Abstract
Different types of cancer take advantage of inflammatory components to improve their life-span in the organs. A sustenance of growth factors and cytokines (e.g. interleukin (IL)-1, tumor necrosis factor, IL-6, vascular endothelial growth factor) supports malignant cell progression and contributes to suppress the body immune defense. Strategies to modulate the host micro-environment offer new approaches for anti-cancer therapies. For these reasons new molecules with anti-tumor and anti-inflammatory features (e.g. trabectedin) are looked at with new eyes in the light of the crucial link between inflammation and cancer.
| Abbreviations | ||
| ATP | = | adenosine triphosphate |
| Bcl-2 | = | B-cell lymphoma 2 |
| Bcl-XL | = | B-cell lymphoma XL |
| CAPS | = | cold auto-inflammatory syndrome |
| COX-2 | = | cyclo-oxygenase-2 isoform |
| CSC | = | cancer stem cell |
| FAK | = | focal adhesion kinase |
| FOXP3 | = | Forkhead Box P3 |
| HCC | = | hepatocellular carcinoma |
| HNSCC | = | squamous cell carcinoma of the head and neck |
| ICAM-1 | = | inter-cellular adhesion molecule 1 |
| IL-1 | = | interleukin 1 |
| IL-10 | = | interleukin 10 |
| IL-10R | = | interleukin 10 receptor |
| IL-13 | = | interleukin 13 |
| IL-1Ra | = | IL-1R antagonist |
| IL-4 | = | interleukin 4 |
| IL-6 | = | interleukin 6 |
| IFN | = | γ Interferon |
| JAK | = | Janus kinase |
| LPS | = | lipopolysaccharide |
| Mcl-1 | = | myeloid cell leukemia 1 |
| M-CSF | = | macrophage colony-stimulating factor |
| MIF-1 | = | macrophage migration inhibitory factor 1 |
| MMPs | = | matrix Metalloproteinases |
| MyD88 | = | myeloid differentiation primary response gene 88 |
| NF-kB | = | nuclear factor-kB |
| NGR | = | Asn-Gly-Arg |
| NK | = | natural killer cells |
| NSAIDs | = | non-steroid anti-inflammatory drugs |
| NSCC | = | non-stem cancer cells |
| PCNA | = | proliferating cell nuclear antigen |
| PI3K | = | phosphoinositide 3-kinase |
| STAT3 | = | signal transducer and activator of transcription 3 |
| TAM | = | tumor-associated macrophages |
| TGFβ | = | transforming growth factor β |
| Th1 | = | T helper 1 |
| Th2 | = | T helper 2 |
| TLR9 | = | Toll-like receptor 9 |
| TNF | = | tumor necrosis factor |
| V-CAM | = | vascular cell adhesion molecule 1 |
| VEGF | = | vascular endothelial growth factor |
Key messages
Cancer-related inflammation is a promising target of innovative therapeutic interventions in oncology.
Drug targeting of the inflammatory cytokines, angiogenesis, and the corrupted immune cells is under development or has already entered clinical trials.
Cancer-related inflammation: Rudolf Virchow was right
Inflammation, known as a complex biological response of tissues to harmful stimuli, can be picked up in the medical field to represent the history of Dr Jekyll and Mr Hyde. A protective shield for the organism in some situations, it can set off abnormal responses against self components or create a substrate for malignancy.
In 1863 the German pathologist Rudolf Virchow noted that neoplastic lesions were infiltrated by leukocytes and postulated the hypothesis of a pro-tumoral role of the inflammatory micro-environment. After 150 years this concept is now established and constitutes the rationale to develop new therapeutic strategies, specifically targeted to inflammatory components, for the treatment and prevention of different types of cancer () (Citation1–3).
Figure 1. The tumor micro-environment is a complex network of inflammatory cells and molecules that are potential targets of anti-cancer treatments. Antibodies or antagonists blocking cytokines (IL-1β, TNFα, IL-6), chemokines (CCL2), or chemokine receptors (CXCR4) or targeting tumor-associated macrophages (TAM) have been developed. Bisphosphonates and trabectedin are cytotoxic for TAM; trabectedin also inhibits macrophage recruitment by inhibiting CCL2.
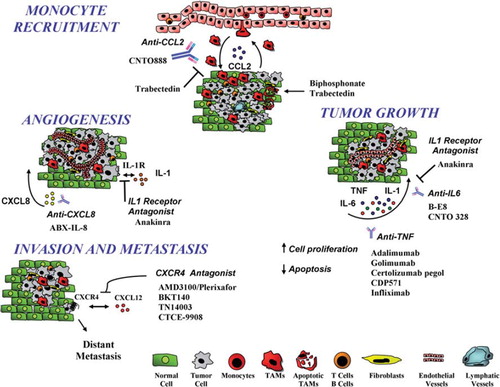
Generally two pathways, not mutually exclusive, can be identified as the major affluent to the inflammatory milieu: 1) the intrinsic one (neoplastic cells), where genetic events (e.g. oncogenes, genetic aberrations), inducing neoplastic transformation, activate the production of inflammatory mediators; and 2) the extrinsic pathway (inflammatory leukocytes and soluble mediators) where chronic infections (e.g. viruses, bacteria, and parasites) significantly increase the risk of cancer. Epidemiological studies estimate that about 8%–17% of global cancer burden is linked to chronic infections. Indeed, different common tumors (e.g. bladder, ovarian, gastric, hepatocellular, and colorectal cancer) are associated with unresolved pathogen infections that fuel inflammation (Citation4). These epidemiological results support the evidence that inflammation-related cells and molecules constitute potential targets of anti-tumor therapies.
Non-steroid anti-inflammatory drugs in tumor prevention
Anti-inflammatory therapies could be an additional soldier in the tumor war. Evidence for this came from population-based studies on the prolonged usage of the most common non-steroid anti-inflammatory drugs (NSAIDs): aspirin.
In 1988 a first report from the Melbourne Colorectal Cancer Study demonstrated an inverse association between the use of aspirin and the risk of colorectal cancer. Results from randomized clinical trials have demonstrated that the daily use of aspirin for 5 years is effective in the prevention of colorectal cancer with a latency of about 10 years (Citation5,Citation6). Moreover aspirin reduced colonic malignancy also in different chemically induced animal models. NSAIDs are inhibitors of the cyclo-oxygenase enzyme family, which catalyzes the metabolism of arachidonic acid to prostaglandins, prostacyclin, and thromboxanes. The cyclo-oxygenase-1 isoform is constitutively expressed in most tissues, where it mediates physiologic functions such as gastric mucosal cytoprotection and regulation of platelet aggregation. The cyclo-oxygenase-2 isoform (COX-2) is induced in response to cytokines and growth factors and is expressed in inflammatory diseases, premalignant lesions (such as colorectal adenomas), and colon cancer. COX-2 mediates the production of prostaglandin E2 (PGE2) in epithelial tissues, inducing the activation of signaling pathways that promote cell proliferation and inhibit cell death.
Experimental evidence supports the concept that the chemopreventive effects of NSAIDs may be due, at least in part, to inhibition of COX-2. A molecule specifically inhibiting COX-2 (celecoxib) has been developed for the treatment of pain and inflammation (Citation7). In 2000 a study showed a clear anti-tumoral effect: patients with familial adenomatous polyposis treated with celecoxib had a significant reduction in the number of colorectal polyps (Citation8).
Although the use of celecoxib may be hampered by considerable toxicity, these examples constitute a solid demonstration of the key tumor-promoting role of the inflammatory milieu, at least in supporting gastrointestinal cancer.
Anti-cytokine therapy to silence inflammation
Cytokines produced by leukocytes and other cell types are key orchestrators of the cancer-related inflammation. These molecules are involved in important biological processes that govern proliferation and migration of different cells, regulation of innate and adaptive immune responses, and angiogenic and repair programs of the tissues. Interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and IL-6 are prototypic inflammatory cytokines. Their prime role in the initiation of the inflammatory cascade and their multi-faceted functions in the tumor milieu prompted the focus on these molecules, as well as on angiogenic growth factors, as preferential targets of anti-tumoral therapies.
TNFα
Many malignant cells constitutively produce small amounts of TNFα. This molecule plays a crucial role in the persistence of inflammation, and established evidence demonstrates that tumor-derived TNFα sustains the growth and progression of syngenic, xenogenic, and chemically induced tumors of skin, ovary, pancreas, and bowels (Citation3,Citation9).
Malignant cells are not the only producers of TNFα in the tumor micro-environment. Macrophages (Citation10) and lymphocytic CD4 cells (Citation11) secrete TNFα and trigger inflammation, as shown in a genetic model of liver cancer (Citation12). TNFα initiates a complex cross-talk between epithelial ovarian cancer cells and surrounding tissues, promoting the colonization of the peritoneum. Kulbe et al. showed that the constitutive production of TNFα was associated with increased release of chemokines (CCL2, CXCL8, CXCL12), IL-6, vascular endothelial growth factor (VEGF), and macrophage migration inhibitory factor-1 (MIF-1).
The mechanisms of TNFα action includes indirect effects on tumor cell spread, via CXCR4; on tumor cell survival, via CXCR4/CXCL12; but also on stimulation of new blood vessels in the peritoneal tumor colonies, due to induction of CXCL12 and VEGF expression (Citation13).
The possibility to target TNFα with antibodies, small inhibitors, or other TNFα antagonists has therapeutic application against inflammatory as well as malignant diseases. Adalimumab, golimumab, certolizumab pegol, CDP571, and infliximab are examples of anti-TNF monoclonal antibodies developed for therapeutic purposes, while etanercept is a soluble TNFα receptor.
To date, the major clinical experience comes from patients with chronic inflammatory diseases (e.g. rheumatoid arthritis and Crohn's disease), where blocking of TNFα causes a reduction of inflammatory cells and of the angiogenic network and extracellular matrix deposition. The monoclonal antibody infliximab was the pioneer among TNFα-neutralizing molecules to be used in the treatment of rheumatoid arthritis. It down-regulates the production of T helper (Th)-1 and inflammatory cytokines such as interferon γ (IFNγ), IL-1, IL-6 as well as adhesion molecules like soluble E-selectin, inter-cellular adhesion molecule (ICAM)-1, and metalloproteinases like MMP-1, MMP-3, and MMP-9 (Citation14,Citation15). Infliximab is currently used in phase II studies for the treatment of breast cancer and in advanced renal cell carcinoma (Citation16).
The efficacy of anti-TNF therapies, however, remains controversial due to emerging toxicities and possible association with development of new malignancies. The primary problem concerns the systemic administration of anti-TNF therapy for the occurrence of allergic reactions, fever, and rigors. Other diseases linked to anti-TNF therapies have been reported, in particular cardiopulmonary toxicity, opportunistic infections, autoimmune diseases (e.g. psoriasis), and nervous system diseases (e.g. multiple sclerosis, Guillain–Barré syndrome) (Citation17). Moreover, an increased susceptibility for the onset of cancer was noted: patients treated with infliximab had increased risk of lymphoma and leukemia (Citation18). Also etanercept administration to patients treated for autoimmune diseases was associated with Hodgkin's lymphoma or other hematological malignancies (Citation19,Citation20). Overall these data show an unclear state of the art for systemic anti-TNF therapies in cancer treatment. Other approaches (e.g. lentiviral vectors for a stable expression of TNF) or use of compounds able to modulate TNF production could open a new scenario (Citation21).
The old drug thalidomide, synthesized in 1954 and abandoned because of severe teratogenic effects, is now currently used for inflammatory and malignant diseases. It showed down-modulatory effects on the release of TNFα, IL-6, CXCL8, and VEGF and reduced tumor progression and vascular construction (Citation17,Citation22). In 1999 thalidomide was first used in clinical trials in patients with myeloma, and significant regressions were achieved (Citation23). Other possible anti-neoplastic features of thalidomide are the inhibition of the transcription factor nuclear factor-kB (NF-kB), through the suppression of Ikappa B kinase activity, and consequent pro-apoptotic activity in tumor cells (Citation24); the stimulation and proliferation of CD8 T cells (Citation25); and the increased therapeutic efficacy in metastatic melanoma patients when combined with cytotoxic chemotherapy (Citation26). On the basis of these promising results, thalidomide is being used for the treatment of other solid tumors, for example prostate, renal, breast, and colorectal cancer, and Kaposi's sarcoma. (Citation27,Citation28). A derivative compound, lenalidomide, with lower neural toxicity has been developed and is now in phase II for the treatment of prostate cancer and multiple myeloma.
Finally, in the perspective of exploiting the ability of TNF to improve vascular permeability and therefore to increase chemotherapeutic drug penetration at tumor sites, a derivative molecule has been designed. NGR-TNF is a TNF molecule fused to the tumor-homing peptide Asn-Gly-Arg (NGR); in experimental mouse models it was administered in combination with classical anti-cancer drugs and increased the efficacy of chemotherapy (Citation29). Human clinical trials in patients with advanced diseases are now on-going with this molecule (Citation30,Citation31).
IL-1β
The involvement of IL-1β in tumor progression is well established. IL-1β expression is elevated in human breast, colon, lung, head, and neck cancers and melanomas, and patients with IL-1-producing tumors have generally poor prognosis (Citation32–35). IL-1β promotes tumor growth and metastasis by inducing several pro-metastatic genes such as metalloproteinases, chemokines, growth factors and transforming growth factor (TGF)-β (Citation36). IL-1β is also able to stimulate the expression of endothelial adhesion molecules such as ICAM-1 and VCAM-1 (Citation37,Citation38). Of particular importance are its effects on angiogenesis: IL-1β is a potent pro-angiogenic cytokine, and VEGF release is also IL-1-dependent (Citation34,Citation39,Citation40). In a pancreatic islet tumor model, a first wave of myc-driven angiogenesis is induced by IL-1. In an unexpected twist, IL-1α was also shown to play a pivotal role in the pathogenesis of liver cancer (Citation41).
In 1993 the first study using IL-1R antagonist (IL-1Ra) treatment in mice transplanted with human melanoma cells demonstrated a strong reduction in the number of lung metastases (Citation42). Similar findings were reported in a model of hepatic metastasis: a single injection of IL-1Ra reduced tumor colonies by 50% and tumor volume by 70% (Citation43).
There is evidence of therapeutic efficacy in blocking the IL-1β pathway in humans with IL-1Ra and IL-1 inhibitors. Anakinra is an antagonist that interferes with the cross-talk between bone-marrow and myeloma cells, where IL-1β and IL-6 are the major effector molecules. In myeloma treated with dexamethasone and anakinra long progression-free intervals of over 3 or 4 years were obtained in some patients, indicating a long stabilization of the disease (Citation44).
More potent IL-1 inhibitors are reaching clinical evaluation, in particular for autoimmune diseases, opening a possible future use in anti-cancer therapies. An IL-1-blocking antibody (canakinumab) has been approved for the treatment of a group of rare familial cold auto-inflammatory syndromes (CAPS) (Citation45). Also rilonacept (a dimeric fusion protein consisting of the extracellular domain of IL-1R and the Fc domain of human IgG1) has been approved for the treatment of CAPS (Citation46). For these IL-1-blocking molecules no hematopoietic and/or gastrointestinal toxicities have been reported. In particular—and differently from what is observed with anti-TNF therapy—there is no increase in opportunistic infections (Citation37).
IL-6
IL-6 is a key growth-regulating and anti-apoptotic inflammatory cytokine and is also one of the effector signals of activated NF-kB and oncogenic Ras signaling in the promotion of neoplasia (Citation47–49). IL-6 has tumor-inducing activities on both malignant and stromal cells. A clear pro-tumoral role has been demonstrated in multiple myeloma (Citation50,Citation51); accordingly, IL-6-deficient mice were resistant to the development of murine plasmocytoma (Citation52). IL-6 also enhances angiogenesis and vascular remodeling and increases the transcriptional up-regulation of VEGF in tumor cells; conversely, VEGF induces up-regulation of IL-6 in endothelial and stromal cells (Citation53,Citation54).
In the last years, the involvement of IL-6 in colorectal cancer has been deeply investigated, highlighting NF-kB as a link (Citation55,Citation56). In mouse models of colitis, IL-6 is mainly produced by innate immune cells in response to intestinal injury and in an NF-kB-dependent manner (Citation57).
IL-6 activates the JAK–STAT3 (Janus kinase–signal transducer and activator of transcription 3) pathway which induces the expression of genes important for cell cycle progression (such as cyclin D and PCNA), and suppression of apoptosis (Bcl-XL, Bcl-2, and Mcl-1) (Citation58,Citation59). Therefore IL-6 is considered an important regulator of intestinal epithelial cell proliferation during colitis-associated tumorigenesis.
Another example of IL-6-dependent tumor is the hepatocellular carcinoma (HCC), the most common type of liver cancer, frequently occurring after years of viral-induced chronic inflammation or sustained alcohol consumption. IL-6 serum levels in these patients at risk of HCC are usually elevated (Citation60–62). Naugler et al. explored the molecular mechanisms to explain the higher incidence of liver cancer in males. It turned out that in females estrogens block IL-6 production in local immune cells (liver Kupffer cells). In IL-6-deficient mice, and in mice lacking the adaptor protein MyD88 (myeloid differentiation primary response gene 88) (linked to NF-kB signaling), the sex difference in susceptibility to cancer was absent. Once more, IL-6 released by innate immune cells stimulated by tissue injury triggers local inflammation, epithelial cell proliferation, and carcinogenesis (Citation63–65).
IL-6 is a protagonist also in prostate cancer where its reduction delays tumor growth and ameliorates cancer cell susceptibility to docetaxel and bortezomib (Citation66). In squamous cell carcinoma of the head and neck (HNSCC), expression of IL-6 is associated with increased disease invasiveness as well as with poor patient prognosis and recurrence (Citation67). Moreover, IL-6 may tip the balance between cancer stem cell (CSC) and non-stem cancer cells (NSCC). IL-6 is sufficient to convert NSCCs into CSCs in genetically different tumor cell lines and human breast tumors (Citation68).
Antibodies to IL-6 have been safely administered to patients with lymphoproliferative diseases or solid cancer. CNTO 328 is a mouse–human chimeric anti-IL-6 antibody in phase II development for the potential treatment of multiple myeloma and renal cell carcinoma. In lung cancer, CNTO 328 inhibits STAT3 tyrosine phosphorylation, inducing a reduction of cancer cell growth in vivo (Citation69). A murine anti-IL-6 monoclonal antibody, elsilimomab (also known as B-E8) is in clinical phase II for multiple myeloma and metastatic renal cell carcinoma, showing preliminary efficacy (Citation70). Tocilizumab, a humanized anti-IL-6 receptor antibody (also known as MRA, R-1569, or humanized PM-1), was recently approved in Japan for the treatment of Castleman's disease, a rare lymphoproliferative disease associated with high IL-6 levels (Citation71). This reagent is also in clinical development for rheumatoid arthritis, Crohn's disease, myeloproliferative disorders, and systemic lupus erythematosus (Citation72). Overall, these clinical data show that IL-6 is a promising cytokine to target in oncology.
Chemokines and their involvement in tumor progression
Chemokines and their receptors are key players of the cancer-related inflammation and are considered important potential targets of future anti-tumor treatments. They have long been associated with the recruitment of leukocytes in tumors and therefore with the sustained presence of inflammatory cells (Citation73,Citation74). Tumor-produced chemokines are downstream of genetic events that cause neoplastic transformation but are also abundantly expressed by infiltrating leukocytes and stromal cells. These molecules affect multiple pathways of tumor progression, including angiogenesis, tumor cell proliferation and survival, invasion, and metastasis. Both ligands and receptors have been considered for the development of innovative therapeutic strategies, with CCL2 (see below) and CXCR4 being the preferential targets.
CXCR4 is the most studied chemokine receptor in tumors. The CXCL12/CXCR4 axis is involved in several aspects of tumor progression (e.g. angiogenesis, metastasis, and cell survival) in an ample panel of cancer types, including breast, prostate, pancreatic, colorectal, and non-small cell lung cancer as well as ovarian, esophageal, and bladder carcinoma (Citation75–78).
CXCR4 has been targeted by small antagonists, like the bicyclam AMD3100 (plerixafor), and peptides designed to the N-terminal region: TN14003 and CTCE-9908. In experimental mouse tumor models TN14003 reduced pulmonary metastasis, whereas CTCE-9908 showed inhibitory effects on primary as well as metastatic melanoma, osteosarcoma, and prostate tumors (Citation79–81). Recently Hassans et al. published evidence that also primary breast tumor growth and metastasis were reduced by CTCE-9908 with concomitant enhanced efficacy of anti-VEGF treatment, in a transgenic mouse model (Citation82).
Due to its effect in disrupting the adhesion of hematopoietic stem cells (HSC) within the bone-marrow niche, AMD3100/plerixafor is in clinical use for HSC mobilization in patients with non-Hodgkin's lymphoma and multiple myeloma, receiving autologous transplantation. Furthermore, this drug enhances the sensitivity of multiple myeloma or acute myeloid leukemia blasts to cytotoxic chemotherapy (Citation83). Plerixafor and BKT 140 (another CXCR4 antagonist) are both being investigated in human clinical trials in different types of cancer (Citation84).
The CXC chemokines bearing the ELR motif (e.g. CXCL1, CXCL2, and the most popular CXCL8) have potent angiogenic function and activate the proliferation, migration, and invasion of endothelial cells (Citation85). In different experimental cancer models, expression of CXC ELR + chemokines has been associated with neo-angiogenesis and tumor progression; conversely, depletion or inhibition of chemokines in these models resulted in attenuation of tumor growth and spontaneous metastases and reduced angiogenesis (Citation86,Citation87).
Several human studies confirmed that the high expression of CXCL8 and the extent of angiogenesis were associated with more aggressive diseases and poor prognosis in different tumor types (Citation88,Citation89). In an autocrine loop, melanoma cells produce CXCL8 to increase their proliferation (Citation74). Patients with melanoma metastases have elevated serum levels of CXCL8 (Citation90). Similarly, high levels of CXCL5 and CXCL8 were found in human non-small cell lung cancer (NSCLC) (Citation91,Citation92).
CXCL8 inhibitory molecules are being studied for anti-cancer therapies. Treatment of nude mice bearing human tumor xenografts with ABX-IL-8, a neutralizing anti-CXCL8 antibody, reduced tumor growth and inhibited neo-vascularization (Citation93).
Few other chemokine receptors have been targeted for clinical use. Cutaneous T cell lymphoma and T cell acute lymphoblastic leukemia express the chemokine receptor CCR4 (Citation94,Citation95). A humanized defucosylated antibody to CCR4 has been developed and showed anti-tumoral activity in lymphoma-bearing mice (Citation96). Of interest, this antibody also increased the number of tumor-infiltrating CD56 + natural killer cells mediating antibody-dependent cytotoxicity, and reduced the number of FOXP3+ Treg cells.
Targeting the corrupted immune system
Solid tumors are composed of malignant cells but also of bone-marrow-derived leukocytes, vessels, and resident stromal cells, forming a complex organ-like structure. The relationship between the immune system and malignancy is particularly complex. Tumor-infiltrating CD3 lymphocytes are usually associated with more favorable clinical outcome in human melanoma, ovarian, and colorectal cancer (Citation97–100).
On the other hand, the presence of high numbers of tumor-associated macrophages (TAM) is frequently correlated with tumor progression (Citation2,Citation101–104). It is believed that the tumor micro-environment influences blood-derived monocytes to acquire an immunosuppressive and pro-tumor phenotype (Citation105,Citation106). Along a current concept, macrophage activities are influenced by specific factors to which they are exposed during terminal differentiation. For instance, in the presence of bacterial products (e.g. lipopolysaccharide) and Th1 cytokines (e.g. IFNγ) they become M1 or classically activated macrophages with immunostimulatory activity. In the presence of Th2 cytokines (e.g. IL-4 and IL-13) or immunosuppressive factors (e.g. TGFβ and IL-10) they differentiate into M2 macrophages, whose physiological function is to promote tissue regeneration and to dampen Th1 immune responses. In the tumor micro-environment, where M1-inducing stimuli are absent, macrophage polarization is shifted to an M2 type.
Indeed TAM resemble M2 macrophages and display several pro-tumor functions: they produce growth factors for cancer cells and vessels that contribute to tumor proliferation, sustain matrix remodeling and tumor invasion by releasing activated proteases, and suppress adaptive anti-tumor immunity (Citation102,Citation105,Citation107–109). Thus, TAM are key players of the smoldering inflammation present in the tumor micro-environment that eventually promotes disease progression. Inhibition of their recruitment and/or survival at the tumor site could result in improved prognosis. In fact, in experimental mouse models of neoplasia, tumors grew less in macrophage-depleted animals (Citation1,Citation110,Citation111).
Earlier and more recent studies have shown that the bisphosphonate clodronate, encapsulated in liposomes, is an efficient reagent for the depletion of macrophages; this molecule has been developed and successfully applied in several immunological studies (Citation112,Citation113). Clodronate depletion of TAM in tumor-bearing mice resulted in reduced angiogenesis, tumor growth, and metastasis (Citation114,Citation115). Moreover, the combination of clodronate with sorafenib significantly increased the efficacy of sorafenib alone in a xenograft model of hepatocellular carcinoma (Citation115).
In clinical practice, bisphosphonates are commonly used to treat osteoporosis. Current applications in cancer treatment include their use to treat skeletal metastases in multiple myeloma, prostate, and breast cancers. Treatment with zoledronic acid was associated with a significant reduction of skeletal-related events and, possibly, with direct pro-apoptotic effects in tumor cells (Citation116).
Recruitment of circulating monocytes within the tumor mass is regulated by several chemokines produced locally, as well as other factors such as VEGF and macrophage colony-stimulating factor (M-CSF). Among the many chemokines, CCL2 occupies a prominent role and has been selected for therapeutic purposes. In addition, CCL2 has been shown to have a direct promotional effect on tumor cell proliferation, migration, and survival (Citation117). Preclinical studies demonstrated that anti-CCL2 antibodies, given in combination with chemotherapy, were able to induce tumor regression and improved survival in prostate mouse cancer models (Citation118,Citation119). Antibodies or antagonists to the specific receptor (CCR2) were also able to reduce tumor burden in mice bearing prostatic cancer (Citation120,Citation121).
On the basis of this experimental evidence clinical studies with CNTO 888, a human anti-CCL2 monoclonal antibody, have been initiated and are now in phase I and phase II clinical trials. Combined therapies with standard cytoreductive treatments are also being evaluated.
The possibility to block macrophage recruitment was obtained also with modified chemokines and a CCR5 antagonist. Treatment of mice with Met-CCL5 significantly reduced tumor volume and weight as well as the percentage of infiltrating TAM in experimental breast cancer (Citation122).
We have reported that the anti-tumor agent of marine origin, trabectedin, in addition to directly inhibiting tumor cell proliferation, has unique modulatory effects on the tumor micro-environment that can be related to targeting of myeloid tumor-associated cells. Trabectedin has been recently registered in Europe for the second-line treatment of soft tissue sarcoma and, in combination with cisplatin, for ovarian cancer. We reported that trabectedin is selectively cytotoxic to macrophages and TAM at therapeutic concentrations and also inhibits the transcription of selected inflammatory mediators such as CCL2, IL-6, and CXCL8, both in TAM and tumor cells (Citation123). Other chemokines involved in monocyte recruitment are also transcriptionally affected by trabectedin treatment, for example CCL7, CCL3, and CCL14 (Germano, unpublished data). Treatment with trabectedin in mice bearing human liposarcoma xenografts significantly reduced the chemokines CCL2 and CXCL8, IL-6, the angiogenic factor VEGF, and the number of vessels and TAM (Citation124). The anti-inflammatory properties of trabectedin on the tumor micro-environment, its selective cytotoxicity on monocytes/macrophages, combined with an efficient anti-proliferative effect on tumor cells, render this compound particularly promising in oncology (Citation125).
As mentioned above, tumor macrophages are major effectors of the neo-angiogenic switch occurring in growing tumors (Citation126). Further, it has been demonstrated that TAM and other myeloid cells are responsible for the resistance to anti-VEGF therapy (Citation127). Anti-angiogenic treatments have clearly demonstrated efficacy in various malignancies, especially when combined with conventional cytotoxic chemotherapy. Bevacizumab is a humanized anti-VEGF antibody used in patients with advanced colorectal, breast, ovarian, and renal cancer (Citation126). Therapeutic strategies that combine anti-angiogenic treatments and targeting of tumor-associated myeloid cells are likely to have increased clinical efficacy.
Tumor-infiltrating macrophages are potentially able to induce anti-tumor innate responses when appropriately stimulated. A distinct immunotherapeutic approach is to revert their immune-suppressive phenotype and thus overcome tumor escape mechanisms. Activation of the Toll-like receptor 9 (TLR9) receptor with agonists (e.g. CpG oligodeoxynucleotides) increases antigen presentation and boosts anti-tumor immunity. In preclinical studies, treatment with a combination of CpG, the chemokine CCL16, and an IL-10R antibody, achieved major responses in mice bearing large tumors. This treatment has been shown to shift resident and newly recruited macrophages from an M2 into an M1 type, to induce massive hemorrhagic necrosis, tumor shrinkage, and also the generation of tumor-specific immune responses (Citation128). TLR9 agonists are, therefore, a promising approach to overcome immune suppression in cancer patients (Citation129).
Still in the area of tumor-induced immune escape, critical enzymes regulating amino acid metabolic processes are importantly implicated in the deregulation of T cell activation. Elevated levels of the enzymes indoleamine 2,3-dioxygenase (IDO) or arginases are responsible for the tumor-induced tolerance observed in a number of experimental models (Citation130). Treatment with inhibitors of these enzymes may thus restore T cell surveillance and also improve responses to conventional chemotherapy or vaccination strategies (Citation131,Citation132).
Cell adhesion: targeting focal adhesion kinase
The communication of tumor tissues with the surrounding micro-environment is a crucial requirement for cell proliferation and survival to apoptotic stimuli. Contact points between extra-cellular matrix (ECM) and cells are largely mediated by adhesive integrins and focal adhesion kinase (FAK). FAK forms a complex network with membrane integrins and cytoplasmic proteins, thus building a plaque that links the ECM to the actin cytoskeleton (Citation133–135). FAK activity is crucial for cell motility, and this kinase has been investigated in malignancy for its potential involvement in metastasis dissemination (Citation136–138), as well as for other important activities such as continuous matrix degradation (Citation138), neo-formation of vessels (Citation139–141), and resistance to cytotoxic chemotherapy (Citation142,Citation143).
FAK is frequently over-expressed in tumors, and its expression correlates with negative prognosis in hepatocellular carcinoma (Citation144,Citation145), invasive breast carcinoma (Citation146), endometrial cancer (Citation147), and acute myelogenous leukemia (Citation148). A recent study showed that FAK supports Ras- and PI3K-dependent initiation of mammary tumors and progression to metastasis by orchestrating multiple cellular functions, including proliferation, survival, and avoidance of senescence (Citation149). However, no prognostic value has been found in other tumors such as colon carcinoma (Citation150), node-negative breast cancer (Citation146), and pancreatic cancer (Citation151). These contradictory results reflect the multiple roles of FAK as well as the high dynamic regulation of this molecule. The role of FAK in tumors can also be indirect, as shown recently for a novel regulator of FAK signaling (RGNEF) that was strongly involved in colon carcinoma motility and invasion (Citation152).
Inhibition of FAK activity may have effects on distant tumor spread by limiting cell migration, angiogenesis, and chemo-resistance (Citation137,Citation139,Citation142). Different synthetic molecules have been developed, in particular ATP-competitive FAK inhibitors, which showed promising results in preclinical settings and are now in phase I and II clinical trials (Citation135). Furthermore, the clinical use of FAK inhibitors in combination with cytotoxic therapies deserves additional consideration.
Conclusion
In the last century anti-cancer therapies concentrated on natural or engineered molecules having the ability to target solely the neoplastic cells. Often these strategies do not completely eradicate the tumor mass and select for resistant clones of cancer cells able to repopulate tumor sites. The key pro-tumoral role of the inflammatory micro-environment is now established. A new and distinct point of view is to develop therapies capable of creating a hostile niche for neoplastic cells. Therapies able to restrain inflammatory factors together with the elimination of the corrupted immune system could be a ‘good’ starting-point. The possibility to contrast cancer cells with a single molecule is obviously naive. A set of different molecules able to target tumor cells, able to normalize the angiogenic network and to restore the correct function of immune cells, could increase the chance of success (). Although anti-inflammatory therapies have yet to make a breakthrough in clinical oncology—and much further work must be done—they constitute a great opportunity to have an add-on therapy toward cancer solution.
Declaration of interest: This work was supported by the Italian Association for Cancer Research (AIRC) and by grant from Regione Lombardia NEPENTE.
References
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7.
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44.
- Balkwill F, Mantovani A. Cancer and inflammation: implications for pharmacology and therapeutics. Clin Pharmacol Ther. 2010;87:401–6.
- Porta C, Riboldi E, Sica A. Mechanisms linking pathogens-associated inflammation and cancer. Cancer Lett. 2010; 305:250–62.
- Flossmann E, Rothwell PM. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet. 2007;369:1603–13.
- Flossmann E, Rothwell PM. Commentary: aspirin and colorectal cancer an epidemiological success story. Int J Epidemiol. 2007;36:962–5.
- Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, . Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006;355:873–84.
- Steinbach G, Lynch PM, Phillips RK, Wallace MH, Hawk E, Gordon GB, . The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342:1946–52.
- Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9:361–71.
- Hagemann T, Wilson J, Kulbe H, Li NF, Leinster DA, Charles K, . Macrophages induce invasiveness of epithelial cancer cells via NF-kappa B and JNK. J Immunol. 2005;175:1197–205.
- Charles KA, Kulbe H, Soper R, Escorcio-Correia M, Lawrence T, Schultheis A, . The tumor-promoting actions of TNF-alpha involve TNFR1 and IL-17 in ovarian cancer in mice and humans. J Clin Invest. 2009;119:3011–23.
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, . NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6.
- Kulbe H, Thompson R, Wilson JL, Robinson S, Hagemann T, Fatah R, . The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res. 2007;67:585–92.
- Shealy DJ, Visvanathan S. Anti-TNF antibodies: lessons from the past, roadmap for the future. Handb Exp Pharmacol. 2008(181):101–29.
- Nakada MT, Tam SH, Woulfe DS, Casper KA, Swerlick RA, Ghrayeb J. Neutralization of TNF by the antibody cA2 reveals differential regulation of adhesion molecule expression on TNF-activated endothelial cells. Cell Adhes Commun. 1998;5:491–503.
- Larkin JM, Ferguson TR, Pickering LM, Edmonds K, James MG, Thomas K, . A phase I/II trial of sorafenib and infliximab in advanced renal cell carcinoma. Br J Cancer. 2010;103:1149–53.
- Zidi I, Mestiri S, Bartegi A, Amor NB. TNF-alpha and its inhibitors in cancer. Med Oncol. 2010;27:185–98.
- Askling J, Fored CM, Baecklund E, Brandt L, Backlin C, Ekbom A, . Haematopoietic malignancies in rheumatoid arthritis: lymphoma risk and characteristics after exposure to tumour necrosis factor antagonists. Ann Rheum Dis. 2005;64:1414–20.
- Nair B, Raval G, Mehta P. TNF-alpha inhibitor etanercept and hematologic malignancies: report of a case and review of the literature. Am J Hematol. 2007;82:1022–4.
- Sandborn WJ, Hanauer SB. Antitumor necrosis factor therapy for inflammatory bowel disease: a review of agents, pharmacology, clinical results, and safety. Inflamm Bowel Dis. 1999;5:119–33.
- Kianmanesh A, Hackett NR, Lee JM, Kikuchi T, Korst RJ, Crystal RG. Intratumoral administration of low doses of an adenovirus vector encoding tumor necrosis factor alpha together with naive dendritic cells elicits significant suppression of tumor growth without toxicity. Hum Gene Ther. 2001;12:2035–49.
- Kumar S, Witzig TE, Rajkumar SV. Thalidomide as an anti-cancer agent. J Cell Mol Med. 2002;6:160–74.
- Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P, . Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341:1565–71.
- Keifer JA, Guttridge DC, Ashburner BP, Baldwin AS Jr. Inhibition of NF-kappa B activity by thalidomide through suppression of IkappaB kinase activity. J Biol Chem. 2001;276:22382–7.
- Haslett PA, Corral LG, Albert M, Kaplan G. Thalidomide costimulates primary human T lymphocytes, preferentially inducing proliferation, cytokine production, and cytotoxic responses in the CD8 + subset. J Exp Med. 1998;187:1885–92.
- Laber DA, Khan MI, Kloecker GH, Schonard C, Taft BS, Salvador C. A phase I study of thalidomide, capecitabine and temozolomide in advanced cancer. Cancer Biol Ther. 2007;6:840–5.
- D'Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A. 1994;91:4082–5.
- Teo SK. Properties of thalidomide and its analogues: implications for anticancer therapy. AAPS J. 2005;7:E14–9.
- Curnis F, Sacchi A, Borgna L, Magni F, Gasparri A, Corti A. Enhancement of tumor necrosis factor alpha antitumor immunotherapeutic properties by targeted delivery to aminopeptidase N (CD13). Nat Biotechnol. 2000;18: 1185–90.
- Gregorc V, Citterio G, Vitali G, Spreafico A, Scifo P, Borri A, . Defining the optimal biological dose of NGR-hTNF, a selective vascular targeting agent, in advanced solid tumours. Eur J Cancer. 2010;46:198–206.
- Santoro A, Pressiani T, Citterio G, Rossoni G, Donadoni G, Pozzi F, . Activity and safety of NGR-hTNF, a selective vascular-targeting agent, in previously treated patients with advanced hepatocellular carcinoma. Br J Cancer. 2010;103:837–44.
- Naldini A, Filippi I, Miglietta D, Moschetta M, Giavazzi R, Carraro F. Interleukin-1beta regulates the migratory potential of MDAMB231 breast cancer cells through the hypoxia-inducible factor-1alpha. Eur J Cancer. 2010;46:3400–8.
- Lewis AM, Varghese S, Xu H, Alexander HR. Interleukin-1 and cancer progression: the emerging role of interleukin-1 receptor antagonist as a novel therapeutic agent in cancer treatment. J Transl Med. 2006;4:48.
- Voronov E, Shouval DS, Krelin Y, Cagnano E, Benharroch D, Iwakura Y, . IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci U S A. 2003;100:2645–50.
- Dinarello CA. The paradox of pro-inflammatory cytokines in cancer. Cancer Metastasis Rev. 2006;25:307–13.
- Apte RN, Dotan S, Elkabets M, White MR, Reich E, Carmi Y, . The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006;25:387–408.
- Dinarello CA. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 2010;29:317–29.
- Arguello F, Baggs RB, Graves BT, Harwell SE, Cohen HJ, Frantz CN. Effect of IL-1 on experimental bone/bone-marrow metastases. Int J Cancer. 1992;52:802–7.
- Song X, Voronov E, Dvorkin T, Fima E, Cagnano E, Benharroch D, . Differential effects of IL-1 alpha and IL-1 beta on tumorigenicity patterns and invasiveness. J Immunol. 2003;171:6448–56.
- Carmi Y, Voronov E, Dotan S, Lahat N, Rahat MA, Fogel M, . The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. J Immunol. 2009;183:4705–14.
- Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, . Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–65.
- Chirivi RG, Garofalo A, Padura IM, Mantovani A, Giavazzi R. Interleukin 1 receptor antagonist inhibits the augmentation of metastasis induced by interleukin 1 or lipopolysaccharide in a human melanoma/nude mouse system. Cancer Res. 1993;53:5051–4.
- Vidal-Vanaclocha F, Amezaga C, Asumendi A, Kaplanski G, Dinarello CA. Interleukin-1 receptor blockade reduces the number and size of murine B16 melanoma hepatic metastases. Cancer Res. 1994;54:2667–72.
- Lust JA, Lacy MQ, Zeldenrust SR, Dispenzieri A, Gertz MA, Witzig TE, . Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1{beta}-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin Proc. 2009;84:114–22.
- Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, Quartier P, . Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med. 2009;360:2416–25.
- Hoffman HM, Throne ML, Amar NJ, Sebai M, Kivitz AJ, Kavanaugh A, . Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 2008;58:2443–52.
- Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–83.
- Naugler WE, Karin M. The wolf in sheep's clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med. 2008;14:109–19.
- Ancrile B, Lim KH, Counter CM. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007;21:1714–9.
- van Zaanen HC, Koopmans RP, Aarden LA, Rensink HJ, Stouthard JM, Warnaar SO, . Endogenous interleukin 6 production in multiple myeloma patients treated with chimeric monoclonal anti-IL6 antibodies indicates the existence of a positive feed-back loop. J Clin Invest. 1996;98:1441–8.
- Pal R, Janz M, Galson DL, Gries M, Li S, Johrens K, . C/EBPbeta regulates transcription factors critical for proliferation and survival of multiple myeloma cells. Blood. 2009;114:3890–8.
- Lattanzio G, Libert C, Aquilina M, Cappelletti M, Ciliberto G, Musiani P, . Defective development of pristane-oil-induced plasmacytomas in interleukin-6-deficient BALB/c mice. Am J Pathol. 1997;151:689–96.
- Ara T, Declerck YA. Interleukin-6 in bone metastasis and cancer progression. Eur J Cancer. 2010;46:1223–31.
- Dankbar B, Padro T, Leo R, Feldmann B, Kropff M, Mesters RM, . Vascular endothelial growth factor and interleukin-6 in paracrine tumor-stromal cell interactions in multiple myeloma. Blood. 2000;95:2630–6.
- Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, . Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–30.
- Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, . Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–44.
- Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, . IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009 Feb 3;15(2): 103–13.
- Lamb J, Ramaswamy S, Ford HL, Contreras B, Martinez RV, Kittrell FS, . A mechanism of cyclin D1 action encoded in the patterns of gene expression in human cancer. Cell. 2003;114:323–34.
- Liu Y, Li PK, Li C, Lin J. Inhibition of STAT3 signaling blocks the anti-apoptotic activity of IL-6 in human liver cancer cells. J Biol Chem. 2010;285:27429–39.
- Khoruts A, Stahnke L, McClain CJ, Logan G, Allen JI. Circulating tumor necrosis factor, interleukin-1 and interleukin-6 concentrations in chronic alcoholic patients. Hepatology. 1991;13:267–76.
- Kakumu S, Shinagawa T, Ishikawa T, Yoshioka K, Wakita T, Ito Y, . Serum interleukin 6 levels in patients with chronic hepatitis B. Am J Gastroenterol. 1991;86:1804–8.
- Malaguarnera M, Di Fazio I, Laurino A, Ferlito L, Romano M, Trovato BA. Serum interleukin 6 concentrations in chronic hepatitis C patients before and after interferon-alpha treatment. Int J Clin Pharmacol Ther. 1997;35:385–8.
- Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marme D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 1996;87:3336–43.
- Gregg SD, Campbell JL, Fisher JW, Bartlett MG. Methods for the characterization of Jet Propellent-8: vapor and aerosol. Biomed Chromatogr. 2007;21:463–72.
- Dossus L, Kaaks R, Canzian F, Albanes D, Berndt SI, Boeing H, . PTGS2 and IL6 genetic variation and risk of breast and prostate cancer: results from the Breast and Prostate Cancer Cohort Consortium (BPC3). Carcinogenesis. 2010;31:455–61.
- Sakai I, Miyake H, Terakawa T, Fujisawa M. Inhibition of tumor growth and sensitization to chemotherapy by RNA interference targeting interleukin-6 in the androgen-independent human prostate cancer PC3 model. Cancer Sci. 2011;102:769–75.
- Duffy SA, Taylor JM, Terrell JE, Islam M, Li Y, Fowler KE, . Interleukin-6 predicts recurrence and survival among head and neck cancer patients. Cancer. 2008;113:750–7.
- Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A. 2011;108:1397–402.
- Song L, Rawal B, Nemeth JA, Haura EB. JAK1 activates STAT3 activity in non-small-cell lung cancer cells and IL-6 neutralizing antibodies can suppress JAK1-STAT3 signaling. Mol Cancer Ther. 2011;10:481–94.
- Trikha M, Corringham R, Klein B, Rossi JF. Targeted anti-interleukin-6 monoclonal antibody therapy for cancer: a review of the rationale and clinical evidence. Clin Cancer Res. 2003;9:4653–65.
- Ahmed B, Tschen JA, Cohen PR, Zaki MH, Rady PL, Tyring SK, . Cutaneous castleman's disease responds to anti interleukin-6 treatment. Mol Cancer Ther. 2007;6:2386–90.
- Naka T, Nishimoto N, Kishimoto T. The paradigm of IL-6: from basic science to medicine. Arthritis Res. 2002;4 Suppl 3:S233–42.
- Mantovani A. Molecular pathways linking inflammation and cancer. Curr Mol Med. 2010;10:369–73.
- Allavena P, Germano G, Marchesi F, Mantovani A. Chemokines in cancer related inflammation. Exp Cell Res. 2010;317:664–73.
- Zlotnik A. Chemokines and cancer. Int J Cancer. 2006;119:2026–9.
- Kryczek I, Wei S, Keller E, Liu R, Zou W. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. Am J Physiol Cell Physiol. 2007;292:C987–95.
- Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16:2927–31.
- Mantovani A, Savino B, Locati M, Zammataro L, Allavena P, Bonecchi R. The chemokine system in cancer biology and therapy. Cytokine Growth Factor Rev. 2010;21:27–39.
- Kim SY, Lee CH, Midura BV, Yeung C, Mendoza A, Hong SH, . Inhibition of the CXCR4/CXCL12 chemokine pathway reduces the development of murine pulmonary metastases. Clin Exp Metastasis. 2008;25:201–11.
- Porvasnik S, Sakamoto N, Kusmartsev S, Eruslanov E, Kim WJ, Cao W, . Effects of CXCR4 antagonist CTCE-9908 on prostate tumor growth. Prostate. 2009;69: 1460–9.
- Richert MM, Vaidya KS, Mills CN, Wong D, Korz W, Hurst DR, . Inhibition of CXCR4 by CTCE-9908 inhibits breast cancer metastasis to lung and bone. Oncol Rep. 2009;21:761–7.
- Hassan S, Buchanan M, Jahan K, Aguilar-Mahecha A, Gaboury L, Muller WJ, . CXCR4 peptide antagonist inhibits primary breast tumor growth, metastasis and enhances the efficacy of anti-VEGF treatment or docetaxel in a transgenic mouse model. Int J Cancer. 2010;129: 225–32.
- De Clercq E. The AMD3100 story: the path to the discovery of a stem cell mobilizer (Mozobil). Biochem Pharmacol. 2009;77:1655–64.
- Burger JA, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2009;23:43–52.
- Raman D, Baugher PJ, Thu YM, Richmond A. Role of chemokines in tumor growth. Cancer Lett. 2007;256: 137–65.
- Arenberg DA, Keane MP, DiGiovine B, Kunkel SL, Morris SB, Xue YY, . Epithelial-neutrophil activating peptide (ENA-78) is an important angiogenic factor in non-small cell lung cancer. J Clin Invest. 1998;102:465–72.
- Bonecchi R, Savino B, Borroni EM, Mantovani A, Locati M. Chemokine decoy receptors: structure-function and biological properties. Curr Top Microbiol Immunol. 2010;341:15–36.
- Bizzarri C, Beccari AR, Bertini R, Cavicchia MR, Giorgini S, Allegretti M. ELR + CXC chemokines and their receptors (CXC chemokine receptor 1 and CXC chemokine receptor 2) as new therapeutic targets. Pharmacol Ther. 2006;112:139–49.
- Desbaillets I, Diserens AC, Tribolet N, Hamou MF, Van Meir EG. Upregulation of interleukin 8 by oxygen-deprived cells in glioblastoma suggests a role in leukocyte activation, chemotaxis, and angiogenesis. J Exp Med. 1997;186:1201–12.
- Scheibenbogen C, Mohler T, Haefele J, Hunstein W, Keilholz U. Serum interleukin-8 (IL-8) is elevated in patients with metastatic melanoma and correlates with tumour load. Melanoma Res. 1995;5:179–81.
- Smith DR, Polverini PJ, Kunkel SL, Orringer MB, Whyte RI, Burdick MD, . Inhibition of interleukin 8 attenuates angiogenesis in bronchogenic carcinoma. J Exp Med. 1994;179:1409–15.
- Belperio JA, Keane MP, Arenberg DA, Addison CL, Ehlert JE, Burdick MD, . CXC chemokines in angiogenesis. J Leukoc Biol. 2000;68:1–8.
- Huang S, Mills L, Mian B, Tellez C, McCarty M, Yang XD, . Fully humanized neutralizing antibodies to interleukin-8 (ABX-IL8) inhibit angiogenesis, tumor growth, and metastasis of human melanoma. Am J Pathol. 2002;161:125–34.
- Ishida T, Ueda R. CCR4 as a novel molecular target for immunotherapy of cancer. Cancer Sci. 2006;97:1139–46.
- Ishida T, Utsunomiya A, Iida S, Inagaki H, Takatsuka Y, Kusumoto S, . Clinical significance of CCR4 expression in adult T-cell leukemia/lymphoma: its close association with skin involvement and unfavorable outcome. Clin Cancer Res. 2003;9(10 Pt 1):3625–34.
- Ito A, Ishida T, Yano H, Inagaki A, Suzuki S, Sato F, . Defucosylated anti-CCR4 monoclonal antibody exercises potent ADCC-mediated antitumor effect in the novel tumor-bearing humanized NOD/Shi-scid, IL-2Rgamma(null) mouse model. Cancer Immunol Immunother. 2009;58:1195–206.
- Taylor RC, Patel A, Panageas KS, Busam KJ, Brady MS. Tumor-infiltrating lymphocytes predict sentinel lymph node positivity in patients with cutaneous melanoma. J Clin Oncol. 2007;25:869–75.
- Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, . Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–13.
- Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, . Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353:2654–66.
- Laghi L, Bianchi P, Miranda E, Balladore E, Pacetti V, Grizzi F, . CD3 + cells at the invasive margin of deeply invading (pT3-T4) colorectal cancer and risk of post-surgical metastasis: a longitudinal study. Lancet Oncol. 2009;10:877–84.
- Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–45.
- Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55.
- Mantovani A. Cancer: an infernal triangle. Nature. 2007;448:547–8.
- Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196: 254–65.
- Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–8.
- Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–6.
- Mantovani A, Allavena P, Sica A. Tumour-associated macrophages as a prototypic type II polarised phagocyte population: role in tumour progression. Eur J Cancer. 2004;40:1660–7.
- Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42:717–27.
- Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86: 1065–73.
- Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, . Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010;70:5728–39.
- Nagai T, Tanaka M, Tsuneyoshi Y, Xu B, Michie SA, Hasui K, . Targeting tumor-associated macrophages in an experimental glioma model with a recombinant immunotoxin to folate receptor beta. Cancer Immunol Immunother. 2009;58:1577–86.
- Seiler P, Aichele P, Odermatt B, Hengartner H, Zinkernagel RM, Schwendener RA. Crucial role of marginal zone macrophages and marginal zone metallophils in the clearance of lymphocytic choriomeningitis virus infection. Eur J Immunol. 1997;27:2626–33.
- Tate MD, Pickett DL, van Rooijen N, Brooks AG, Reading PC. Critical role of airway macrophages in modulating disease severity during influenza virus infection of mice. J Virol. 2010;84:7569–80.
- Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjallman AH, Ballmer-Hofer K, Schwendener RA. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. Br J Cancer. 2006;95:272–81.
- Zhang W, Zhu XD, Sun HC, Xiong YQ, Zhuang PY, Xu HX, . Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res. 2010;16:3420–30.
- Morgan GJ, Davies FE, Gregory WM, Cocks K, Bell SE, Szubert AJ, . First-line treatment with zoledronic acid as compared with clodronic acid in multiple myeloma (MRC Myeloma IX): a randomised controlled trial. Lancet. 2010;376:1989–99.
- Hembruff SL, Jokar I, Yang L, Cheng N. Loss of transforming growth factor-beta signaling in mammary fibroblasts enhances CCL2 secretion to promote mammary tumor progression through macrophage-dependent and -independent mechanisms. Neoplasia. 2010;12:425–33.
- Loberg RD, Ying C, Craig M, Day LL, Sargent E, Neeley C, . Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007;67:9417–24.
- Rozel S, Galban CJ, Nicolay K, Lee KC, Sud S, Neeley C, . Synergy between anti-CCL2 and docetaxel as determined by DW-MRI in a metastatic bone cancer model. J Cell Biochem. 2009;107:58–64.
- Li X, Loberg R, Liao J, Ying C, Snyder LA, Pienta KJ, . A destructive cascade mediated by CCL2 facilitates prostate cancer growth in bone. Cancer Res. 2009;69:1685–92.
- Popivanova BK, Kostadinova FI, Furuichi K, Shamekh MM, Kondo T, Wada T, . Blockade of a chemokine, CCL2, reduces chronic colitis-associated carcinogenesis in mice. Cancer Res. 2009;69:7884–92.
- Robinson SC, Scott KA, Wilson JL, Thompson RG, Proudfoot AE, Balkwill FR. A chemokine receptor antagonist inhibits experimental breast tumor growth. Cancer Res. 2003;63:8360–5.
- Allavena P, Signorelli M, Chieppa M, Erba E, Bianchi G, Marchesi F, . Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): inhibition of macrophage differentiation and cytokine production. Cancer Res. 2005;65:2964–71.
- Germano G, Frapolli R, Simone M, Tavecchio M, Erba E, Pesce S, . Antitumor and anti-inflammatory effects of trabectedin on human myxoid liposarcoma cells. Cancer Res. 2010;70:2235–44.
- D'Incalci M, Galmarini CM. A review of trabectedin (ET-743): a unique mechanism of action. Mol Cancer Ther. 2010;9:2157–63.
- Roodink I, Leenders WP. Targeted therapies of cancer: angiogenesis inhibition seems not enough. Cancer Lett. 2010;299:1–10.
- Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, . G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A. 2009;106:6742–7.
- Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005;65:3437–46.
- Holtick U, Scheulen ME, von Bergwelt-Baildon MS, Weihrauch MR. Toll-like receptor 9 agonists as cancer therapeutics. Expert Opin Investig Drugs. 2011;20:361–72.
- Grohmann U, Bronte V. Control of immune response by amino acid metabolism. Immunol Rev. 2010;236:243–64.
- Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, . Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–74.
- Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005; 11:312–9.
- Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, . Nanoscale architecture of integrin-based cell adhesions. Nature. 2011;468:580–4.
- Schwock J, Dhani N, Hedley DW. Targeting focal adhesion kinase signaling in tumor growth and metastasis. Expert Opin Ther Targets. 2010;14:77–94.
- Schultze A, Fiedler W. Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer. Expert Opin Investig Drugs. 2010;19:777–88.
- McLean GW, Komiyama NH, Serrels B, Asano H, Reynolds L, Conti F, . Specific deletion of focal adhesion kinase suppresses tumor formation and blocks malignant progression. Genes Dev. 2004;18:2998–3003.
- Sieg DJ, Hauck CR, Schlaepfer DD. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J Cell Sci. 1999;112 (Pt 16):2677–91.
- Hauck CR, Hsia DA, Puente XS, Cheresh DA, Schlaepfer DD. FRNK blocks v-Src-stimulated invasion and experimental metastases without effects on cell motility or growth. EMBO J. 2002;21:6289–302.
- Mitra SK, Mikolon D, Molina JE, Hsia DA, Hanson DA, Chi A, . Intrinsic FAK activity and Y925 phosphorylation facilitate an angiogenic switch in tumors. Oncogene. 2006;25:5969–84.
- Naldini A, Leali D, Pucci A, Morena E, Carraro F, Nico B, . Cutting edge: IL-1beta mediates the proangiogenic activity of osteopontin-activated human monocytes. J Immunol. 2006;177:4267–70.
- Skuli N, Monferran S, Delmas C, Favre G, Bonnet J, Toulas C, . Alphavbeta3/alphavbeta5 integrins-FAK-RhoB: a novel pathway for hypoxia regulation in glioblastoma. Cancer Res. 2009;69:3308–16.
- Chen YY, Wang ZX, Chang PA, Li JJ, Pan F, Yang L, . Knockdown of focal adhesion kinase reverses colon carcinoma multicellular resistance. Cancer Sci. 2009; 100:1708–13.
- Huanwen W, Zhiyong L, Xiaohua S, Xinyu R, Kai W, Tonghua L. Intrinsic chemoresistance to gemcitabine is associated with constitutive and laminin-induced phosphorylation of FAK in pancreatic cancer cell lines. Mol Cancer. 2009;8:125.
- Fujii T, Koshikawa K, Nomoto S, Okochi O, Kaneko T, Inoue S, . Focal adhesion kinase is overexpressed in hepatocellular carcinoma and can be served as an independent prognostic factor. J Hepatol. 2004;41: 104–11.
- Yuan Z, Fan J, Wu ZQ, Zhou J, Qiu SJ. [Focal adhesion kinase mRNA overexpression in hepatocellular carcinoma HCC) and correlation thereof with prognosis of HCC]. Zhonghua Yi Xue Za Zhi. 2007;87:1256–9.
- Lark AL, Livasy CA, Dressler L, Moore DT, Millikan RC, Geradts J, . High focal adhesion kinase expression in invasive breast carcinomas is associated with an aggressive phenotype. Mod Pathol. 2005;18:1289–94.
- Gabriel B, Hasenburg A, Waizenegger M, Orlowska-Volk M, Stickeler E, zur Hausen A. Expression of focal adhesion kinase in patients with endometrial cancer: a clinicopathologic study. Int J Gynecol Cancer. 2009;19:1221–5.
- Tavernier-Tardy E, Cornillon J, Campos L, Flandrin P, Duval A, Nadal N, . Prognostic value of CXCR4 and FAK expression in acute myelogenous leukemia. Leuk Res. 2009;33:764–8.
- Pylayeva Y, Gillen KM, Gerald W, Beggs HE, Reichardt LF, Giancotti FG. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J Clin Invest. 2009;119:252–66.
- Theocharis SE, Kouraklis GP, Kakisis JD, Kanelli HG, Apostolakou FE, Karatzas GM, . Focal adhesion kinase expression is not a prognostic predictor in colon adenocarcinoma patients. Eur J Surg Oncol. 2003;29:571–4.
- Furuyama K, Doi R, Mori T, Toyoda E, Ito D, Kami K, . Clinical significance of focal adhesion kinase in resectable pancreatic cancer. World J Surg. 2006;30: 219–26.
- Yu HG, Nam JO, Miller NL, Tanjoni I, Walsh C, Shi L, . p190RhoGEF (Rgnef) Promotes colon carcinoma tumor progression via interaction with focal adhesion kinase. Cancer Res. 2011;71:360–70.