement of endothelial function with dietary flavanols is associated with mobilization of circulating angiogenic cells in patients with coronary artery disease. J Am Coll Cardiol. 2010;56:218–24.
- Tousoulis D, Antoniades C, Vassiliadou C, Toutouza M, Pitsavos C, Tentolouris C, . Effects of combined administration of low dose atorvastatin and vitamin E on inflammatory markers and endothelial function in patients with heart failure. Eur J Heart Fail. 2005;7:1126–32.
- Ellis GR, Anderson RA, Chirkov YY, Morris-Thurgood J, Jackson SK, Lewis MJ, . Acute effects of vitamin C on platelet responsiveness to nitric oxide donors and endothelial function in patients with chronic heart failure. J Cardiovasc Pharmacol. 2001;37:564–70.
- Tousoulis D, Xenakis C, Tentolouris C, Davies G, Antoniades C, Crake T, . Effects of vitamin C on intracoronary L-arginine dependent coronary vasodilatation in patients with stable angina. Heart. 2005;91:1319–23.
- Grebe M, Eisele HJ, Weissmann N, Schaefer C, Tillmanns H, Seeger W, . Antioxidant vitamin C improves endothelial function in obstructive sleep apnea. Am J Respir Crit Care Med. 2006;173:897–901.
- Tam LS, Li EK, Leung VY, Griffith JF, Benzie IF, Lim PL, . Effects of vitamins C and E on oxidative stress markers and endothelial function in patients with systemic lupus erythematosus: a double blind, placebo controlled pilot study. J Rheumatol. 2005;32:275–82.
- Ghiadoni L, Cupisti A, Huang Y, Mattei P, Cardinal H, Favilla S, . Endothelial dysfunction and oxidative stress in chronic renal failure. J Nephrol. 2004;17:512–9.
- Cross JM, Donald AE, Nuttall SL, Deanfield JE, Woolfson RG, Macallister RJ. Vitamin C improves resistance but not conduit artery endothelial function in patients with chronic renal failure. Kidney Int. 2003;63:1433–42.
- Nanayakkara PW, van Guldener C, ter Wee PM, Scheffer PG, van Ittersum FJ, Twisk JW, . Effect of a treatment strategy consisting of pravastatin, vitamin E, and homocysteine lowering on carotid intima-media thickness, endothelial function, and renal function in patients with mild to moderate chronic kidney disease: results from the Anti-Oxidant Therapy in Chronic Renal Insufficiency (ATIC) Study. Arch Intern Med. 2007;167:1262–70.
- Gori T, Saunders L, Ahmed S, Parker JD. Effect of folic acid on nitrate tolerance in healthy volunteers: differences between arterial and venous circulation. J Cardiovasc Pharmacol. 2003;41:185–90.
- Thomas GR, DiFabio JM, Gori T, Parker JD. Once daily therapy with isosorbide-5-mononitrate causes endothelial dysfunction in humans: evidence of a free-radical-mediated mechanism. J Am Coll Cardiol. 2007;49:1289–95.
- Dragoni S, Gori T, Di Stolfo G, Sicuro S, Forconi S, Parker JD. Folic acid does not limit endothelial dysfunction induced by ischemia and reperfusion: a human study. J Cardiovasc Pharmacol. 2005;46:494–7.
- Woodman RJ, Celermajer DE, Thompson PL, Hung J. Folic acid does not improve endothelial function in healthy hyperhomocysteinaemic subjects. Clin Sci (Lond). 2004; 106:353–8.
- Farouque HM, Leung M, Hope SA, Baldi M, Schechter C, Cameron JD, . Acute and chronic effects of flavanol-rich cocoa on vascular function in subjects with coronary artery disease: a randomized double-blind placebo-controlled study. Clin Sci (Lond). 2006;111:71–80.
- Sesso HD, Buring JE, Christen WG, Kurth T, Belanger C, MacFadyen J, . Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians’ Health Study II randomized controlled trial. JAMA. 2008;300:2123–33.
- Hennekens CH, Buring JE, Manson JE, Stampfer M, Rosner B, Cook NR, . Lack of effect of long-term supplementation with beta carotene on the incidence of malignant neoplasms and cardiovascular disease. N Engl J Med. 1996;334:1145–9.
- Omenn GS, Goodman GE, Thornquist MD, Balmes J, Cullen MR, Glass A, . Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N Engl J Med. 1996;334:1150–5.
- Yusuf S, Dagenais G, Pogue J, Bosch J, Sleight P. Vitamin E supplementation and cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:154–60.
- Cook NR, Albert CM, Gaziano JM, Zaharris E, MacFadyen J, Danielson E, . A randomized factorial trial of vitamins C and E and beta carotene in the secondary prevention of cardiovascular events in women: results from the Women's Antioxidant Cardiovascular Study. Arch Intern Med. 2007;167:1610–8.
- Armitage JM, Bowman L, Clarke RJ, Wallendszus K, Bulbulia R, Rahimi K, . Effects of homocysteine-lowering with folic acid plus vitamin B12 vs placebo on mortality and major morbidity in myocardial infarction survivors: a randomized trial. JAMA. 2010;303:2486–94.
- Ebbing M, Bonaa KH, Arnesen E, Ueland PM, Nordrehaug JE, Rasmussen K, . Combined analyses and extended follow-up of two randomized controlled homocysteine-lowering B-vitamin trials. J Intern Med. 2010;268:367–82.
- Ebbing M, Bleie O, Ueland PM, Nordrehaug JE, Nilsen DW, Vollset SE, . Mortality and cardiovascular events in patients treated with homocysteine-lowering B vitamins after coronary angiography: a randomized controlled trial. JAMA. 2008;300:795–804.
- Albert CM, Cook NR, Gaziano JM, Zaharris E, MacFadyen J, Danielson E, . Effect of folic acid and B vitamins on risk of cardiovascular events and total mortality among women at high risk for cardiovascular disease: a randomized trial. JAMA. 2008;299:2027–36.
- Brown BG, Zhao XQ, Chait A, Fisher LD, Cheung MC, Morse JS, . Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345:1583–92.
- Hodis HN, Mack WJ, LaBree L, Mahrer PR, Sevanian A, Liu CR, . Alpha-tocopherol supplementation in healthy individuals reduces low-density lipoprotein oxidation but not atherosclerosis: the Vitamin E Atherosclerosis Prevention Study (VEAPS). Circulation. 2002;106:1453–9.
- Davidson MH, Maki KC, Dicklin MR, Feinstein SB, Witchger M, Bell M, . Effects of consumption of pomegranate juice on carotid intima-media thickness in men and women at moderate risk for coronary heart disease. Am J Cardiol. 2009;104:936–42.
- Aviram M, Rosenblat M, Gaitini D, Nitecki S, Hoffman A, Dornfeld L, . Pomegranate juice consumption for 3 years by patients with carotid artery stenosis reduces common carotid intima-media thickness, blood pressure and LDL oxidation. Clin Nutr. 2004;23:423–33.
- Waters DD, Alderman EL, Hsia J, Howard BV, Cobb FR, Rogers WJ, . Effects of hormone replacement therapy and antioxidant vitamin supplements on coronary atherosclerosis in postmenopausal women: a randomized controlled trial. JAMA. 2002;288:2432–40.
- Wang JG, Staessen JA, Li Y, Van Bortel LM, Nawrot T, Fagard R, . Carotid intima-media thickness and antihypertensive treatment: a meta-analysis of randomized controlled trials. Stroke. 2006;37:1933–40.
- Napoli C, Bruzzese G, Ignarro LJ, Crimi E, de Nigris F, Williams-Ignarro S, . Long-term treatment with sulfhydryl angiotensin-converting enzyme inhibition reduces carotid intima-media thickening and improves the nitric oxide/oxidative stress pathways in newly diagnosed patients with mild to moderate primary hypertension. Am Heart J. 2008;156:1154.e1–8.
- Hosomi N, Mizushige K, Ohyama H, Takahashi T, Kitadai M, Hatanaka Y, . Angiotensin-converting enzyme inhibition with enalapril slows progressive intima-media thickening of the common carotid artery in patients with non-insulin-dependent diabetes mellitus. Stroke. 2001;32:1539–45.
- Ono H, Minatoguchi S, Watanabe K, Yamada Y, Mizukusa T, Kawasaki H, . Candesartan decreases carotid intima-media thickness by enhancing nitric oxide and decreasing oxidative stress in patients with hypertension. Hypertens Res. 2008;31:271–9.
- Zanchetti A, Crepaldi G, Bond MG, Gallus G, Veglia F, Mancia G, . Different effects of antihypertensive regimens based on fosinopril or hydrochlorothiazide with or without lipid lowering by pravastatin on progression of asymptomatic carotid atherosclerosis: principal results of PHYLLIS—a randomized double-blind trial. Stroke. 2004; 35:2807–12.