Abstract
Cardiovascular diseases (CVD) and infectious diseases represent the two most important causes of death in patients with chronic kidney disease (CKD). The traditional risk factors of CVD do not appear to account sufficiently for the increased risk of CVD in patients with CKD, and vitamin D deficiency appears to be an important non-traditional, and potentially modifiable, CVD risk factor in this patient population. 25-Hydroxyvitamin D (25(OH)D) is converted to its biologically active form, 1,25-dihydroxyvitamin D (1,25(OH)2D), by the enzyme 1α-hydroxylase in the kidneys. The recent discovery that many extrarenal tissues also possess both the 1α-hydroxylase enzyme and the vitamin D receptors has provided new insights into the important physiologic autocrine and paracrine roles of vitamin D in various tissues and organs that are mainly dependent on the availability of 25(OH)D from the circulating plasma. Accordingly, the present review focuses on the rapidly expanding body of clinical and experimental evidence that supports a strong association between 25(OH)D deficiency/insufficiency and the risk of adverse CVD outcomes and infectious diseases as well as on the non-calcemic autocrine and paracrine actions of vitamin D both in the general population and in patients with CKD.
| Abbreviations | ||
| 1,25(OH)2D | = | 1,25 dihydroxyvitamin D |
| 25(OH)D | = | 25-hydroxyvitamin D |
| AMPs | = | antimicrobial peptides |
| CKD | = | chronic kidney disease |
| CVD | = | cardiovascular disease |
| GFR | = | glomerular filtration rate |
| RAAS | = | renin-angiotensin-aldosterone system |
| TLR | = | toll-like receptor |
| VDR | = | vitamin D receptor |
Key messages
Vitamin D deficiency and insufficiency are very common in people with chronic kidney disease (CKD).
Low serum levels of 25-hydroxy vitamin D have been associated with important co-morbidities like infectious diseases and cardiovascular disease as well as death both in the general population and in patients with CKD.
Large placebo-controlled randomized clinical trials are urgently needed to determine whether vitamin D supplementation could have any potential benefit in reducing future cardiovascular events and mortality risk in people with CKD.
Introduction
Patients with chronic kidney disease (CKD) have a reduced life-span compared to the general population, and cardiovascular disease (CVD) accounts for the majority of deaths (Citation1). Rates of CVD morbidity and mortality increase when the estimated glomerular filtration rate (GFR) falls below 60 mL/min/ 1.73 m2 and rise sharply when the GFR falls below 45 mL/min/1.73 m2 (Citation2). Patients with stage 3 and 4 CKD have at least a 2–3-fold increased risk of future CVD events compared to the general population after adjusting for traditional CVD risk factors (Citation2). Although patients with CKD have a high prevalence of traditional risk factors for CVD, the severity and extent of their CVD complications appear to be disproportionate to the risk factor profile(s) (Citation3). Furthermore, pharmacological interventions aimed primarily at reducing traditional risk factors have significantly decreased the risk of CVD in the general population but do not appear to have a similar cardiovascular benefit in the CKD population (Citation4). It is, therefore, imperative to identify other non-traditional and emerging CVD risk factors that may contribute to the increased risk of CVD in patients with CKD such as chronic inflammation, enhanced oxidative stress, insulin resistance, and abnormalities in mineral metabolism like vitamin D deficiency.
To date, the experimental and clinical evidence that supports a strong link between vitamin D insufficiency or deficiency and the risk of CVD and infectious diseases, both in the general population and in patients with CKD, is rapidly accumulating (Citation5–8) and will constitute the main focus of this review, as these two co-morbidities represent the main causes of death in patients with CKD (Citation9,Citation10)
Sources of vitamin D
Vitamin D refers to two biologically inert precursors or prohormones: vitamin D2 (ergocalciferol) and vitamin D3 (cholecalciferol) (Citation11). Vitamin D3 is the naturally occurring form in man, although vitamin D2 has been widely used as the oral supplement (Citation11). The potential differences in the impact on clinical outcomes between the two forms of vitamin D and their respective metabolites are poorly known (Citation12,Citation13). Both forms of vitamin D can undergo hydroxylation to their respective 25-hydroxyvitamin D (25(OH)D) and 1,25-dihydroxyvitamin D (1,25(OH)2D) derivatives. Humans derive vitamin D mostly from the exposure to sunlight and, to a lesser extent, from the diet and dietary supplements (Citation14–17). During the exposure to sunlight, ultraviolet B radiation penetrates skin where it is absorbed by 7-dehydrocholesterol that is present in the plasma membranes of epidermal keratinocytes and dermal fibroblasts (Citation17,Citation18), resulting in the formation of pre-vitamin D3. Once formed, the pre-vitamin D3 rapidly undergoes transformation to vitamin D3 (cholecalciferol) (Citation17,Citation18). Certain foods (e.g. oily fish) also contain vitamin D2 and vitamin D3. Nonetheless, vitamin D insufficiency or deficiency persists in most of the world, including North America and Europe, possibly due to nutritional deficits, avoidance of sunlight exposure, and use of sunscreens (Citation16–18). However, a myriad of other risk factors and important co-morbidities may also contribute to vitamin D deficiency/insufficiency such as advanced age, dark skin tone, living in northern latitudes, being institutionalized, malabsorption syndromes, obesity, liver dysfunction, CKD, and use of drugs that may accelerate vitamin D metabolism (e.g. anticonvulsants, ketoconazole, corticosteroids) (Citation16–18).
Vitamin D as a hormone regulating calcium and bone metabolism
Dietary vitamin D is incorporated into chylomicrons and transported by the lymphatic system into the venous circulation (Citation15,Citation18). Vitamin D in the circulation is bound to the vitamin D-binding protein and transported to the liver, where it is converted by the 25-hydroxylase enzyme to 25(OH)D (Citation16–18). This form of vitamin D is biologically inactive and must be converted, largely in the kidneys, by 1α-hydroxylase to its active form 1,25(OH)2D (as schematically reported in ) (Citation16–18). Hence, the vitamin D hormonal system consists of multiple forms, ranging from cutaneous precursors or dietary components to the most active metabolite, 1,25(OH)2D, which acts upon the target organ receptors to maintain calcium homeostasis and bone health. The best indicator of the overall vitamin D storage or status is, however, the serum concentration of 25(OH)D, which is the precursor form of the biologically active vitamin D and is commonly measured in non-CKD individuals but is much less often measured in patients with CKD (Citation16–19).
Figure 1. Vitamin D metabolism.
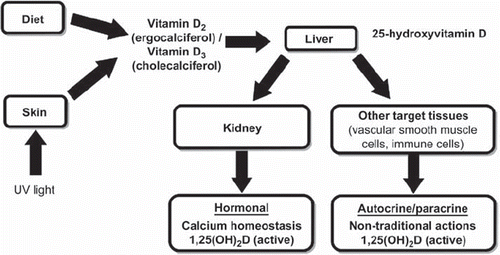
Renal 1α-hydroxylase activity is tightly regulated, in keeping with the potent activity of its product on calcium homeostasis (Citation16–18). In fact, the renal production of 1,25(OH)2D is tightly regulated by serum calcium, phosphorus, parathyroid hormone (PTH), fibroblast growth factor-23, and other factors (Citation16–18). The 1,25(OH)2D enters the circulation and is transported to the small intestine where it enhances intestinal calcium absorption. The 1,25(OH)2D also interacts with its nuclear vitamin D receptor (VDR) on bone pre-osteoblasts, resulting in their transformation into mature osteoclasts. Mature osteoclasts release calcium and phosphorus from the bone, maintaining the appropriate levels of these two minerals in the plasma (Citation16–18). Thus, the endocrine effects of vitamin D appear to be strictly dependent on the renal production of 1,25(OH)2D. When the GFR falls to 30 mL/min/1.73 m2, the renal reserve of 1α-hydroxylase becomes inadequate, resulting in reduced serum concentrations of 1,25(OH)2D (Citation17) and, consequently, decreased serum calcium levels; secondary hyperparathyroidism ensues. The compensatory increase in serum levels of PTH maintains serum calcium by enhancing osteoclastic activity (Citation18). The deficiency of 1,25(OH)2D also reduces osteoblast function (Citation18). Hence, the deficiency of 1,25(OH)2D in combination with an enhanced PTH-driven osteoclastic activity results in the classic osteitis fibrosa cystica. Patients with advanced CKD often require the supplementation of 1,25(OH)2D or its analogs to maintain calcium homeostasis and suppress PTH levels in order to avoid bone disease (Citation16,Citation17). Given the increasing recognition that 25(OH)D can be also hydroxylated outside of the kidneys, including at the level of the parathyroid glands, several small reports suggest that native vitamin D3 may be effective at suppressing serum PTH levels in patients with stage 3 CKD, although it appears to be only marginally effective in those with stage 4 CKD (Citation20–22). A post-hoc analysis of the vitamin D, Calcium, Lyon Study II (DECALYOS II) examined the effect of a 2-year treatment with 800 IU cholecalciferol plus 1,200 mg calcium daily versus placebo in 610 elderly institutionalized women, of whom 322 (52.7%) had an eGFR of less than 60 mL/min/1.73 m2 (Citation22). The proportion of women with ≥30% reduction in serum PTH levels at the end of the study was ∼50% in the active treatment group versus ∼6% for the placebo group (P < 0.001). The effect of the intervention on serum intact PTH levels did not significantly differ according to base-line estimated GFR of participants. Moreover, no significant changes in serum calcium levels were observed among patients on treatment during the 2 years of the study follow-up (Citation22). Thus, the use of active vitamin D analogs appears to be important in replacing the endocrine effects of vitamin D, but it does not appear to address adequately the paracrine and autocrine effects of vitamin D, which will be discussed below.
Prevalence of vitamin D deficiency in patients with chronic kidney disease
At present, there is no consensus about what represents vitamin D deficiency. Most of the considerations for defining the presence of vitamin D deficiency both in the general population and in patients with CKD are based mainly on the effects of vitamin D on calcium homeostasis and bone health. Although there is no consensus on the optimal levels of 25(OH)D, vitamin D deficiency has been defined by most experts as a serum 25(OH)D level of less than 20 ng/mL (<50 nmol/L) for the general population (Citation16). Serum 25(OH)D level is inversely associated with serum PTH level both in patients with CKD (Citation23) and in those without this disease (Citation24–26), until serum 25(OH)D level increases to 30–40 ng/mL (75–100 nmol/L), at which point PTH level plateaus at its nadir (Citation26). Furthermore, the intestinal calcium transport increases by 45%–65% in women when serum 25(OH)D levels are increased from 20 to 32 ng/mL (50–80 nmol/L) (Citation27). Such data provide the basis for defining a relative insufficiency of vitamin D for bone health as serum 25(OH)D levels of 21–29 ng/mL (52–72 nmol/L) in the general adult population (Citation16,Citation27).
A series of investigations have estimated the prevalence and magnitude of 25(OH)D deficiency in the non-dialysis CKD population (Citation28–30). In a cross-sectional study, Levin et al. reported no significant differences in the mean serum 25(OH)D levels across deciles of estimated GFR >30 mL/min/1.73 m2 (Citation28). The prevalence of 25(OH)D deficiency (defined as a serum 25(OH)D level of less than 15 ng/mL) also remained relatively stable until the GFR fell below 30 mL/min/1.73 m2 (Citation28). In another cross-sectional study, among 14,679 US adult participants in the Third National Health and Nutrition Examination Survey (NHANES III), the adjusted mean serum 25(OH)D level was lower in patients with estimated GFR < 30 mL/min/1.73 m2 compared to those with normal kidney function (24.6 versus 29.3 ng/mL; P < 0.001) (Citation29). A cross-sectional study by LaClair et al. (Citation30) measured serum 25(OH)D levels in approximately 200 patients with a mean estimated GFR of 27 ± 11 mL/min/1.73 m2 (GFR range 6–69 mL/min/1.73 m2). The overall mean serum levels of 25(OH)D were 19 ± 14 ng/mL. Only 29% of the 65 patients with stage 3 CKD and only 17% of the 113 patients with stage 4 CKD had a serum 25(OH)D level above 30 ng/mL. Furthermore, 14% of patients with stage 3 CKD and 26% of those with stage 4 CKD had a severely decreased 25(OH)D level (i.e. <10 ng/mL) (Citation30). These findings were essentially comparable with those reported by Gonzalez et al. who found that, in 43 patients with CKD (estimated GFR range 10–111 mL/min/1.73 m2) and 103 chronic dialysis patients, only 14% and 3% of them, respectively, had a serum 25(OH)D level of 30 ng/mL or greater (Citation23). Collectively, these data indicate that vitamin D deficiency/insufficiency is an extremely frequent condition in patients with CKD, especially in those with an estimated GFR of less than 30 mL/min/1.73 m2.
The strongest evidence, to date, on the magnitude of vitamin D deficiency in chronic dialysis patients comes from a recent report by Wolf and colleagues (Citation8). In that study, the investigators have estimated the prevalence of both 25(OH)D and 1,25(OH)2D deficiency in a large cross-sectional data set of United States patients with CKD at the initiation of hemodialysis. The study sample included 825 US consecutive patients from 569 dialysis centers in 37 states. The overall mean serum levels of 25(OH)D were 21 ± 13 ng/mL. Only 22% of these chronic dialysis patients had a serum 25(OH)D level above 30 ng/mL, whereas 60% of them had levels of 10–30 ng/mL, and the remaining 18% were severely vitamin D-deficient (i.e. 25(OH)D < 10 ng/mL). Compared to men, women were more likely to be severely 25(OH)D-deficient (23% versus 15%; P < 0.01). There were significant variations in serum 25(OH)D levels between races (24 ± 14 ng/mL in Caucasians versus 17 ± 10 ng/mL in African-Americans; P < 0.01). Furthermore, chronic dialysis patients who had diabetes were also more likely to be severely 25(OH)D-deficient compared to their counterparts who did not have diabetes. In the same study, the mean serum level of 1,25(OH)2D was 11 ± 10 pg/mL (Citation8) compared to a median of 44 pg/mL in subjects with estimated GFR > 80 mL/min/ 1.73 m2 as reported in earlier studies (Citation28). In contrast to 25(OH)D levels, serum 1,25(OH)2D levels did not significantly differ between men and women, between Caucasians and African-American individuals, or between patients with and without diabetes. Both serum 25(OH)D and 1,25(OH)2D levels were greater among patients who initiated hemodialysis during summer months compared to those who initiated hemodialysis during winter months (25(OH)D: 25 ± 14 versus 19 ± 11 ng/mL; P < 0.01; 1,25(OH)2D: 13 ± 11 versus 10 ± 9 pg/mL; P < 0.01) (Citation8). Thus, it is conceivable that a part of the variability in serum 25(OH)D levels reported in the literature could be likely due to differences in gender distribution, age, and race/ethnicity among the different study populations as well as to differences in the season of the year that might affect the sunlight exposure.
Overall, the published data support the two following notions: 1) in the general population (as specified in ), serum 25(OH)D levels of 30 ng/mL (75 nmol/L) or greater can be considered to indicate sufficient vitamin D, serum 25(OH)D levels of 21–29 ng/mL (52–72 nmol/L) represent a relative insufficiency of vitamin D, while serum 25(OH)D levels of less than 20 ng/mL (<50 nmol/L) represent an absolute vitamin D deficiency (Citation16,Citation27), and 2) patients with estimated GFR < 30 mL/min/ 1.73 m2 are more likely to have vitamin D deficiency compared to subjects with no documented kidney disease (Citation16,Citation17,Citation28,Citation29). Moreover, patients with advanced CKD are even prone to have a severe vitamin D deficiency, defined as a serum 25(OH)D level less than 10 ng/mL (<25 nmol/L) (Citation30).
Table I. Vitamin D status.
Several plausible mechanisms have been proposed to explain the 25(OH)D deficiency in the CKD population (Citation16,Citation17). Among these, we can remember that: 1) the ingestion of foods rich in vitamin D (e.g. fish, cream, milk, and butter) by patients with CKD is relatively low, because the progression of kidney disease is associated with a spontaneous decrease in dietary intake in general, and phosphorus-restricted diets are often imposed; 2) many CKD patients have limited outdoor physical activities with a reduced exposure to sunlight, and the endogenous synthesis of vitamin D3 in the skin, following exposure to sunlight, is reduced in patients with CKD; 3) the main cause of CKD worldwide is diabetes which, in many cases, is associated with nephrotic-range proteinuria. Therefore, lower values of 25(OHD in patients with diabetic nephropathy is likely, as greater urinary levels of vitamin D metabolites have been found in patients with overt proteinuria (Citation31); and 4) a loss of vitamin D2 or D3 through the dialysis membranes has been reported in uremic patients receiving chronic dialysis (Citation32).
Vitamin D on bone health
The beneficial effects of 25(OH)D levels on bone health have been largely attributed to the endocrine actions of its active metabolite, 1,25(OH)2D, on the intestine, parathyroid glands, and bone (Citation16,Citation33,Citation34). The 25(OH)D, however, appears to exert beneficial effects on the bone that are independent of circulating levels of 1,25(OH)2D. Consistent with this hypothesis is the observation of Van Driel et al., who showed that 1α-hydroxylase was also expressed in human osteoblasts. After incubation with the substrate 25(OH)D, the osteoblasts produced sufficient 1,25(OH)2D to modulate their own osteoblastic activity, resulting in bone mineralization (Citation35). Of note, osteomalacia is a generalized bone disorder typically characterized by an impairment of mineralization, leading to accumulation of unmineralized matrix or osteoid in the skeleton, of which the most common cause is vitamin D deficiency. In addition, Ritter et al. demonstrated that 25(OH)D suppressed PTH synthesis in primary cultures of bovine parathyroid cells by converting 25(OH)D to other 1-hydroxylated metabolites without requiring the conversion to 1,25(OH)2D (Citation36). Hence, these autocrine/paracrine actions implicate a potential role of 25(OH)D in the regulation of both calcium and PTH metabolism, independent of the hormonal effects of 1,25(OH)2D that is synthesized by the kidneys.
Non-calcemic autocrine and paracrine actions of vitamin D
The adverse consequences of vitamin D deficiency on bone health represent only the ‘tip of the iceberg’. The discovery that most tissues in the body possess the VDRs has provided new patho-physiological insights into the broad functions of vitamin D and the non-skeletal consequences of its deficiency (Citation16,Citation17).
The VDRs are ubiquitously distributed in the heart, vasculature, brain, skeletal muscle, pancreas, and immune cells, such as T and B lymphocytes and monocytes (Citation37,Citation38). Although 25(OH)D has a relatively low affinity for the VDR (Citation36), many cells including vascular smooth muscle cells and endothelial cells also express 1α-hydroxylase that converts 25(OH)D to the active form 1,25(OH)2D locally (Citation37,Citation38).
The ability to generate 1,25(OH)2D in various cell types is dependent upon the availability of the precursor molecule 25(OH)D derived from the circulating plasma and the availability of the converting enzyme 1α-hydroxylase. In cultured vascular smooth muscle cells, the production of 1,25(OH)2D increased in a dose-dependent manner with increasing availability of 25(OH)D, reaching a plateau at a substrate concentration of 200 ng/mL (500 nmol/L), with a Km of 25 ng/mL (50 nmol/L) (Citation39). A serum 25(OH)D concentration of 30 ng/mL (75 nmol/L) or greater appears to provide adequate substrate for the 1,25(OH)2D production in various extrarenal tissues (Citation16–18,Citation33,Citation34,Citation38–40). The extrarenally produced 1,25(OH)2D primarily serves autocrine or paracrine cell-specific functions, instead of endocrine functions. Thus, contrary to the kidney, the extrarenal production of 1,25(OH)2D does not contribute to the circulating pool of 1,25(OH)2D (Citation18). The regulation of 1α-hydroxylase at these extrarenal sites is quite different from that of the renal enzyme. In keeping with its autocrine and paracrine functions, the rates of 1,25(OH)2D synthesis and degradation in these tissues are under the control of several local factors, such as cytokines and growth factors that may optimize the intracellular 1,25(OH)2D levels for cell-specific actions (Citation18).
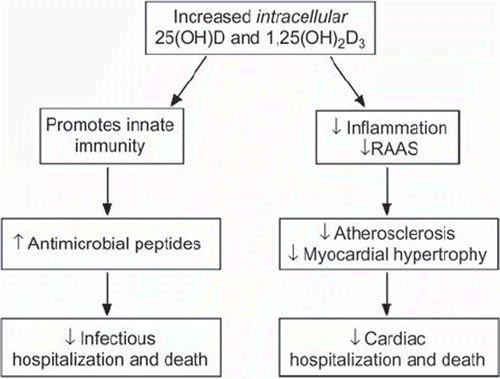
Locally synthesized 1,25(OH)2D in these tissues binds to the nuclear VDR in an autocrine/paracrine manner (Citation18). The binding of 1,25(OH)2D to its nuclear receptor has been found to 1) suppress the renin-angiotensin-aldosterone system (Citation41,Citation42); 2) promote or prevent apoptosis as required for the tissue development (Citation16,Citation18); and 3) modulate the inflammatory response and the immunogenic state of monocyte-macrophages, antigen-presenting cells, dendritic cells, and lymphocytes, which are critical determinants of the susceptibility to infections and autoimmune diseases (Citation16–18,Citation43,Citation44). The ubiquitous availability of both 1α-hydroxylase and VDRs, in addition to the wide scopes of the 25(OH)D effects on extrarenal tissues, supports the possibility that the autocrine/paracrine vitamin D system is primarily designed to fulfill the immediate local needs by a complex and co-ordinated local regulation (Citation16–18), thus circumventing the dependence on the circulating 1,25(OH)2D pool, which is tightly regulated by systemic calcium homeostatic factors. It is important to note that chronic dialysis patients receiving an active vitamin D analog, such as 1,25(OH)2D, for the management of secondary hyperparathyroidism and/or renal osteodystrophy may still be deficient in serum 25(OH)D levels (Citation16,Citation17). Given the important physiologic autocrine/paracrine roles of vitamin D in multiple tissues and organs (Citation45), it is likely that 25(OH)D deficiency would lead to various systemic disorders in patients with CKD. The following discussion will focus on the putative adverse effects of 25(OH)D deficiency on CVD outcomes and infectious diseases. A schema that summarizes the proposed sequence of events in the pathogenesis of CVD and infectious diseases secondary to 25(OH)D deficiency is presented in .
Figure 2. Sequence of events in the pathogenesis of cardiovascular disease and infectious diseases secondary to vitamin D deficiency.
Vitamin D and adverse cardiovascular outcomes
In 1997, Rostand reported that people living at higher latitudes throughout the world were at higher risk of developing hypertension (Citation46). He further hypothesized that this phenomenon might have been due to vitamin D deficiency secondary to decreased sun exposure. This hypothesis was confirmed in a randomized study of ultraviolet (UV) B light exposure in hypertensive patients. In this study participants were randomized to whole-body UV-A (wavelengths of 315–400 nm) or UV-B radiation (3 times/week) for 6 weeks at the end of the winter season. Plasma levels of 25(OH)D increased by 162% and plasma PTH decreased by 15% during UV-B therapy, and UV-B had a beneficial effect on systolic and diastolic blood pressure (−6 and −8 mmHg, respectively; P < 0.001) (Citation47).
More recently, Martins et al. demonstrated a significant association between low serum 25(OH)D levels and various CVD risk factors, including hypertension and diabetes, in a nationally representative sample of US adults (Citation45). Additionally, we found that lower serum 25(OH)D levels were independently associated with increased carotid artery intimal-medial thickness (Citation48) and that type 2 diabetic individuals with clinically manifest CVD had remarkably lower serum 25(OH)D concentrations than their vitamin D-sufficient diabetic counterparts without CVD (Citation49). Serum 25(OH)D levels less than 20 ng/mL (<50 nmol/L) have been also associated with decreased pancreatic beta cell function, while insulin sensitivity was ∼60% higher in individuals with serum 25(OH)D levels of 30 ng/mL (75 nmol/L) or greater compared to those with 25(OH)D levels less than 10 ng/mL (<25 nmol/L) (Citation50). Similarly, in patients with CKD not requiring dialysis, both decreased kidney function and low serum 25(OH)D levels have been independently associated with insulin resistance (Citation29). In addition, the finding of 1α-hydroxylase activity in the pancreatic beta cells raises the possibility of an autocrine control of insulin secretion by 1,25(OH)2D (Citation51). Through a VDR-mediated modulation of calbindin expression, 1,25(OH)2D appears to control the calcium influx in the beta islet cells of the pancreas, which in turn enhances insulin release (Citation51). Thus, these data suggest a potential role for increased serum 25(OH)D levels in the promotion of insulin sensitivity and the prevention of diabetes mellitus. Insulin resistance has been also associated with increased CVD mortality in chronic dialysis patients (Citation52).
Other observations that may have clinical implications in terms of CVD risk assessment in patients with CKD include the strong association between low vitamin D status and abnormalities of the renin-angiotensin-aldosterone system (RAAS). The RAAS plays a key role in the regulation of blood pressure, electrolytes, volume homeostasis, endothelial function, vascular remodeling, and fibrogenesis (Citation53,Citation54). The 1,25(OH)2D acts as a negative endocrine regulator of the RAAS (Citation42). In VDR-null mice, there were marked increases in the intrarenal messenger RNA levels of renin and in the plasma angiotensin II concentrations, associated with hypertension and cardiac hypertrophy. Corroborative studies in wild-type mice demonstrated that the inhibition of 1,25(OH)2D synthesis increased the renin expression by the kidneys (Citation41). Moreover, several epidemiologic studies further suggested a strong association between inadequate sunlight exposure and high plasma renin activity (Citation47,Citation54).
Not only is vitamin D insufficiency/deficiency associated with various CVD risk factors in the US adult population (Citation46), but it has also been associated with an increased risk of incident CVD events in other large population-based cohort studies. Indeed, the number of clinical studies has steadily grown in recent years, with the largest number comprising observational studies showing a significant and independent association between low 25(OH)D levels and a variety of adverse CVD outcomes, including CVD events, CVD mortality, and cerebrovascular mortality (Citation5,Citation6,Citation55–60). For instance, Ginde et al. studied approximately 3,500 older US individuals enrolled in the NHANES III and found that those with vitamin D deficiency had 1.8-fold and 2.4-fold excess all-cause and CVD mortality, respectively, in multivariable-adjusted analyses (Citation60).
A few small, randomized, controlled studies have been published, but these have been largely inconclusive (Citation61). Thus, although low vitamin D status seems to be associated with an increased risk of incident CVD, it remains uncertain whether vitamin D supplementation may positively affect clinical CVD outcomes.
Low serum 25(OH)D levels may also be a contributing factor in the pathogenesis of congestive heart failure, as low circulating levels of 25(OH)D have been demonstrated in patients with this disease (Citation53). The exact mechanisms by which 25(OH)D may protect against the development of congestive heart failure and CVD have not been fully delineated. It is, however, known that 1,25(OH)2D is one of the most potent hormones for down-regulating renin expression in the kidneys (Citation41). In addition to increasing blood pressure, the RAAS plays important pathogenic roles in various types of CVD (Citation54). The RAAS directly contributes to coronary ischemic events through atherosclerosis, altered post-infarct remodeling, and reduced fibrinolysis. Conversely, the inhibition of the RAAS may exert anti-atherosclerotic effects both in animal and in human studies (Citation54,Citation62). Furthermore, vascular smooth muscle cells express VDRs and relax in the presence of 1,25(OH)2D (Citation39). London et al. (Citation63) examined the cross-sectional relationship of arterial alterations with circulating levels of 25(OH)D and 1,25(OH)2D in chronic dialysis patients. Multivariate regression analysis revealed that serum levels of 25(OH)D and 1,25(OH)2D were positively associated with brachial artery distensibility and negatively associated with aortic pulse wave velocity after adjusting for several potential confounders. These results suggest that vitamin D deficiency may be associated with atherosclerosis and endothelial dysfunction in chronic dialysis patients (Citation63).
Data on the relationship between vitamin D status and the risk of incident CVD events in patients with advanced CKD are scant. In a retrospective study by Teng et al. (Citation64), chronic dialysis patients who received the 1,25(OH)2D analog paracalcitol were less likely to die from CVD causes after 2 years of follow-up than those who did not receive paracalcitol. This observation further supports the importance of vitamin D for cardiovascular health. In the same study, the 2-year all-cause mortality rates were 13.8/100 patient-years among patients who received paracalcitol compared to 28.6/100 patient-years among those who did not (P < 0.001). After adjusting for potential confounding factors, the 2-year survival advantage among chronic dialysis patients who received paracalcitol was still 20% lower (hazard ratio for all-cause mortality 0.80; 95% confidence interval 0.76–0.83) (Citation64). Recently, lower serum levels of 25(OH)D in incident chronic dialysis patients were also shown to be associated with an increased risk of all-cause and CVD mortality during a short-term follow-up of 3 months (Citation8). Collectively, these data suggest a potential cardioprotective effect of vitamin D supplementation in patients with advanced CKD. However, no randomized controlled clinical trials have been performed to determine the cardiovascular or survival benefits of treating vitamin D-deficient CKD patients with 25(OH)D, activated vitamin D, or both. More clarity on the potential benefit of vitamin D replacement on CVD outcomes will hopefully come from the recently initiated Vitamin D and Omega-3 Trial (VITAL), a 2 × 2 factorial study of vitamin D and fish oil that is funded by the National Institutes of Health (www.vitalstudy.org; accessed on 1 May 2010). In this study, 20,000 older US men and women will be randomized to daily dietary supplements of vitamin D (about 2,000 IU) or placebo and followed-up for the occurrence of CVD events and other pathologic conditions, including hypertension.
Vitamin D and infectious diseases
In the present review, we postulated that 25(OH)D deficiency also predisposes to infectious diseases through the impairment of the autocrine and paracrine vitamin D system. Recent insights into the functions of 1,25(OH)2D as an immunomodulator have illuminated a large body of previously unexplained associations between alterations in vitamin D and infections (Citation65,Citation66). For instance, low serum 25(OH)D concentrations have been associated with an increased susceptibility to mycobacterial infections (Citation67,Citation68), and recurrent infections are commonly seen among vitamin D-deficient rickets (Citation16–18). In addition, subtle changes in the VDR function, as the result of the expression of certain VDR alleles, can affect the susceptibility or resistance to mycobacterial or viral infections in humans (Citation18). The molecular basis for the association between vitamin D deficiency and impaired innate immunity is discussed as follows.
An essential element of the innate immune response is the capacity of the body to recognize microbial invasion and stimulate the production of a family of proteins called antimicrobial peptides (AMPs) that kill bacteria, viruses, fungi, protozoa, and other microbes (Citation67–70). In humans, AMPs are synthesized and secreted in large amounts in tissues that are usually exposed to environmental microbes, such as the skin and mucosal epithelia, in order to elicit an early defense against infections (Citation67–70). Some AMPs are expressed constitutively, while others are expressed only in response to stimuli such as tissue injury or microbial components (Citation68,Citation69). One of the major AMP families in mammals is a group of molecules, classified as cathelicidin peptides (Citation67,Citation69), which are products of the vitamin D autocrine system. Thus, when macrophages, lymphocytes, or monocytes are stimulated through the mammalian toll-like receptors (TLR) by various infectious agents, the expression of 1α-hydroxylase and VDRs are both up-regulated (Citation18,Citation67). As discussed above, a sufficient substrate concentration, however, is required for the conversion of 25(OH)D to its biologically active form 1,25(OH)2D. Once formed, 1,25(OH)2D is translocated into the nucleus, where it binds to its nuclear receptor and then induces the transcription and production of cathelicidin peptides (Citation66,Citation67). The human cathelicidin peptides are chemotactants for neutrophils, monocytes, mast cells, and T cells (Citation68,Citation69).
The importance of the vitamin D autocrine system in ‘host’ defense can be illustrated by the clinical study of Liu and colleagues on cathelicidin (Citation68). These investigators observed that serum 25(OH)D levels in African-Americans were significantly lower than those in Caucasian individuals. Accordingly, the TLR 2/1 activation and subsequent cathelicidin expression in macrophages were lower in the presence of serum obtained from African-Americans than that obtained from Caucasians. Strikingly, in-vitro supplementation of the African-American serum with 25(OH)D to a physiologic range restored the TLR 2/1 induction of cathelicidin expression in macrophages (Citation68).
Besides the enhancement of cathelicidin production, there are a number of other potential mechanisms by which the autocrine/paracrine vitamin D system can regulate the innate immunity. The VDRs are constitutively expressed in monocyte-macrophages as well as B and T lymphocytes (Citation71). Interactions between the locally produced 1,25(OH)2D and VDRs in these immune cells may produce pro-differentiating effects on both monocyte-macrophages and lymphocytes (Citation71). In addition, high local levels of macrophage-derived 1,25(OH)2D induce Th2 helper T lymphocyte-mediated responses to cutaneous antigens, which are integral components of the defense mechanisms against the colonization of infectious agents (Citation44,Citation73). Collectively, these data support a critical role for the autocrine/paracrine vitamin D system in the systemic immune modulation. Defects in this system as a result of vitamin D deficiency are thus expected to lead to an impaired innate immunity and increased risk of infections.
Summary
Low serum levels of 25(OH)D have been associated with important co-morbid conditions like infectious diseases and CVD as well as death both in the general population and in patients with CKD.
The association between low serum levels of 25(OH)D and adverse CVD outcomes appears to be independent of traditional CVD risk factors and other co-morbidities and is supported by biologic plausibility from various experimental studies both in animals and humans. Several plausible mechanisms explain how vitamin D may modify risk for CVD outcomes. Vitamin D regulates the RAAS, suppresses proliferation of vascular cell smooth muscle, improves insulin resistance and endothelial cell-dependent vasodilation, inhibits anticoagulant activity and myocardial cell hypertrophy, and modulates macrophage activity and cytokine generation.
Vitamin D deficiency/insufficiency is easy to screen for and easy to treat with supplementation. Vitamin D deficiency/insufficiency is an extremely common condition in the CKD population. In patients with any stage of CKD, serum 25(OH)D levels should be measured annually. Given the lack of large randomized clinical trials in patients with CKD, however, the current recommendations are to attempt to maintain serum 25(OH)D concentrations above the sufficient level of 30 ng/mL (75 nmol/L). Hence, due to the high and growing prevalence of mild-to- moderate CKD worldwide, the escalating rates of associated CVD morbidity and mortality, and the resulting high costs of the treatment, placebo- controlled randomized clinical trials are urgently needed to determine whether vitamin D supplementation could have any potential benefit in reducing future CVD events and mortality risk in people with CKD.
Declaration of interest: The authors state no conflict of interest and have received no payment in preparation of this manuscript.
References
- Keith DS, Nichols GA, Gullion CM, Brown JB, Smith DH. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med. 2004;164:659–63.
- Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351: 1296–305.
- Weiner DE, Tighiouart H, Elsayed EF, Griffith JL, Salem DN, Levey AS, . The relationship between nontraditional risk factors and outcomes in individuals with stage 3 to 4 CKD. Am J Kidney Dis. 2008;51:212–23.
- Wanner C, Krane V, März W, Olschewski M, Mann JF, Ruf G. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N Engl J Med. 2005;353: 238–48.
- Giovannucci E, Liu Y, Hollis BW, Rimm EB. 25-hydroxyvitamin D and risk of myocardial infarction in men: a prospective study. Arch Intern Med. 2008;168:1174–80.
- Dobnig H, Pilz S, Scharnagl H, Renner W, Seelhorst U, Wellnitz B, . Independent association of low serum 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D levels with all-cause and cardiovascular mortality. Arch Intern Med. 2008;168:1340–9.
- Mehrotra R, Kermah DA, Salusky IB, Wolf MS, Thadhani RI, Chiu YW, . Chronic kidney disease, hypovitaminosis D, and mortality in the United States. Kidney Int. 2009; 76:977–83.
- Wolf M, Shah A, Gutierrez O, Ankers E, Monroy M, Tamez H, . Vitamin D levels and early mortality among incident hemodialysis patients. Kidney Int. 2007;72: 1004–13.
- Eknoyan G, Beck GJ, Cheung AK, Daugirdas JT, Greene T, Kusek JW, . Effect of dialysis dose and membrane flux in maintenance hemodialysis. N Engl J Med. 2002;347: 2010–9.
- Menon V, Sarnak MJ. The epidemiology of chronic kidney disease stages 1 to 4 and cardiovascular disease: a high-risk combination. Am J Kidney Dis. 2005;45:223–32.
- Lensmeyer GL, Wiebe DA, Binkley N, Drezner MK. HPLC method for 25-hydroxyvitamin D measurement: comparison with contemporary assays. Clin Chem. 2006;2:1120–6.
- Armas LAG, Hollis BW, Heaney RP. Vitamin D2 is much less effective than vitamin D3 in humans. J Clin Endocrinol Metab. 2004;89:5387–91.
- Trang HM, Cole DE, Rubin LA, Pierratos A, Siu S, Vieth R. Evidence that vitamin D3 increases serum 25-hydroxyvitamin D more efficiently than does vitamin D2. Am J Clin Nutr. 1998;68:854–8.
- Holick MF. Resurrection of vitamin D deficiency and rickets. J Clin Invest. 2006;116:2062–72.
- De Luca HF. Overview of general physiologic features and functions of vitamin D. Am J Clin Nutr. 2004;80(Suppl 1): S1689–96.
- Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357: 266–81.
- Holick MF. Vitamin D for health and in chronic kidney disease. Semin Dial. 2005;18:266–75.
- Dusso AS, Brown AJ, Slatopolsky E. Vitamin D. Am J Physiol Renal Physiol. 2005;289:F8–F28.
- Gómez-Alonso C, Naves-Díaz ML, Fernández-Martín JL, Díaz-López JB, Fernández-Coto MT, Cannata-Andía JB. Vitamin D status and secondary hyperparathyroidism: the importance of 25-hydroxyvitamin D cut-off levels. Kidney Int. 2003;(Suppl 85):S44–48.
- Al-Aly Z, Qazi RA, Gonzalez EA, Zeringue A, Martin KJ. Changes in serum 25-hydroxyvitamin D and plasma intact PTH levels following treatment with ergocalciferol in patients with CKD. Am J Kidney Dis. 2007;50:59–68.
- Zisman AL, Ghantous W, Schinleber P, Roberts L, Sprague SM. Inhibition of parathyroid hormone: a dose equivalency study of paricalcitol and doxercalciferol. Am J Nephrol. 2005;25:591–5.
- Kooienga L, Fried L, Scragg R, Kendrick J, Smits G, Chonchol M. The effect of combined calcium and vitamin D3 supplementation on serum intact parathyroid hormone in moderate CKD. Am J Kidney Dis. 2009;53:408–16.
- González EA, Sachdeva A, Oliver DA, Martin KJ. Vitamin D insufficiency and deficiency in chronic kidney disease. A single center observational study. Am J Nephrol. 2004;24: 503–10.
- Chapuy MC, Preziosi P, Maamer M, Arnaud S, Galan P, Hercberg S, . Prevalence of vitamin D insufficiency in an adult normal population. Osteoporosis Int. 1997;7:439–43.
- Thomas MK, Lloyd-Jones DM, Thadhani RI, Shaw AC, Deraska DJ, Kitch BT, . Hypovitaminosis D in medical inpatients. N Engl J Med. 1998;338:777–83.
- Dawson-Hughes B, Harris SS, Dallal GE. Plasma calcidiol, season, and serum parathyroid hormone concentrations in healthy elderly men and women. Am J Clin Nutr. 1997;65: 67–71.
- Heaney RP, Dowell MS, Hale CA, Bendich A. Calcium absorption varies within the reference range for serum 25-hydroxyvitamin D. J Am Coll Nutr. 2003;22:142–6.
- Levin A, Bakris GL, Molitch M, Smulders M, Tian J, Williams LA, . Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int. 2007;71:31–8.
- Chonchol M, Scragg R. 25-Hydroxyvitamin D, insulin resistance, and kidney function in the Third National Health and Nutrition Examination Survey. Kidney Int. 2007;71: 134–9.
- LaClair RE, Hellman RN, Karp SL, Kraus M, Ofner S, Li Q, . Prevalence of calcidiol deficiency in CKD: a cross-sectional study across latitudes in the United States. Am J Kidney Dis. 2005;45:1026–33.
- Sato KA, Gray RW, Lemann J Jr. Urinary excretion of 25-hydroxyvitamin D in health and the nephrotic syndrome. J Lab Clin Med. 1982;99:325–30.
- Aloni Y, Shany S, Chaimovitz C. Losses of 25-hydroxyvitamin D in peritoneal fluid: possible mechanism for bone disease in uremic patients treated with chronic ambulatory peritoneal dialysis. Miner Electrolyte Metab. 1983;9:82–6.
- Holick MF. The vitamin D epidemic and its health consequences. J Nutr. 2005;35:S2739–48.
- Bischoff-Ferrari HA, Giovannucci E, Willett WC, Dietrich T, Dawson-Hughes B. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84:18–28.
- van Driel M, Koedam M, Buurman CJ, Roelse M, Weyts F, Chiba H, . Evidence that both 1alpha,25-dihydroxyvitamin D3 and 24-hydroxylated D3 enhance human osteoblast differentiation and mineralization. J Cell Biochem. 2006;99: 922–35.
- Ritter CS, Armbrecht HJ, Slatopolsky E, Brown AJ. 25- Hydroxyvitamin D(3) suppresses PTH synthesis and secretion by bovine parathyroid cells. Kidney Int. 2006;70: 654–9.
- Holick MF. Sunlight and vitamin D for bone health and prevention of autoimmune diseases, cancers, and cardiovascular disease. Am J Clin Nutr. 2004;80 (suppl):S1678–88.
- Stumpf WE, Sar M, Reid FA, Tanaka Y, DeLuca HF. Target cells for 1,25-dihydroxyvitamin D3 in intestinal tract, stomach, kidney, skin, pituitary, and parathyroid. Science. 1979;206:1188–90.
- Somjen D, Weisman Y, Kohen F, Gayer B, Limor R, Sharon O, . 25-hydroxyvitamin D3-1alpha-hydroxylase is expressed in human vascular smooth muscle cells and is up-regulated by parathyroid hormone and estrogenic compounds. Circulation. 2005;111:1666–71.
- Hewison M, Zehnder D, Chakraverty R, Adams JS. Vitamin D and barrier function: a novel role for extra-renal 1α-hydroxylase. Mol Cell Endocrinol. 2004;215:31–8.
- Li YC. Vitamin D regulation of the renin-angiotensin system. J Cell Biochem. 2003;88:327–31.
- Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25- Dihydroxyvitamin D3 is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110:229–38.
- Asaka M, Iida H, Izumino K, Sasayama S. Depressed natural killer cell activity in uremia. Evidence for immunosuppressive factor in uremic sera. Nephron. 1988;49:291–5.
- Hayes CE, Nashold FE, Spach KM, Pedersen LB. The immunological functions of the vitamin D endocrine system. Cell Mol Biol. 2003;49:277–300.
- Martins D, Wolf M, Pan D, Zadshir A, Tareen N, Thadhani R, . Prevalence of cardiovascular risk factors and the serum levels of 25-hydroxyvitamin D in the United States: data from the Third National Health and Nutrition Examination Survey. Arch Intern Med. 2007;167:1159–65.
- Rostand SG. Ultraviolet light may contribute to geographic and racial blood pressure differences. Hypertension. 1997; 30: 150–6.
- Krause R, Buhring M, Hopfenmuller W, Holick MF, Sharma AM. Ultraviolet B and blood pressure. Lancet. 1998;352:709–10.
- Targher G, Bertolini L, Padovani R, Zenari L, Scala L, Cigolini M, . Serum 25-hydroxyvitamin D3 concentrations and carotid artery intima-media thickness among type 2 diabetic patients. Clin Endocrinol (Oxf). 2006;65:593–7.
- Cigolini M, Iagulli MP, Miconi V, Galiotto M, Lombardi S, Targher G. Serum 25-hydroxyvitamin D3 concentrations and prevalence of cardiovascular disease among type 2 diabetic patients. Diabetes Care. 2006;29:722–4.
- Chiu KC, Chu A, Go VL, Saad MF. Hypovitaminosis D is associated with insulin resistance and β cell dysfunction. Am J Clin Nutr. 2004;79:820–5.
- Schwartz GG, Eads D, Rao A, Cramer SD, Willingham MC, Chen TC, . Pancreatic cancer cells express 25-hydroxyvitamin D-1α-hydroxylase and their proliferation is inhibited by the prohormone 25-hydroxyvitamin D3. Carcinogenesis. 2004;25:1015–26.
- Shinohara K, Shoji T, Emoto M, Tahara H, Koyama H, Ishimura E, . Insulin resistance as an independent predictor of cardiovascular mortality in patients with end-stage renal disease. J Am Soc Nephrol. 2002;13:1894–900.
- Zittermann A, Schleithoff SS, Tenderich G, Berthold HK, Körfer R, Stehle P. Low vitamin D status: a contributing factor in the pathogenesis of congestive heart failure. J Am Coll Cardiol. 2003;41:105–12.
- Jacoby DS, Rader DJ. Renin-angiotensin system and athero-thrombotic disease: from genes to treatment. Arch Intern Med. 2003;163:1155–64.
- Wang TJ, Pencina MJ, Booth SL, Jacques PF, Ingelsson E, Lanier K, . Vitamin D deficiency and risk of cardiovascular disease. Circulation. 2008;117:503–11.
- Kilkkinen A, Knekt P, Aro A, Rissanen H, Marniemi J, Heliovaara M, . Vitamin D status and the risk of cardiovascular disease death. Am J Epidemiol. 2009;170:1032–9.
- Pilz S, Dobnig H, Nijpels G, Heine RJ, Stehouwer CD, Snijder MB, . Vitamin D and mortality in older men and women. Clin Endocrinol (Oxf). 2009;71:666–72.
- Kendrick J, Targher G, Smits G, Chonchol M. 25-Hydroxyvitamin D deficiency is independently associated with cardiovascular disease in the Third National Health and Nutrition Examination Survey. Atherosclerosis. 2009;205:255–60.
- Pittas AG, Chung M, Trikalinos T, Mitri J, Brendel M, Patel K, . Systematic review. Vitamin D and cardiometabolic outcomes. Ann Intern Med. 2010;152:307–14.
- Ginde AA, Scragg R, Schwartz RS, Camargo CA Jr. Prospective study of serum 25-hydroxyvitamin d level, cardiovascular disease mortality, and all-cause mortality in older US adults. J Am Geriatr Soc. 2009;57:1595–603.
- Wang L, Manson J, Song Y, Sesso HD. Systematic review. Vitamin D and calcium supplementation in prevention of cardiovascular events. Ann Intern Med. 2010;152:315–23.
- Candido R, Jandeleit-Dahm KA, Cao Z, Nesteroff SP, Burns WC, Twigg SM, . Prevention of accelerated atherosclerosis by angiotensin-converting enzyme inhibition in diabetic apolipoprotein E-deficient mice. Circulation. 2002;106: 246–53.
- London GM, Guérin AP, Verbeke FH, Pannier B, Boutouyrie P, Marchais SJ, . Mineral metabolism and arterial functions in end-stage renal disease: potential role of 25-hydroxyvitamin D deficiency. J Am Soc Nephrol. 2007;8:613–20.
- Teng M, Wolf M, Ofsthun MN, Lazarus JM, Hernán MA, Camargo CA Jr, . Activated injectable vitamin D and hemodialysis survival: a historical cohort study. J Am Soc Nephrol. 2005;16:1115–25.
- Roth DE, Soto G, Arenas F, Bautista CT, Ortiz J, Rodriguez R, . Association between vitamin D receptor gene polymorphisms and response to treatment of pulmonary tuberculosis. J Infect Dis. 2004;190:920–7.
- Wayse V, Yousafzai A, Mogale K, Filteau S. Association of subclinical vitamin D deficiency with severe acute lower respiratory infection in Indian children under 5 yrs. Eur J Clin Nutr. 2004;58:563–7.
- Zasloff M. Fighting infections with vitamin D. Nat Med. 2006;2:388–90.
- Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, . Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770–3.
- Zanetti M. Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol. 2004;75:39–48.
- Zaiou M, Gallo RL. Cathelicidins, essential gene-encoded mammalian antibiotics. J Mol Med. 2002;80:549–61.
- Mathieu C, Adorini L. The coming of age of 1,25- dihydroxyvitamin D3 analogs as immunomodulatory agents. Trends Mol Med. 2002;8:174–9.
- Gorgoni B, Maritano D, Marthyn P, Righi M, Poli V. C/EBPβ gene inactivation causes both impaired and enhanced gene expression and inverse regulation of IL-12 p40 and p35 mRNAs in macrophages. J Immunol. 2002;168:4055–62.
- Lemire JM. Immunomodulatory actions of 1,25-dihydroxyvitamin D3. J Steroid Biochem Mol Biol. 1995;53:599–602.