Abstract
Glucocorticoid therapy is used in the treatment of moderate to severe inflammatory bowel disease (IBD). However, IBD patients display varying degrees of glucocorticoid sensitivity: some respond rapidly to the given treatment, whereas others show no response, or develop steroid therapy-related side-effects. At present, we cannot foresee whether the patient will benefit from the administered glucocorticoids or not. During the past 10 years, numerous attempts have been made to provide the means to identify and predict steroid therapy-sensitive patients in advance. This would be vital to avoid unnecessary glucocorticoid exposure in patients that do not respond to treatment with steroids. Here we provide a concise review of recent developments regarding the molecular basis of glucocorticoid sensitivity in IBD patients and the methods employed to assess it.
| Abbreviations | ||
| CD | = | Crohn's disease |
| DBD | = | DNA-binding domain |
| ELISA | = | enzyme-linked immunosorbent assay |
| FOXP3 | = | forkhead box P3 |
| GC | = | glucocorticoid |
| G-CSF | = | granulocyte colony-stimulating factor |
| GITR | = | glucocorticoid-induced tumour necrosis factor inhibitor |
| GM-CSF | = | granulocyte-macrophage colony-stimulating factor |
| GR | = | glucocorticoid receptor |
| GRE | = | glucocorticoid response element |
| HR | = | hinge region |
| 11β-HSD | = | 11β-hydroxysteroid dehydrogenase |
| Imax | = | maximum inhibition |
| IBD | = | inflammatory bowel disease |
| IFN | = | interferon |
| IL | = | interleukin |
| IP-10 | = | interferon gamma-induced protein 10 |
| Kd | = | dissociation constant |
| LBD | = | ligand-binding domain |
| LPS | = | lipopolysaccharide |
| MDR1 | = | multi-drug resistance 1 |
| mRNA | = | messenger ribonucleic acid |
| NTD | = | N-terminal domain |
| PBMC | = | peripheral blood mononuclear cell |
| PCR | = | polymerase chain reaction |
| RT-PCR | = | reverse transcriptase PCR |
| SNP | = | single nucleotide polymorphism |
| TNF-α | = | tumour necrosis factor α |
| UC | = | ulcerative colitis |
Key messages
Glucocorticoid response is unpredictable and thus a common clinical problem encountered in the treatment of inflammatory bowel disease.
Being able to predict the patients’ response to glucocorticoid therapy would improve the treatment of patients.
Here we review the recent developments regarding the molecular basis of glucocorticoid sensitivity in inflammatory bowel disease (IBD) patients and the methods employed to assess it.
Introduction
Synthetic glucocorticoids are employed in the induction of remission in moderate to severe inflammatory bowel disease (IBD) (Citation1,Citation2). The response rates vary, and around 20%–30% of patients show resistance to the therapeutic actions of the administered steroids (Citation1,Citation3–5). Furthermore, glucocorticoid-related side-effects are common and range from cosmetic to life-threatening (Citation6).
Glucocorticoids employed in the treatment of IBD include parenteral hydrocortisone and methylprednisolone and oral prednisone, prednisolone, and budesonide () (Citation7). Hydrocortisone corresponds to the natural glucocorticoid hormone cortisol, and the other administered steroids are its synthetic analogues. Only a few studies have addressed the optimal glucocorticoid dosage and dose tapering in IBD patients (Citation10,Citation11). At present, the recommended treatment strategy for traditional steroids (prednisone, prednisolone) is 1 mg/kg/day of prednisone equivalents (max 60 mg/day) followed by a tapering period of 12–16 weeks (Citation7). No direct comparisons with conventional steroids (prednisone, prednisolone) regarding the efficacy or appearance of glucocorticoid-related side-effects have been made. In severe disease, however, parenteral treatment is recommended (Citation7). Budesonide, which has low systemic bioavailability (10%) and high affinity for glucocorticoid receptor (GR), has been studied in the treatment of Crohn's disease (CD). A recent Cochrane review, however, stated that budesonide seems to be less effective than traditional steroids in the treatment of CD, specifically if the patient has more extensive colonic involvement or severe disease (Citation12,Citation13).
Table I. Synthetic glucocorticoids employed in the treatment of inflammatory bowel disease, adapted from Rang et al. (Citation8).
In IBD the patient's sensitivity to glucocorticoids cannot be predicted from disease extent or severity, even though colectomy rates are higher in patients with severe colonic disease (Citation14,Citation15), which might imply that heavy inflammatory activity impairs glucocorticoid sensitivity in these patients and leads to a poorer response. In addition, the absorption and protein binding of glucocorticoids do not explain why some patients respond to glucocorticoid therapy and others do not (Citation16,Citation17). At tissue level, the interconversion between active and inert cortisol metabolites by the 11β-hydroxysteroid dehydrogenases (11β-HSDs) 1 and 2 is now considered to be a significant regulator of hormone action (Citation18,Citation19). In IBD-related inflammation the interconversion of cortisol metabolites in the intestine is predominantly from inactive to active form (Citation18,Citation19), and it is unlikely to be directly affected by the therapeutic regimens (Citation18). Glucocorticoids enter the cells mostly via diffusion, but an active mechanism, drug-efflux pump P-glycoprotein 170, exists that transports steroids and other agents out of intestinal epithelial cells and lymphocytes. Previously, the expression of the multi-drug resistance (MDR1) gene that codes for the pump protein was found to be elevated in IBD patients who had undergone bowel resection for failed medical therapy (Citation20). More recently, however, the impact of the MDR1 gene on glucocorticoid sensitivity in IBD patients has been questioned (Citation21). Therefore, variable pharmacokinetics of the administered steroids does not seem to explain the variation in the glucocorticoid responsiveness.
The difference in the interindividual glucocorticoid sensitivity is a clinical problem encountered daily in the treatment of IBD. Progress in the fields of molecular and cell biology has dramatically increased our knowledge of the GR signalling pathway and has enabled us to study the structure, expression, ligand binding, and affinity of the GR. In addition, it is now possible to measure the bioactivity of the administered glucocorticoids with different methods. Here we review the recent studies on glucocorticoid sensitivity in IBD patients.
Glucocorticoid receptor structure
Glucocorticoids enter the cells via diffusion and bind to their cognate receptor GR that resides in the cytoplasm as part of a multi-protein complex. After ligand binding, the protein complex dissociates: the ligand–receptor complex dimerizes and translocates to the nucleus where it affects the transcription of glucocorticoid target genes (Citation22).
GR belongs to the superfamily of nuclear receptors together with receptors for oestrogen, testosterone, and progesterone (Citation23). The GR gene is localized in the long arm of chromosome 5q31-32 (Citation24). It contains nine exons, the first of which is an untranslated region (). The first exon has at least 13 splice variants that do not alter the amino acid sequence of the receptor (). However, these variants have been reported to show different tissue expression profiles and thus might be a significant control mechanism in the transcription of GR () (Citation25,Citation26).
Figure 1. The structure of the glucocorticoid receptor (GR). A: The GR gene consists of at least nine alternative exon 1s (A–J) and eight other exons (Citation2–9). The polymorphisms of the GR are presented at their respective positions in the gene. B: The alternative mRNA transcripts of the GR gene. The 13 different exon 1 splice variants do not alter the amino acid sequence of the gene. For GRα the presence of the different translation initiation sites for A-D3 isoforms is schematically presented. Arg = arginine. C: The full-length GRα, its main functional domains, and the sites of post-translational covalent modifications: S = sumoylation; Ub = ubiquitination.
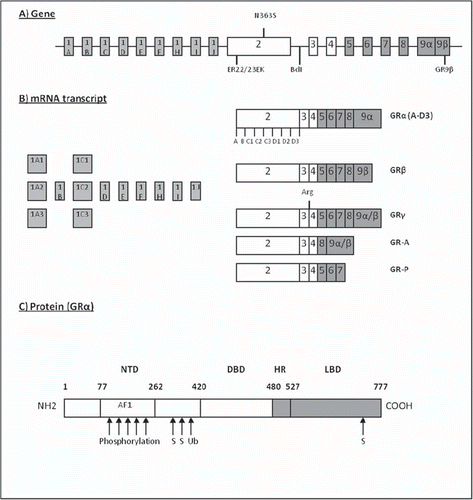
Exon 2 is the site for translation initiation. In 2005, it was reported that eight alternative translation initiation sites exist for human GRα () (Citation27). These GR isoforms differ from their amino-terminal region and have been reported to vary from one another in terms of transactivation capacity from synthetic glucocorticoid response element (GRE)-driven promoter, tissue expression, and target-gene induction profile (Citation27). It has been hypothesized that similar isoforms could also exist for GRβ, which would further increase the capacity of the GR to regulate its signal transduction.
Exons 3 to 9 contain the sequence for DNA- and ligand-binding domains of the GR (Citation28). Receptor isoforms that can affect the glucocorticoid sensitivity arising from this area are the GRα/β isoforms generated by the alternative splicing of the exon 9, GR-A lacking amino acids encoded by exons 5, 6, and 7, and GR-P lacking amino acids from exons 8 and 9 (). Alternative splicing of exon 9 produces the two receptor isoforms GRα and GRβ, of which the former is considered to be the ‘classical’ glucocorticoid receptor of 777 amino acids (Citation28). The latter, GRβ, is 35 amino acids shorter and after its identification was thought to be a dominant negative inhibitor of GRα (Citation29). However, recent research has revealed its potential role as an independent regulator of gene expression, even in the absence of GRα (Citation30). GR-A and GR-P have been identified in glucocorticoid-resistant myeloma patients, but at present it is not clear whether they increase or decrease glucocorticoid sensitivity (Citation31,Citation32). Also, a fifth isoform GRγ with an additional arginine between exons 3 and 4 has for example been reported in childhood lymphoblastic leukaemia. This modification decreases the transactivation potential of the receptor by 50% (Citation33–35).
In addition to the several isoforms, a number of single nucleotide polymorphisms (SNPs) have been characterized in the GR gene that can further modulate the glucocorticoid sensitivity of an individual patient. Two polymorphisms, N363S and BclI, have been associated with increased sensitivity to glucocorticoids (Citation36). Instead, ER22/23EK and GR-9β are linked to a relative glucocorticoid resistance (Citation36,Citation37).
Glucocorticoid receptor in inflammatory bowel disease
Since the GR was cloned in the 1980s, it has become evident that even carrying a normal GR gene allele allows for rich variation in the GR characteristics in otherwise healthy individuals. In IBD, alterations in the number and structure of GR that may affect therapeutic response to GCs have been reported (see below).
Altered numbers of glucocorticoid receptors
Studies in patients with asthma and rheumatoid arthritis have suggested that glucocorticoids can down-regulate the expression of the GR in target tissues, which has been considered to be a protective mechanism against long-term exposure to the hormone (Citation38,Citation39). After glucocorticoid treatment, the receptor expression levels return to base-line, but the results regarding the association between GR numbers and glucocorticoid sensitivity have remained contradictory (Citation38,Citation39). In IBD patients, the number of GRs has been evaluated in peripheral blood mononuclear cells (PBMCs) and colonic mucosal cells (). However, the results have been confusing, as the GR numbers in IBD patients have been reported to be lower, similar, or higher than in the control patients (Citation40–42) (). Therefore, no consensus can be drawn from these findings. This might reflect different study designs applied in these studies, different technical methods (dexamethasone-binding assay versus mRNA quantitation), and the small number of patients employed that does not allow definitive conclusions, as well as the cells’ potential to up- and down-regulate GR in different situations (Citation43).
Table II. Studies assessing the number and affinity of glucocorticoid receptors (GR) in colonic mucosa and peripheral blood mononuclear cells (PBMC) in inflammatory bowel disease.
Altered affinity to the ligand
Another possible explanation for the varying glucocorticoid sensitivity of IBD patients could be the altered affinity of the GR to its ligand. Indeed, IBD patients have been shown to have lower affinity to GR than normal controls () (Citation44). In addition, in another study, patients with steroid-resistant IBD were reported to have a lower affinity to GR than glucocorticoid responders () (Citation45). These studies, although small, do support the idea that systemic inflammation could alter the affinity of the GR to its ligand, as seen for example in asthma (Citation39).
Glucocorticoid receptor isoforms α and β
The elevated expression of GRβ has been implicated as being one of the causes behind decreased sensitivity to glucocorticoid therapy (Citation46). In IBD, a number of groups reported that elevated GRβ expression was detected in colonic mucosal cells and PBMCs of patients resistant to steroid treatment () (Citation47–51). However, the largest study on this subject (86 patients) arrived at opposing results, showing no difference in the GRβ levels of glucocorticoid responders and non-responders () (Citation52). In addition, a small study (n = 17) in patients with Crohn's disease concluded that GRα and GRβ levels were lower in PBMCs of CD patients than in healthy controls (Citation53). Therefore, more studies would be needed to clarify the contribution of the GRβ to the glucocorticoid sensitivity of IBD patients.
Table III. Studies assessing the association between the number of glucocorticoid receptors (GR) GRα and GRβ in peripheral blood mononuclear cells (PBMC) and colonic mucosa, and the response to glucocorticoid (GC) therapy in adult patients with inflammatory bowel disease.
Glucocorticoid receptor polymorphisms
An Italian group investigated the presence of the GR polymorphisms in paediatric patients with IBD and compared the results with glucocorticoid therapy success (Citation54). They reported that the presence of the BclI polymorphism between exons 2 and 3 associated significantly with glucocorticoid therapy responsiveness. However, another study on glucocorticoid receptor haplotypes in adult IBD patients did not find the BclI polymorphism to be significant in IBD, nor could they find any association between different GR haplotypes and steroid therapy response in IBD patients (Citation55). Also the ER22/23EK and N363S polymorphisms have been recently studied in IBD, but these polymorphisms did not show any allele or haplotype difference between paediatric CD patients and healthy controls (Citation56).
Glucocorticoid bioactivity
Our group has studied the possibility of using the circulating bioactivity of the administered glucocorticoids as a marker of therapy outcome or the appearance of treatment-related side-effects () (Citation57). Glucocorticoid bioactivity was measured with a COS-1 cell bioassay in which the cells are transfected with GR and reporter genes (Citation58). In this technique, a 10 µL sample of patient serum is added to the cell culture medium, and after an overnight incubation the relative glucocorticoid bioactivity can be measured. Unfortunately, no associations were found between clinical response to glucocorticoids and circulating glucocorticoid bioactivity () (Citation57). Our findings were confirmed in another study on 50 patients receiving intravenous steroids for severe IBD (Citation59). These findings support the hypothesis that glucocorticoid treatment response is dependent on glucocorticoid sensitivity at the target cell level and not on the circulating bioactivity of the drug. So far, there is no clear explanation why some patients failing oral glucocorticoid therapy respond to intravenous administration of steroids. In search of other biomarkers that could be associated with steroid therapy outcome, we found, however, that serum adiponectin, a hormone secreted by white adipose tissue, associated with glucocorticoid treatment-related early side-effects () (Citation60). If this finding is confirmed in larger studies, serum adiponectin might be used as a readily available biomarker of steroid therapy-associated adverse events.
Table IV. Other methods employed to study the glucocorticoid (GC) response in patients with inflammatory bowel disease (IBD).
Lymphocyte activation
We have also recently developed a novel bioassay in which the effect of the patient serum on healthy donor lymphocytes is measured with a panel of T cell activation markers () (Citation61,Citation62). The markers analysed include the expression of FOXP3 and glucocorticoid-induced tumour necrosis factor inhibitor (GITR), reflecting the actions of regulatory T cells and the secretion of interferon (IFN) γ mirroring effector T cells. In paediatric IBD patients glucocorticoid therapy decreased the potential of the patient serum to induce expression of these activation markers in donor cells (Citation61). In addition, in adult CD patients the immunological activation potency in patient serum before anti-tumour necrosis factor-α (TNF-α) therapy associated with endoscopic CD activity score and with anti-TNF-α treatment response () (Citation62). Therefore, this novel assay can be used in monitoring the individual responses to IBD therapy, including glucocorticoids. However, the assay has not yet been standardized for commercial cell lines.
Cytokines and glucocorticoid sensitivity in IBD
In IBD, a few studies have concentrated on the impact of the cytokine profile on glucocorticoid therapy response. In 1999, Ishiguro reported that the levels of TNF-α, interleukin (IL) 6, and IL-8 were elevated in steroid-resistant ulcerative colitis (UC) patients (Citation63). Interestingly, almost 10 years later a –308A allele of the TNF-α gene was associated with steroid resistance in paediatric CD patients (Citation64). Also, in another study several cytokines (IFN-γ, granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), TNF-α, IL-1α, IL-10, IL-17, and IP-10) were reported to correlate with in-vitro steroid response: in this report, IL-2 seemed to convey steroid resistance, both in healthy controls and in patients with UC (Citation65). Indeed, IL-2 has been associated with increased GRβ expression, decreased affinity of GR to its ligand, and decreased GR translocation to the cell nucleus (Citation66). A third study on the subject, however, arrived at contradicting results and did not find any associations between cytokine mRNA expression and response to glucocorticoid therapy (Citation67). Extracellular cytokines are able to regulate transcriptional processes via phosphorylation of transcription factors, e.g. the STAT family. At tissue level, the presence of STAT-5 has been related to steroid-resistance in UC in a preliminary study in adults (Citation68).
Other methods employed to study glucocorticoid sensitivity in patients with inflammatory bowel disease
During the past 10 years, a number of other methods have also been employed in the study of glucocorticoid sensitivity in IBD patients (). These include studying lymphocytes and other genes and cellular markers from IBD patients in relation to glucocorticoid sensitivity, as well as assessing different methods that could be employed in the prediction of the steroid response (Citation69–72). However, none of these has emerged as a clinically applicable means for monitoring steroid therapy. Thus, the steroid response in severe disease is currently monitored with clinical variables predicting response failures on days 3–7 of therapy to guide the need for second-line therapy or surgery (Citation14,Citation15,Citation73–77).
Also other inflammatory diseases, such as asthma and rheumatoid arthritis that are treated with glucocorticoids, face the problem of glucocorticoid insensitivity (Citation78). In asthma, genetic factors might play a role in glucocorticoid responsiveness as glucocorticoid insensitivity has been found within families and microarray studies have revealed a panel of genes that discriminate between glucocorticoid-responsive and non-responsive groups (Citation78). Recently, a similar study was conducted in paediatric patients with severe ulcerative colitis that resulted in the characterization of 41 genes that expressed differentially between glucocorticoid responders and non-responders (Citation79). However, so far the total number of steroid non-responsive patients studied is low, challenging the recruitment of patients to provide clear answers on the underlying mechanisms.
Conclusions
Being able to predict in advance the patients’ response to glucocorticoid therapy would be a powerful tool in the treatment of IBD. With the gradual unfolding of the molecular basis of glucocorticoid actions in different tissues, this might be possible in the future. In IBD, numerous attempts have been made to characterize patients according to their sensitivity to glucocorticoids with respect to ligand availability, GR structure, cytokine profile, and other glucocorticoid sensitivity-assessing methods and biomarkers. At present, however, the data on the glucocorticoid signalling cascade are still rather fragmentary, and clinical studies seem to reach contradictory conclusions—from bench to bedside is still a long way. Small studies with heterogeneous study designs might be one cause for the controversy, but, as recent studies have shown, the GR splice and translational isoforms identified until now can form up to 256 different homo- and heterodimers (Citation80). These receptor complex subtypes might all express distinct patterns of intracellular localization, coactivator recruitment, and transactivation and -repression properties. Therefore, the contradictions we see in the results might only be a reflection of the complexity of the steroid receptor signalling network that has just begun to reveal itself. It is more than likely that no single cytokine, polymorphism, or isoform alone can respond to the question of whether or not a person is sensitive to glucocorticoid therapy. Rather, it is possible that the combined information from all of these variables might one day provide us with an estimate of the probable response.
Acknowledgements
This study was funded by the Emil Aaltonen Foundation, the Finnish Cultural Foundation, the Helsinki University Central Hospital Grant, the Finnish Paediatric Research Foundation, the Päivikki and Sakari Sohlberg Foundation, the Finnish Medical Society Duodecim, the Maud Kuistila Memorial Foundation, and the Biomedicum Helsinki Foundation.
Declaration of interest: The authors report no conflict of interest. The authors alone are responsible of the content and writing of the paper.
References
- Truelove SC, Witts LJ. Cortisone in ulcerative colitis; final report on a therapeutic trial. Br Med J. 1955;4947:1041–8.
- Benchimol EI, Seow CH, Steinhart AH, Griffiths AM. Traditional corticosteroids for induction of remission in Crohn's disease. Cochrane Database Syst Rev. 2008;2: CD006792.
- Truelove SC, Willoughby CP, Lee EG, Kettlewell MG. Further experience in the treatment of severe attacks of ulcerative colitis. Lancet. 1978;2:1086–8.
- Munkholm P, Langholz E, Davidsen M, Binder V. Frequency of glucocorticoid resistance and dependency in Crohn's disease. Gut. 1994;35:360–2.
- Faubion WA Jr, Loftus EV Jr, Harmsen WS, Zinsmeister AR, Sandborn WJ. The natural history of corticosteroid therapy for inflammatory bowel disease: a population-based study. Gastroenterology. 2001;121:255–60.
- Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. 2002; 96:23–43.
- Teshima C, Fedorak RN. Are there differences in type, dosage, and method of administration for the systemic steroids in IBD treatment? Inflamm Bowel Dis. 2008;14 (Suppl 2):S216–8.
- Rang HP, Dale MM, Ritter JM, Flower R. Rang and Dale's Pharmacology. 6th. Edinburgh: Churchill Livingstone; 2007.
- Edsbacker S, Andersson T. Pharmacokinetics of budesonide (Entocort EC) capsules for Crohn's disease. Clin Pharmacokinet. 2004;43:803–21.
- Baron JH, Connell AM, Kanaghinis TG, Lennard-Jones JE, Jones AF. Out-patient treatment of ulcerative colitis. Comparison between three doses of oral prednisone. Br Med J. 1962;5302:441–3.
- Rutgeerts PJ. Review article: the limitations of corticosteroid therapy in Crohn's disease. Aliment Pharmacol Ther. 2001; 15:1515–25.
- Friend DR. Review article: issues in oral administration of locally acting glucocorticosteroids for treatment of inflammatory bowel disease. Aliment Pharmacol Ther. 1998;12: 591–603.
- Seow CH, Benchimol EI, Griffiths AM, Otley AR, Steinhart AH. Budesonide for induction of remission in Crohn's disease. Cochrane Database Syst Rev. 2008;3: CD000296.
- Turner D, Walsh CM, Benchimol EI, Mann EH, Thomas KE, Chow C, . Severe paediatric ulcerative colitis: incidence, outcomes and optimal timing for second-line therapy. Gut. 2008;57:331–8.
- Dinesen LC, Walsh AJ, Protic MN, Heap G, Cummings F, Warren BF, . The pattern and outcome of acute severe colitis. J Crohns Colitis. 2010;4:431–7.
- Frey BM, Frey FJ. Clinical pharmacokinetics of prednisone and prednisolone. Clin Pharmacokinet. 1990;19: 126–46.
- Schwab M, Klotz U. Pharmacokinetic considerations in the treatment of inflammatory bowel disease. Clin Pharmacokinet. 2001;40:723–51.
- Zbankova S, Bryndova J, Leden P, Kment M, Svec A, Pacha J. 11beta-Hydroxysteroid dehydrogenase 1 and 2 expression in colon from patients with ulcerative colitis. J Gastroenterol Hepatol. 2007;22:1019–23.
- Stegk JP, Ebert B, Martin HJ, Maser E. Expression profiles of human 11beta-hydroxysteroid dehydrogenases type 1 and type 2 in inflammatory bowel diseases. Mol Cell Endocrinol. 2009;301:104–8.
- Farrell RJ, Murphy A, Long A, Donnelly S, Cherikuri A, O’Toole D, . High multidrug resistance (P-glycoprotein 170) expression in inflammatory bowel disease patients who fail medical therapy. Gastroenterology. 2000;118:279–88.
- Annese V, Valvano MR, Palmieri O, Latiano A, Bossa F, Andriulli A. Multidrug resistance 1 gene in inflammatory bowel disease: a meta-analysis. World J Gastroenterol. 2006; 12:3636–44.
- Duma D, Jewell CM, Cidlowski JA. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J Steroid Biochem Mol Biol. 2006;102:11–21.
- Nuclear Receptors Nomenclature Committee. A unified nomenclature system for the nuclear receptor superfamily. Cell. 1999;97:161–3.
- Francke U, Foellmer BE. The glucocorticoid receptor gene is in 5q31-q32 [corrected]. Genomics. 1989;4:610–2.
- Turner JD, Muller CP. Structure of the glucocorticoid recetor (NR3C1) gene 5’ untranslated region: identification, and tissue distribution of multiple new human exon 1. J Mol Endocrinol. 2005;35:283–92.
- Presul E, Schmidt S, Kofler R, Helmberg A. Identification, tissue expression, and glucocorticoid responsiveness of alternative first exons of the human glucocorticoid receptor. J Mol Endocrinol. 2007;38:79–90.
- Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. 2005;18: 331–42.
- Encio IJ, Detera-Wadleigh SD. The genomic structure of the human glucocorticoid receptor. J Biol Chem. 1991;266: 7182–8.
- de Castro M, Elliot S, Kino T, Bamberger C, Karl M, Webster E, . The non-ligand binding beta-isoform of the human glucocorticoid receptor (hGR beta): tissue levels, mechanism of action, and potential physiologic role. Mol Med. 1996;2:597–607.
- Kino T, Manoli I, Kelkar S, Wang Y, Su YA, Chrousos GP. Glucocorticoid receptor (GR) beta has intrinsic, GRalpha-independent transcriptional activity. Biochem Biophys Res Commun. 2009;381:671–5.
- Krett NL, Pillay S, Moalli PA, Greipp PR, Rosen ST. A variant glucocorticoid receptor messenger RNA is expressed in multiple myeloma patients. Cancer Res. 1995; 55:2727–9.
- Moalli PA, Pillay S, Krett NL, Rosen ST. Alternatively spliced glucocorticoid receptor messenger RNAs in glucocorticoid-resistant human multiple myeloma cells. Cancer Res. 1993;53:3877–9.
- Ray DW, Davis JR, White A, Clark AJ. Glucocorticoid receptor structure and function in glucocorticoid-resistant small cell lung carcinoma cells. Cancer Res. 1996;56: 3276–80.
- Rivers C, Levy A, Hancock J, Lightman S, Norman M. Insertion of an amino acid in the DNA-binding domain of the glucocorticoid receptor as a result of alternative splicing. J Clin Endocrinol Metab. 1999;84:4283–6.
- Beger C, Gerdes K, Lauten M, Tissing WJ, Fernandez-Munoz I, Schrappe M, . Expression and structural analysis of glucocorticoid receptor isoform gamma in human leukaemia cells using an isoform-specific real-time polymerase chain reaction approach. Br J Haematol. 2003;122: 245–52.
- van Rossum EF, Lamberts SW. Polymorphisms in the glucocorticoid receptor gene and their associations with metabolic parameters and body composition. Recent Prog Horm Res. 2004;59:333–57.
- Derijk RH, Schaaf MJ, Turner G, Datson NA, Vreugdenhil E, Cidlowski J, . A human glucocorticoid receptor gene variant that increases the stability of the glucocorticoid receptor beta-isoform mRNA is associated with rheumatoid arthritis. J Rheumatol. 2001;28:2383–8.
- Pujols L, Mullol J, Picado C. Alpha and beta glucocorticoid receptors: relevance in airway diseases. Curr Allergy Asthma Rep. 2007;7:93–9.
- Buttgereit F, Saag KG, Cutolo M, da Silva JA, Bijlsma JW. The molecular basis for the effectiveness, toxicity, and resistance to glucocorticoids: focus on the treatment of rheumatoid arthritis. Scand J Rheumatol. 2005;34:14–21.
- Rogler G, Meinel A, Lingauer A, Michl J, Zietz B, Gross V, . Glucocorticoid receptors are down-regulated in inflamed colonic mucosa but not in peripheral blood mononuclear cells from patients with inflammatory bowel disease. Eur J Clin Invest. 1999;29:330–6.
- Flood L, Lofberg R, Stierna P, Wikstrom AC. Glucocorticoid receptor mRNA in patients with ulcerative colitis: a study of responders and nonresponders to glucocorticosteroid therapy. Inflamm Bowel Dis. 2001;7:202–9.
- Raddatz D, Middel P, Bockemuhl M, Benohr P, Wissmann C, Schworer H, . Glucocorticoid receptor expression in inflammatory bowel disease: evidence for a mucosal down-regulation in steroid-unresponsive ulcerative colitis. Aliment Pharmacol Ther. 2004;19:47–61.
- Geng CD, Vedeckis WV. A new, lineage specific, autoup-regulation mechanism for human glucocorticoid receptor gene expression in 697 pre-B-acute lymphoblastic leukemia cells. Mol Endocrinol. 2011;25:44–57.
- Schottelius A, Wedel S, Weltrich R, Rohde W, Buttgereit F, Schreiber S, . Higher expression of glucocorticoid receptor in peripheral mononuclear cells in inflammatory bowel disease. Am J Gastroenterol. 2000;95:1994–9.
- Shimada T, Hiwatashi N, Yamazaki H, Kinouchi Y, Toyota T. Relationship between glucocorticoid receptor and response to glucocorticoid therapy in ulcerative colitis. Dis Colon Rectum. 1997;40(Suppl 10):S54–8.
- Kino T, Su YA, Chrousos GP. Human glucocorticoid receptor isoform beta: recent understanding of its potential implications in physiology and pathophysiology. Cell Mol Life Sci. 2009;66:3435–48.
- Honda M, Orii F, Ayabe T, Imai S, Ashida T, Obara T, . Expression of glucocorticoid receptor beta in lymphocytes of patients with glucocorticoid-resistant ulcerative colitis. Gastroenterology. 2000;118:859–66.
- Orii F, Ashida T, Nomura M, Maemoto A, Fujiki T, Ayabe T, . Quantitative analysis for human glucocorticoid receptor alpha/beta mRNA in IBD. Biochem Biophys Res Commun. 2002;296:1286–94.
- Zhang H, Ouyang Q, Wen ZH, Fiocchi C, Liu WP, Chen DY, . Significance of glucocorticoid receptor expression in colonic mucosal cells of patients with ulcerative colitis. World J Gastroenterol. 2005;11:1775–8.
- Towers R, Naftali T, Gabay G, Carlebach M, Klein A, Novis B. High levels of glucocorticoid receptors in patients with active Crohn's disease may predict steroid resistance. Clin Exp Immunol. 2005;141:357–62.
- Fujishima S, Takeda H, Kawata S, Yamakawa M. The relationship between the expression of the glucocorticoid receptor in biopsied colonic mucosa and the glucocorticoid responsiveness of ulcerative colitis patients. Clin Immunol. 2009;133:208–17.
- Hausmann M, Herfarth H, Scholmerich J, Rogler G. Glucocorticoid receptor isoform expression does not predict steroid treatment response in IBD. Gut. 2007;56:1328–9.
- Hori T, Watanabe K, Miyaoka M, Moriyasu F, Onda K, Hirano T, . Expression of mRNA for glucocorticoid receptors in peripheral blood mononuclear cells of patients with Crohn's disease. J Gastroenterol Hepatol. 2002;17:1070–7.
- De Iudicibus S, Stocco G, Martelossi S, Drigo I, Norbedo S, Lionetti P, . Association of BclI polymorphism of the glucocorticoid receptor gene locus with response to glucocorticoids in inflammatory bowel disease. Gut. 2007; 56:1319–20.
- Mwinyi J, Wenger C, Eloranta JJ, Kullak-Ublick GA. Glucocorticoid receptor gene haplotype structure and steroid therapy outcome in IBD patients. World J Gastroenterol. 2010;16:3888–96.
- Sanchez R, Levy E, Costea F, Sinnett D. IL-10 and TNF-alpha promoter haplotypes are associated with childhood Crohn's disease location. World J Gastroenterol. 2009;15: 3776–82.
- Vihinen MK, Raivio T, Verkasalo M, Janne OA, Kolho KL. Circulating glucocorticoid bioactivity during peroral glucocorticoid treatment in children and adolescents with inflammatory bowel disease. J Clin Gastroenterol. 2008;42: 1017–24.
- Raivio T, Palvimo JJ, Kannisto S, Voutilainen R, Janne OA. Transactivation assay for determination of glucocorticoid bioactivity in human serum. J Clin Endocrinol Metab. 2002; 87:3740–4.
- Turner D, Kolho KL, Mack DR, Raivio T, Leleiko N, Crandall W, . Glucocorticoid bioactivity does not predict response to steroid therapy in severe pediatric ulcerative colitis. Inflamm Bowel Dis. 2010;16:469–73.
- Vihinen MK, Kolho KL, Janne OA, Andersson S, Raivio T. Circulating adiponectin as a marker for glucocorticoid-related side effects in children and adolescents with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2009; 48:504–6.
- Rintamaki H, Salo HM, Vaarala O, Kolho KL. New means to monitor the effect of glucocorticoid therapy in children. World J Gastroenterol. 2010;16:1104–9.
- Rintamaki H, Sipponen T, Salo HM, Vaarala O, Kolho KL. Serum immune-activation potency and response to anti-TNF-alpha therapy in Crohn's disease. World J Gastroenterol. 2010;16:5845–51.
- Ishiguro Y. Mucosal proinflammatory cytokine production correlates with endoscopic activity of ulcerative colitis. J Gastroenterol. 1999;34:66–74.
- Cucchiara S, Latiano A, Palmieri O, Canani RB, D’Inca R, Guariso G, . Polymorphisms of tumor necrosis factor-alpha but not MDR1 influence response to medical therapy in pediatric-onset inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2007;44:171–9.
- Creed TJ, Lee RW, Newcomb PV, di Mambro AJ, Raju M, Dayan CM. The effects of cytokines on suppression of lymphocyte proliferation by dexamethasone. J Immunol. 2009;183:164–71.
- Smith MA, Marinaki AM, Sanderson JD. Pharmacogenomics in the treatment of inflammatory bowel disease. Pharmacogenomics. 2010;11:421–37.
- Raddatz D, Bockemuhl M, Ramadori G. Quantitative measurement of cytokine mRNA in inflammatory bowel disease: relation to clinical and endoscopic activity and outcome. Eur J Gastroenterol Hepatol. 2005;17:547–57.
- Löwenberg M, Scheffer M, Verhaar A, Peppelenbosch M, Hommes D. A role for STAT5 in steroid-resistant UC? Inflamm Bowel Dis. 2006;12:665.
- Hearing SD, Norman M, Probert CS, Haslam N, Dayan CM. Predicting therapeutic outcome in severe ulcerative colitis by measuring in vitro steroid sensitivity of proliferating peripheral blood lymphocytes. Gut. 1999;45:382–8.
- Franchimont D, Louis E, Dupont P, Vrindts-Gevaert Y, Dewe W, Chrousos G, . Decreased corticosensitivity in quiescent Crohn's disease: an ex vivo study using whole blood cell cultures. Dig Dis Sci. 1999;44:1208–15.
- Jinno Y, Ohtani H, Nakamura S, Oki M, Maeda K, Fukushima K, . Infiltration of CD19 + plasma cells with frequent labeling of Ki-67 in corticosteroid-resistant active ulcerative colitis. Virchows Arch. 2006;448:412–21.
- Nakahara S, Arimura Y, Saito K, Goto A, Motoya S, Shinomura Y, . Association of SLC22A4/5 polymorphisms with steroid responsiveness of inflammatory bowel disease in Japan. Dis Colon Rectum. 2008;51:598–603.
- Turner D, Otley AR, Mack D, Hyams J, de Bruijne J, Uusoue K, . Development and evaluation of a Pediatric Ulcerative Colitis Activity Index (PUCAI): A prospective multicenter study. Gastroenterology. 2007; 133:423–32.
- Seo M, Okada M, Yao T, Matake H, Maeda K. Evaluation of the clinical course of acute attacks in patients with ulcerative colitis through the use of an activity index. J Gastroenterol. 2002;37:29–34.
- Ho GT, Mowat C, Goddard CJ, Fennell JM, Shah NB, Prescott RJ, . Predicting the outcome of severe ulcerative colitis: development of a novel risk score to aid early selection of patients for second-line medical therapy or surgery. Aliment Pharmacol Ther. 2004;19:1079–87.
- Travis SP, Farrant JM, Ricketts C, Nolan DJ, Mortensen NM, Kettlewell MG, . Predicting outcome in severe ulcerative colitis. Gut. 1996;38:905–10.
- Lindgren SC, Flood LM, Kilander AF, Löfberg R, Persson TB, Sjödahl RI. Early predictors of glucocorticosteroid treatment failure in severe and moderately severe attacks of ulcerative colitis. Eur J Gastroenterol Hepatol. 1998; 10:831–5.
- Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet. 2009;373:1905–17.
- Kabakchiev B, Turner D, Hyams J, Mack D, Leleiko N, Crandall W, . Gene expression changes associated with resistance to intravenous corticosteroid therapy in children with severe ulcerative colitis. PLoS One. 2010;5: pii: e13085.
- Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E. The human glucocorticoid receptor: molecular basis of biologic function. Steroids. 2010;75:1–12.