Abstract
Reactive oxygen species (ROS) are signaling molecules that influence many physiological processes. Increased ROS bioavailability and altered redox signaling (oxidative stress) have been implicated in chronic diseases including hypertension. Although oxidative stress may not be the sole cause of hypertension, it amplifies blood pressure elevation in the presence of other prohypertensive factors (salt, renin-angiotensin system, sympathetic hyperactivity). A major source for cardiovascular ROS is a family of non-phagocytic NADPH oxidases (Nox1, Nox2, Nox4, Nox5). Other sources of ROS involve mitochondrial electron transport enzymes, xanthine oxidase, and uncoupled nitric oxide synthase. Although evidence from experimental and animal studies supports a role for oxidative stress in the pathogenesis of hypertension, there is still no convincing proof that oxidative stress is a cause of human hypertension. However, what is clear is that oxidative stress is important in the molecular mechanisms associated with cardiovascular and renal injury in hypertension and that hypertension itself can contribute to oxidative stress. The present review addresses the putative function of ROS in the pathogenesis of hypertension and focuses on the role of Noxs in ROS generation in vessels and the kidney. Implications of oxidative stress in human hypertension are discussed, and clinical uncertainties are highlighted.
Key messages
NADPH oxidases are a family of enzymes that generate reactive oxygen species (ROS), which contribute to the physiological regulation of vascular function.
Oxidative stress, through redox signaling, plays a role in cardiovascular and renal vascular inflammation and injury.
Extensive experimental evidence indicates that oxidative stress is involved in the pathogenesis of hypertension.
Clinical data have not yet demonstrated a causal association between oxidative stress and hypertension.
Development of isoform-specific NADPH oxidase inhibitors may have promise as therapeutic modalities in the management of patients with NADPH oxidase-related diseases. The therapeutic potential in hypertension is unclear.
Introduction
Hypertension is a major modifiable risk factor for renal failure, cardiovascular disease, and stroke (Citation1,Citation2). Hypertension affects 30% of adults in the Western world and is the leading cause of morbidity and mortality worldwide (Citation1). Although the exact etiology still remains largely unknown, it is clear that hypertension is a multifactorial, complex polygenic disorder with many interacting mechanisms contributing to its pathophysiology and involving many organ systems, including the heart, kidney, brain, vessels, and possibly the immune system (Citation2,Citation3). Factors implicated in the pathophysiology of hypertension include activation of the sympathetic nervous system, up-regulation of the renin-angiotensin-aldosterone system, altered G protein-coupled receptor signaling, and inflammation (Citation4,Citation5). Common to these processes is oxidative stress (increased bioavailability of reactive oxygen species (ROS)) due, primarily, to excess ROS generation, decreased nitric oxide (NO) levels, and reduced antioxidant capacity in the cardiovascular and renal systems (Citation6,Citation7).
Reactive oxygen species (ROS), including superoxide (•O2 − ) and hydrogen peroxide (H2O2), were originally considered to be injurious metabolic cellular by-products. However, ROS are now recognized to have important physiological actions, including the induction of host defense genes, stimulation of transcription factors, and activation of ion transporters (Citation8,Citation9). In the vascular system ROS play a physiological role in controlling endothelial function and vascular tone and a pathophysiological role in processes underlying endothelial dysfunction, hyperreactivity, and vascular remodeling in cardiovascular diseases, including hypertension. Superoxide anion and H2O2 influence signaling pathways that regulate vascular hypertrophy, inflammation, and contraction, including mitogen-activated protein kinases (MAPK), tyrosine kinases, Rho kinase, transcription factors (NFκB, AP-1, and HIF-1), and protein tyrosine phosphatases (PTP) (Citation10–12). http://hyper.ahajournals.org/cgi/content/ full/44/3/248 - R21 - 032003#R21 - 032003ROS also increase intracellular free Ca2 + concentration ([Ca2 + ]i) and up-regulate proto-oncogene and proinflammatory gene expression and activity (Citation13,Citation14).
The relationship between free radicals and hypertension was first suggested in the 1960s (Citation15), but it was some 40 years later that this association was investigated in detail when it was shown that Ang II-mediated hypertension in rats increases vascular ROS production via non-phagocytic NAD(P)H oxidase activation (Citation16). Almost all experimental models of hypertension have evidence of oxidative excess including genetic forms (SHR, SHR-SP), surgically induced (2K1C, aortic banding), hormone-induced (Ang II, aldosterone, DOCA), and diet-induced hypertension (salt, fat) (Citation17–20). Mice deficient in ROS-generating enzymes have lower blood pressure versus wild-type counterparts, and Ang II infusion fails to induce hypertension in these mice (Citation21,Citation22).
Since inhibition of ROS-generating enzymes, antioxidants, and ROS-scavengers lowers blood pressure whereas pro-oxidants increase blood pressure, it has been suggested that ROS are causally associated with hypertension, at least in animal models. However, in clinical hypertension the evidence is not so convincing. Most human studies examining ROS are based on associations between plasma markers of oxidative stress and blood pressure. Biomarkers of systemic oxidative stress, including levels of plasma thiobarbituric acid-reactive substances (TBARS) and 8-epi-isoprostanes, are elevated in patients with hypertension (Citation23,Citation24). Factors implicated in oxidative stress in human hypertension include decreased antioxidant activity, reduced levels of ROS scavengers, and a http://hyper.ahajournals.org/cgi/content/full/44/3/248 - R36 - 032003#R36 - 032003ctivation of ROS-generating enzymes (Citation25–28). A causal link between ROS and high blood pressure has not yet been definitively established in humans. Only a few small clinical studies showed a blood pressure-lowering effect of antioxidants (Citation25,Citation29), whereas many large antioxidant clinical trials failed to demonstrate any cardiovascular benefit and blood pressure reduction (Citation30,Citation31).
Production of ROS in the cardiovascular system
Reactive oxygen species are formed as intermediates in reduction–oxidation (redox) reactions leading from O2 to H2O. Of the ROS produced in the vascular system, •O2 − and H2O2 are particularly important. In biological systems, •O2 − is short-lived and unstable owing to its rapid reduction to H2O2 by superoxide dismutase (SOD) (Citation32). The charge on the superoxide anion makes it unable to cross cellular membranes except possibly through ion channels. H2O2 has a longer life-span than •O2 − , is relatively stable, and is easily diffusible within and between cells. The distinct chemical characteristics between •O2 − and H2O2 and their specific sites of localization mean that different species of ROS activate diverse signaling pathways, leading to divergent, and potentially opposing, cellular responses.
ROS are products of normal cellular metabolism and derive from many sources in different cellular compartments. Enzymatic sources of ROS in cardiovascular disease and hypertension include: xanthine oxidoreductase, uncoupled NO synthase (NOS), mitochondrial respiratory enzymes, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Citation33–36), found in many cell types in vessels, the heart, kidney, and central nervous system. Unlike most ROS-generating enzymes, which produce •O2 − and/or H2O2 either as by-products of their catalytic activity or as a result of abnormal functioning in pathological conditions, NADPH oxidase has as its sole function the generation of ROS, hence termed a ‘professional’ ROS-producer (Citation37). NADPH oxidase appears to be the major source of ROS in the cardiovascular and renal systems and is the focus of the present review.
Nox family NAD(P)H oxidases
NADPH oxidase was originally considered to be expressed only in phagocytic cells involved in host defense and innate immunity. It is now evident that there is a family of NAD(P)H oxidases, based on homologs of the catalytic subunit gp91phox, that are functionally active in non-phagocytic cells. The new homologs, along with gp91phox, are designated the Nox family of NAD(P)H oxidases (Citation38) and are important in vascular ROS production. The prototypical gp91phox-containing phagocytic NAD(P)H oxidase (now termed Nox2) comprises five subunits: p47phox (‘phox’ stands for phagocyte oxidase), p67phox, p40phox, p22phox, and the catalytic subunit gp91phox (Citation39). In basal conditions p47phox, p67phox, and p40phox exist in the cytosol, whereas p22phox and gp91phox are in the membrane, where they occur as a heterodimeric flavoprotein (cytochrome b558). Upon stimulation p47phox and p67phox form a complex that translocates to the membrane, where it associates with cytochrome b558 to assemble the active oxidase, which transfers electrons from the substrate to O2, forming •O2 − (Citation40). Activation also requires Rac2 (or Rac1) and Rap1A.
The mammalian Nox family comprises seven members: Nox1, Nox2, Nox3, Nox4, Nox5, Duox1, and Duox2 (Citation38–42). All are transmembrane proteins that have a core catalytic subunit (Nox) and numerous regulatory subunits. Nox1, Nox2, Nox4, and Nox5 have been identified in cardiovascular and renal tissue. Hyperactivation of Noxs leads to excessive ROS generation that disrupts redox networks, normally regulated by thiol-dependent antioxidant systems. This results in oxidative stress, triggering molecular processes, which, in the vasculature, contributes to vascular injury. The Noxs have been extensively reviewed (Citation43–46), and only an overview of recent developments is discussed here.
Nox1
Nox1, originally identified in colon tumor cells (Citation47), is expressed in vascular and cardiac cells. It localizes to the cell membrane, caveolae/lipid rafts, and endosomes. Nox1 is similar to Nox2 in that it requires p22phox, p47phox (or its homolog NoxO1 (Nox organizer 1)), p67phox (or its homolog NoxA1 (Nox activator 1)), and Rac1 for its activity. New Nox1 regulators have been identified, Tks4 and Tks5, which resemble p47phox and NoxO1, and which interact with NoxA1 (Citation48). Nox1 localizes with p22phox and is expressed at low levels in physiological conditions. Nox1-derived •O2 − is increased in a stimulus-dependent manner, involving complex interactions between regulatory subunits and the redox chaperone protein disulfide isomerase (PDI) (Citation49,Citation50). Nox1 has been implicated in vascular smooth muscle cell migration, proliferation, and extracellular matrix production, effects mediated by cofilin (Citation51).
In cultured endothelial and vascular smooth muscle cells Nox1 is up-regulated by mechanical factors (shear stress), vasoactive agents (Ang II, aldosterone), and growth factors (EGF, PDGF) (Citation52,Citation53). Ang II-induced induction of Nox1 may involve mitochondria, suggesting an interaction between Nox1 and mitochondria, possibly through a Ca2+ -dependent mechanism (Citation54). Nox1 expression/activity is increased in the vasculature in models of cardiovascular disease including hypertension, atherosclerosis, diabetes, and hypercholesterolemia (Citation55). Studies from Nox1 knock-out and transgenic mice suggest a possible role for Nox1 in acute, but not chronic, forms of Ang II-dependent hypertension (Citation56,Citation57), in atherosclerosis, restenosis post injury, endothelial dysfunction, and stroke. Mice genetically deficient in Nox1 also display decreased expression of aortic AT1R (Citation58), which may contribute to blunted hypertensive effects of Ang II infusion in these mice. Although there are extensive experimental data implicating Nox1 in cardiovascular disease, there is little information in humans, although expression of Nox1 and NoxA1 is increased in human atherosclerotic vessels (Citation59).
Nox2
Nox2 is the catalytic subunit of the respiratory burst oxidase in phagocytes but is also expressed in cardiovascular cells (Citation60). Nox2 is unstable without p22phox and requires p47phox, p67phox, and Rac1/2 for its full activation. In neutrophils Nox2 localizes to intracellular and plasma membranes, and in vascular cells it also localizes with the cytoskeleton, lipid rafts/caveolae, and in the perinuclear compartment. The Nox2 gene is inducible and is highly regulated by Ang II and stretch. Vascular Nox2, derived from resident macrophages or vascular cells, is up-regulated in experimental hypertension (Citation61,Citation62), atherosclerosis, ischemia-reperfusion injury, and neointimal formation. Although Nox2 has been shown to be important in models of Ang II-infused hypertension, it does not seem to play a role in blood pressure elevation or cardiac hypertrophy in a model of chronic Ang II-dependent hypertension (Citation63). Nox2 is also implicated in stroke in experimental models. Nox2-deficient mice exhibit significant reduction in cerebral infarct size compared with wild-type controls (Citation64). In humans, NADPH oxidase has been shown to play a role in endothelial function, since patients with chronic granulomatous disease (CGD) who have an X-linked Nox2 mutation exhibit a significant increase in forearm-mediated vasodilation with increased NO bioavailability (Citation65), suggesting that Nox2-based NADPH oxidase influences endothelial function and NO biology in humans. In patients with CGD with mutations in Nox2 or p47phox, endothelial ischemia-reperfusion injury was blunted, indicating a role for NADH oxidase-derived ROS in human ischemia-reperfusion injury (Citation66).
Nox4
Nox4, of which 4 splice variants have been identified (NOX4B, NOX4C, NOX4D and NOX4E), is found in vascular cells, fibroblasts and osteoclasts and is abundantly expressed in the kidney (Citation66,Citation67). In vascular smooth muscle cells, Nox4 co-localizes with p22phox and vinculin in focal adhesions and has been implicated in cell migration, proliferation, tube formation, angiogenesis, and cell differentiation (Citation69). Nox4 has been identified in the endoplasmic reticulum, mitochondria, and nucleus of vascular cells. Nox 4 does not seem to require p47phox, p67phox, p40phox, or Rac for its activation, although polymerase (DNA-directed) delta interacting protein 2 (Poldip2), a Nox4-binding protein, has recently been shown to be important (Citation70). Poldip2 is a Nox4/p22phox-interacting protein and is a potent positive regulator of Nox4 activity in VSMCs. The Nox4/p22phox–Poldip2 complex regulates Rho-dependent cytoskeletal reorganization and cellular migration (Citation70). Another regulator of Nox4, potentially important in vascular fibrosis and differentiation, is transforming growth factor-β (TGF-β), which up-regulates expression and activity of Nox4 in many cell types, including vascular smooth muscle cells (Citation71).
Unlike Nox1 and Nox2, Nox4 is constitutively active, producing primarily H2O2 rather than •O2 − (Citation72). The difference in the species generated may underlie Nox-specific actions in cell signaling. Nox4 contributes to basal ROS production through its constitutive activity and to increased ROS generation when stimulated by Ang II, glucose, TNFα and growth factors. The pathological role of Nox4 is unclear, although it has been implicated in hypertension, atherosclerosis, and cardiovascular and renal complications of diabetes and in remodeling of pulmonary arteries in pulmonary hypertension (Citation73). Nox4-derived ROS has also been suggested in cellular senescence and aging (Citation74) and in insulin-mediated differentiation of adipocytes (Citation75). Recent studies demonstrated that Nox4 may have protective effects. In mice with a genetic deletion of Nox4 or a cardiomyocyte-targeted overexpression of Nox4, basal cardiac function was normal in both models, but Nox4-null animals developed exaggerated contractile dysfunction, hypertrophy, and cardiac dilatation during exposure to chronic overload, whereas Nox4-transgenic mice were protected (Citation76). Nox4-derived H2O2 may act as a vasodilator in some vascular beds, which could explain why mice with targeted endothelial Nox4 overexpression in the endothelium exhibit lower blood pressure and improved endothelium-dependent vasodilation compared with wild-type controls (Citation77). The exact (patho)physiological role of Nox4 in the cardiovascular system remains unclear, because many of the in-vivo studies interrogating Nox4 were performed in transgenic mice where Nox4 was up- or down-regulated.
Nox5
Nox5 is the most recently identified of the Nox enzymes and has unique features compared with other family members. Nox5 is a Ca2 + -sensitive homolog found in testes, spleen, and lymphoid tissue, but also in kidney and vascular cells (Citation78). While all Noxs are present in mice, rats, and man, the rodent genome does not contain the nox5 gene (Citation79). Unlike other vascular Noxs, Nox5 possesses an amino-terminal calmodulin-like domain with four binding sites for Ca2 + (EF hands), and unique to Nox5 is its lack of requirement for p22phox or other subunits for its activation. Nox5 is directly regulated by intracellular Ca2 + ([Ca2 + ]i), the binding of which induces a conformational change leading to enhanced ROS formation (Citation78). The biological significance of vascular Nox5 is unknown, although it has been implicated in cell proliferation, angiogenesis, and migration and in oxidative damage in atherosclerosis (Citation79). Vascular Nox5 is activated by thrombin, PDGF, Ang II, and ET-1 (Citation80), and its expression is regulated by the Ca2 + -sensitive transcription factor CREB. Increased Nox5 expression has been demonstrated in coronary arteries from patients with coronary artery disease (Citation81).
Networking between Noxs and mitochondria
Mitochondrial enzymes and NADPH oxidase are concomitantly activated in pathological processes. Nox4 has been localized within mitochondria, and it has been suggested that cross-talk between mitochondria and Noxs may be involved in dysregulated ROS formation (Citation82–85). Such cross-talk may be important in oxidative stress associated with nitroglycerin-triggered vascular dysfunction, myocardial infarction, cardiac failure, vascular remodeling, and endothelial dysfunction (Citation83). Mitochondrial-Nox interaction may also regulate vascular oxygen-sensing mechanisms (Citation84). Mechanisms linking these ROS-generating systems include ERK1/2, PKC, the mitochondrial permeability transition pore, and ATP-sensitive potassium channels (Citation83–86).
Antioxidant defense systems
In biological systems, enzymatic and non-enzymatic systems protect against injurious oxidative stress. Major enzymatic antioxidants are SOD, catalase, glutathione peroxidases, thioredoxin, and peroxiredoxin (Citation87–89). Non-enzymatic antioxidants include ascorbate, tocopherols, glutathione, bilirubin, and uric acid. SOD catalyzes the dismutation of •O2 − into H2O2 and O2. Of the three SOD isoforms eSOD is the main vascular SOD.
Low antioxidant bioavailability promotes cellular oxidative stress and has been implicated in cardiovascular and renal oxidative damage associated with hypertension (Citation90). Activity of SOD, catalase, and GSH peroxidase is lower, and the GSSG/GSH is higher in plasma and circulating cells from hypertensive patients than normotensive subjects (Citation91). In mice deficient in EC-SOD and in rats in which GSH synthesis is inhibited, blood pressure is significantly elevated, demonstrating that reduced antioxidant capacity is associated with elevated blood pressure (Citation87). In angiotensinogen-overexpressing mice, which exhibit hypertension and renal dysfunction, catalase overexpression prevented blood pressure elevation and protected against kidney damage (Citation92). Failure to up-regulate antioxidant genes and reduced antioxidant capacity are associated with age-accelerated atherosclerosis (Citation93).
Production of ROS in the vascular and renal systems in hypertension
Mechanisms whereby ROS influence the development of hypertension involve oxidative damage of multiple systems including the heart, kidneys, central nervous system, and vasculature (). In pathological conditions ROS are involved in inflammation, endothelial dysfunction, cell proliferation, migration, and activation, extracellular matrix deposition, and fibrosis. These effects are mediated through redox-sensitive signaling pathways including MAPK, PTP, tyrosine kinases, proinflammatory genes, ion channels, and Ca2 + (Citation94–96).
Figure 1. ROS and vascular remodeling in hypertension. In healthy vessels, endothelial cells produce NO and ROS, where they play a role in cellular signaling related to the control of endothelial function and maintenance of vascular integrity. In hypertension, increased ROS production, due to increased activity of pro-oxidant enzymes and decreased antioxidant defense systems, and decreased NO bioavailability, are associated with endothelial dysfunction and vascular remodeling. Exposure of endothelium to high concentrations of ROS induces hypertrophy and expression of adhesion molecules with infiltration of inflammatory cells. In VSMCs, ROS activate profibrotic, proliferative, and apoptotic pathways leading to increased extracellular matrix (ECM) accumulation (fibrosis), hypertrophy, and fibrosis. The adventitia is also responsive to high levels of ROS and, through adventitial fibroblasts and adipocytes, plays a role in the regulation of cellular responses in vascular remodeling.
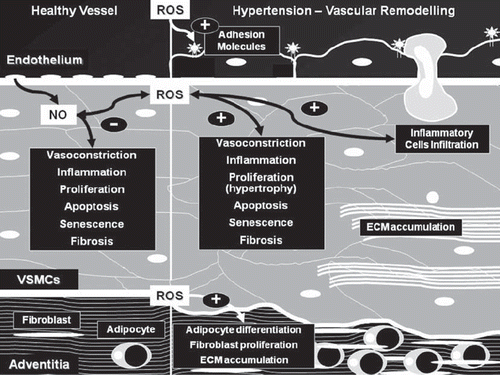
Changes in vascular function and structure probably relate to oxidative stress-induced endothelial dysfunction, reduced vasodilation, increased contraction, vascular inflammation, and structural remodeling, causing increased peripheral resistance and elevated blood pressure () (Citation96). Centrally produced ROS by NADPH oxidase in the hypothalamic and circumventricular organs are implicated in central control of hypertension, in part through sympathetic outflow (Citation97,Citation98).
Figure 2. Potential effects of reactive oxygen species on the development of hypertension—an integrated system. The damaging effects of oxidative stress involve multiple organ systems. Exposure of the central nervous system to increased ROS induces production and release of neurotransmitters responsible for the regulation of vascular resistance, cardiac function, and blood pressure control. Effects of redox-sensitive neurotransmitters may be direct or indirect through activation of the renin-angiotensin-aldosterone system, which influences peripheral resistance by influencing vasoconstriction, volume status (increased sodium reabsorption), target tissue inflammation, and fibrosis. Systemic ROS, lipid peroxidation, and high levels of Ang II/aldosterone lead to increased cardiac contractility, remodeling, inflammation, and fibrosis, reflecting increased cardiac output. ROS also influence the vasculature, where peripheral resistance is increased due to endothelial dysfunction and vasoconstriction as well as to induction of vascular remodeling (as in ). All of these processes could contribute to blood pressure elevation.
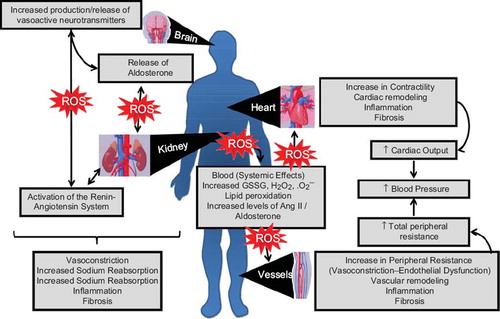
The kidney, and particularly the renal medullary circulation, plays a fundamental role in modulating long-term blood pressure control and fluid balance (Citation99,Citation100). ROS are important regulators of medullary blood flow (Citation100). Elevation of •O2− or reduction of NO in the renal medulla decreases medullary blood flow and Na+ excretion, resulting in sustained hypertension. Oxidative stress within the renal medulla makes the kidney functionally more vulnerable to effects of Ang II and salt and promotes renal dysfunction (Citation101). Superoxide and H2O2 augment afferent arterial tone and reactivity and enhance renal vascular resistance. Renal ROS influence glomerular filtration rate, tubuloglomerular feedback response, and Na + transport. NO inhibits absorption of NaCl in the thick ascending limb, whereas •O2− enhances NaCl reabsorption. Moreover, Ang II and oxidative stress promote mesangial cell proliferation, mesangial matrix accumulation, and podocyte injury, which are hallmarks of glomerulonephritis and diabetic nephropathy (Citation102).
Increased renal ROS production induces up-regulation of proinflammatory genes, such as hypoxia-inducible factor and AP-1, implicated in inflammation, fibrosis, and sclerosis in hypertension-associated kidney damage. Renal ROS generation is especially important in severe and salt-dependent forms of hypertension such as Dahl salt-sensitive rats, DOCA-salt rats, and stroke-prone SHR (SHR-SP) (Citation103). In these models blood pressure-lowering effects of the SOD mimetic tempol were associated with reduced renal excretion of 8-isoprostane PGF2α, decreased vascular resistance, increased GFR, and enhanced diuresis and natriuresis (Citation104).
Renal oxidative stress is also important in kidney disease associated with Ang II-dependent forms of hypertension. In TG(mRen2)27 (Ren2) transgenic rats, which overexpress the mouse renin gene, AT1R blockade reduced blood pressure and tissue oxidative stress, improved glomerular filtration barrier integrity, and prevented albuminuria, thereby slowing progression of kidney disease (Citation105).
A major source of renal •O2− is NAD(P)H oxidase. All of the major Noxs and NAD(P)H oxidase subunits are present in the renal cortex, medulla, and renal vessels (Citation106,Citation107). These proteins are expressed primarily in renal arterioles, glomeruli, and the distal nephron and are abundantly expressed at the luminal border of macula densa cells and in podocytes. In SHR, Ang II-induced hypertension and in salt-sensitive hypertension, renal expression of NAD(P)H oxidase subunits are increased and activity of Nox is enhanced.
Oxidative stress and hypertension—from the bench to the bedside
The link between oxidative stress and increased blood pressure was first suggested in the early 1990s both in patients with essential hypertension (Citation108) and in SHR (Citation109). Oxidative stress precedes development of hypertension in SHR and is implicated in fetal programming and development of hypertension later in life (Citation110). Markers of oxidative stress, such as TBARS and F2α-isoprostanes, tissue concentrations of •O2− and H2O2, and activation of NADPH oxidase and xanthine oxidase are increased, whereas levels of NO and antioxidant enzymes are reduced in experimental hypertension (Citation111–113).
Ang II-dependent hypertension is particularly sensitive to ROS derived from NAD(P)H oxidase (Citation114,Citation115). In Ang II-infused rats and mice, expression of Noxs (Nox1, Nox2, Nox4), oxidase activity, and ROS generation are increased. In p47phox knock-out mice and in gp91phox (Nox2) knock-out mice Ang II infusion failed to induce hypertension, and these animals do not show the same increases in •O22 production, vascular hypertrophy, and endothelial dysfunction observed in Ang II-infused wild-type mice (Citation116). In Ang II-infused mice treated with siRNA targeted to renal p22phox, renal NADPH oxidase activity was blunted, ROS formation was reduced, and blood pressure elevation was attenuated (Citation117). On the other hand, overexpression of vascular p22phox was associated with increased oxidative stress and vascular dysfunction, but no significant increase in blood pressure (Citation118). Treatment with apocynin or diphenylene iodinium, non-specific pharmacological inhibitors of NAD(P)H oxidase, or gp91dstat, a novel specific inhibitor of NADPH oxidase, reduced vascular •O22 production, prevented cardiovascular remodeling, and attenuated development of hypertension in Ang II-treated mice (Citation119,Citation120). Nox1-deficient mice have reduced vascular •O2- production, and blood pressure elevation in response to Ang II is blunted (Citation121), whereas in transgenic mice in which Nox1 is overexpressed in the vascular wall, Ang II-mediated vascular hypertrophy and blood pressure elevation are enhanced (Citation122). In these models, Ang II was infused for a short time period (1–3 weeks), inducing an acute hypertensive response. In a model of chronic Ang II-dependent hypertension, where we crossed transgenic mice expressing human renin (which exhibit an Ang II-sensitive hypertensive phenotype) with Nox2−/− or Nox1−/− mice, development of hypertension was not prevented even though oxidative stress was reduced, suggesting that Noxs may be more important in acute than in chronic hypertension (Citation123,Citation124). It should be stressed that in these Nox knock-out or transgenic studies, baseline cardiovascular phenotypes of mice are surprisingly normal, and it is only in the context of a challenge, such as with Ang II or salt, that mice exhibit vascular and blood pressure alterations (Citation125–128).
Unlike cell-based studies where Ang II alone is sufficient to induce oxidative stress and activation of redox-sensitive pathways that regulate vascular function, growing in-vivo evidence indicates that, in Ang II-dependent models of hypertension, Ang II alone is probably insufficient to induce oxidative stress and that additional stressors are needed. Such co-inducers include, amongst others, salt, activation of the sympathetic and/or central nervous system, humoral factors, and involvement of peripheral T cells and the immune system. As an example are studies demonstrating that redox signaling in the subfornical organ (SFO) in the forebrain is critical in Ang II-mediated regulation of blood pressure and hypertension, because SFO-targeted ablation of SOD3 causes a significant increase in basal blood pressure and enhances the sensitivity to systemic Ang II at a concentration that does not normally affect blood pressure in mice (Citation126,Citation127). Moreover, SOD3 ablation in the SFO is sufficient to increase vascular ROS, an effect associated with increased sympathetic output and increased circulating CD69 + T lymphocytes. Taken together these data suggest that increased •O2− in the forebrain through SOD3 deletion leads to peripheral T cell activation and vascular oxidative stress and inflammation, promoting an increase in blood pressure (Citation128). This paradigm links the central nervous system, immune system, vasculature, and oxidative stress in the pathophysiology of hypertension, at least in experimental models of Ang II-induced hypertension.
Additional sources of NADPH oxidase-derived ROS in Ang II-induced hypertension are cytokines from inflammatory cells, such as T lymphocytes. In RAG1-/- mice, which lack lymphocytes, pressor responses to Ang II are reduced, a response that is restored by adoptive transfer of T lymphocytes, but not of B lymphocytes (Citation129). T cells influence blood pressure elevation by interacting with B7 ligands (CD80 and CD86) and the T cell co-receptor CD28, and through dysregulation of T regulatory and T effector cells (Citation129). Importance of the immune/inflammatory system is further demonstrated by studies showing that T regulatory lymphocytes (Tregs), which suppress T effector lymphocytes, attenuate vascular oxidative stress, inflammation, and blood pressure in Ang II-induced hypertension (Citation130).
There is also evidence for ROS involvement in the pathogenesis of hypertension independent of direct Ang II actions. In SHR, vascular, renal, and cardiac •O2− production is enhanced compared with normotensive controls. In stroke-prone SHR, aortic expression of Nox1 and Nox4 is significantly increased compared with WKY (Citation131). In DOCA salt-induced mineralocorticoid hypertension, vascular •O2− production involving elevated NAD(P)H oxidase activity, uncoupling of endothelial NOS, and mitochondrial sources, in part through the endothelin-1 (ET-1)/ETA receptor pathway, is increased (Citation132). Infusion of ET-1 increases NAD(P)H oxidase-dependent •O2− production; however, preventing this increase in ROS generation does not inhibit development of hypertension in these animals. Overexpression of human ET-1 in mice also induces vascular remodeling and impairs endothelial function, via activation of NAD(P)H oxidase. To support further a role for oxidative stress in experimental hypertension, treatment with antioxidant vitamins, SOD mimetics (tempol (4-hydroxy-2,2,6,6-tetramethyl piperidinoxyl)), free radical scavengers, or tetrahydrobiopterin (BH4) has been shown to attenuate or prevent development of hypertension and associated target organ damage (Citation133–135).
Unlike experimental studies, where there is strong evidence that oxidative stress plays a role in the pathogenesis of hypertension, clinical data are less convincing, and most studies have been based on indirect evidence demonstrating increased plasma markers of oxidative stress in patients with hypertension () (Citation136,Citation137). http://hyper.ahajournals.org/cgi/content/full/44/3/248 - R34 - 032003#R34 - 032003 http://hyper.ahajournals.org/cgi/content/full/44/3/ 248 - R35 - 032003#R35 - 032003. It should be stressed that biomarkers may simply reflect associations, and not cause, between ROS and blood pressure.
Table I. Indirect evidence supporting a role for oxidative stress in human hypertension.
Hypertensive patients exhibit higher levels of plasma H2O2 than normotensive subjects (Citation138). Normotensive subjects with a family history of hypertension have greater H2O2 production than blood pressure-matched normotensives without a family history of hypertension, suggesting that there may be a genetic component that leads to elevated production of hydrogen peroxide (Citation139–141). Racial differences in oxidative stress and inflammation have been demonstrated. Human umbilical venous cells (HUVEC) from African-Americans exhibited higher levels of NO, IL-6, p47phox, Nox2, and Nox4 and lower superoxide dismutase activity than HUVECs from Caucasians (Citation142). Moreover in African men, but not women, plasma ROS was positively associated with systolic blood pressure (Citation142).
We showed that ROS production is increased in vascular smooth muscle cells from resistance arteries of hypertensive patients and that this is associated with up-regulation of vascular NADPH oxidase (Citation143,Citation144). The importance of NADPH oxidase in oxidative stress in human cardiovascular disease is supported by studies showing that polymorphisms in NADPH oxidase subunits are associated with increased atherosclerosis and hypertension. In particular, the −930(A/G) polymorphism in the p22(phox) promoter may be a novel genetic marker associated with hypertension (Citation145). The C242T CYBA polymorphism is associated with essential hypertension, and hypertensive patients carrying the CC genotype of this polymorphism exhibit features of NADPH oxidase-mediated oxidative stress and endothelial damage and are prone to cerebrovascular disease (Citation146). Polymorphisms −337GA and 565 + 64CT of xanthine oxidase gene have been shown to be related to blood pressure and oxidative stress in hypertension, further supporting a role for xanthine oxidase in hypertension (Citation147).
In addition to excess ROS generation, decreased antioxidant defense mechanisms contribute to oxidative stress in patients with hypertension. Hypertensive patients have reduced activity and decreased content of antioxidant enzymes, including SOD, glutathione peroxidase, and catalase (Citation148). Decreased levels of antioxidant vitamins A, C, and E have been demonstrated in newly diagnosed, untreated hypertensive patients compared with normotensive controls (Citation149). Antioxidant vitamins reduced blood pressure and arterial stiffness in patients with diabetes (Citation150), but had no effect in postmenopausal women or in healthy subjects (Citation151). In patients with white-coat hypertension serum protein carbonyl (PCO, indicating protein oxidation) was increased, and endogenous antioxidant proteins (protein thiol, SOD, glutathione) were decreased compared with normotensive individuals, further supporting a relationship between oxidative stress and hypertension (Citation151).
Human studies examining ROS and hypertension are primarily correlative, and there is still no definitive proof that oxidative stress is a cause of human hypertension. There is no direct evidence that Noxs play a pathophysiological role in human hypertension. However, NADPH oxidase-derived ROS have been implicated in the regulation of vascular function, because patients with chronic granulomatous disease due to mutations in genes encoding for NADPH oxidase subunits demonstrate blunted ischemia-reperfusion-induced flow-mediated dilation.
Targeting ROS as a therapeutic strategy in clinical hypertension
The potential of antioxidants in treating conditions associated with oxidative stress is supported by experimental investigations, observational findings, small clinical studies, and epidemiological data. http://hyper.ahajournals.org/cgi/content/full/44/3/248-R44 - 032003#R44 - 032003. http://hyper.ahajournals.org/cgi/content/full/44/3/248-R45 - 032003#R45 - 032003. However, findings are inconsistent, and clinical trial data are inconclusive (Citation152). Recent large clinical trials examining effects of antioxidant vitamins (vitamins C and E) in the prevention of pre-eclampsia and gestational hypertension were also negative (Citation153,Citation154). Possible reasons for these disappointing outcomes relate to 1) type of antioxidants used, 2) patient cohorts included in trials, and 3) the trial design itself. With respect to antioxidants, it is possible that agents examined were ineffective and non-specific and that dosing regimens and duration of therapy were insufficient. It is also possible that orally administered antioxidants may be inaccessible to the source of free radicals, particularly if ROS are generated in intracellular compartments and organelles. Furthermore, antioxidant vitamins do not scavenge H2O2, which may be more important than •O2− in cardiovascular disease. Another factor of importance is that antioxidants do not inhibit ROS production. Regarding cohorts included in large trials, most subjects had significant cardiovascular disease, in which case damaging effects of oxidative stress may be irreversible. Another confounding factor is that most of the enrolled subjects were taking aspirin prophylactically. Since aspirin has intrinsic antioxidant properties, additional antioxidant therapy may be ineffective. Moreover, in patients studied in whom negative results were obtained, it was never proven that these individuals did in fact have increased oxidative stress. To date, there are no large clinical trials in which patients were recruited based on evidence of elevated ROS formation. Also, none of the large clinical trials were designed to examine effects of antioxidants specifically on blood pressure.
Based on the lack of evidence to prove the benefits from use of antioxidants to prevent cardiovascular disease, antioxidant supplementation is not recommended for the prevention or treatment of hypertension. However, most therapeutic guidelines suggest that the general population consumes a diet emphasizing antioxidant-rich fruits and vegetables and whole grains. The low-sodium DASH diet has been shown to reduce oxidative stress and improve vascular function in salt-sensitive individuals (Citation155). Another important life-style modification that may have cardiovascular-protective and blood pressure-lowering effects by reducing oxidative stress is exercise. In experimental models of hypertension and in human patients with coronary artery disease, exercise reduced vascular NADPH oxidase activity and ROS production, ameliorated vascular injury, and reduced blood pressure (Citation156). However, in elderly patients, combining antioxidant therapy with exercise negated beneficial blood pressure-lowering effects of exercise (Citation157).
Recent clinical studies examining effects of xanthine oxidase inhibitors (Citation158), tetrahydrobiopterin (sapropterin dihydrochloride (6r-bh4)) (159), and N-acetylcysteine (160) have demonstrated improved vascular function and blood pressure lowering in patients with hypertension, chronic kidney disease, and pulmonary hypertension. Some of the beneficial effects of classical antihypertensive agents such as β-adrenergic blockers, ACE inhibitors, AT1 receptor antagonists, and Ca2 + channel blockers may be mediated, in part, by decreasing vascular oxidative stress (161,162). http://hyper.ahajournals.org/cgi/content/full/44/3/248-R41 - 032003. These effects have been attributed to direct inhibition of NAD(P)H oxidase activity and to intrinsic antioxidant properties of the drugs. However, some studies failed to show changes in oxidative stress despite significant blood pressure lowering by classical antihypertensive drugs (163,164).
NADPH oxidase and Nox isoforms as therapeutic targets—clinical potential
Based on experimental evidence NADPH oxidase subunits and Nox isoforms are potential therapeutic targets for cardiovascular disease and hypertension. Because of this there has been enormous interest in the development of agents that inhibit NADPH oxidases in an isoform-specific manner (Citation46,165). Different strategies have been employed, including small molecule inhibitors, peptide NADPH oxidase inhibitors, and siRNAs (). Several compounds have been registered as NADPH oxidase inhibitors in the patent literature (165). However, none has gone through clinical trials, and some have not yet completed preclinical studies. To date two different classes of compounds have been claimed as potent and orally active bioavailable NADPH oxidase inhibitors: pyrazolopyridines (GKT136901 and GKT137831) and triazolopyrimidine derivatives (VAS2870 and VAS3947). Although the mechanisms of inhibition have not yet been clarified, GKT compounds may act as competitive substrate inhibitors, since structurally they resemble NADPH. Although much research is still needed to confirm the clinical use of NADPH oxidase inhibitors in humans, these drugs hold promise in the management of patients with Nox-associated pathologies (Citation46,165).
Table II. Compounds that have been registered as NADPH oxidase inhibitors (165). None of these have yet been tested clinically.
Conclusions
In physiological conditions, ROS play an important role in cardiovascular and renal biology through highly regulated redox-sensitive signaling pathways. Uncontrolled production/degradation of ROS results in oxidative stress, which induces cardiovascular and renal injury and activation of the sympathetic nervous system with associated increase in blood pressure. Although oxidative damage may not be the sole cause of blood pressure elevation, together with a constellation of prohypertensive factors, such as salt-loading, activation of the renin-angiotensin system, and sympathetic hyperactivity, it amplifies the development of hypertension. Convincing findings from experimental and animal studies suggest a causative role for oxidative stress in the pathogenesis of hypertension. However, in humans there is still no solid evidence that oxidative stress is fundamentally involved in the pathogenesis of hypertension. Further research in the field of oxidative stress and human hypertension is warranted. In particular, there is an urgent need for the development of sensitive, specific, and reliable biomarkers and assays to assess the redox status of patients. Also needed are clinical trials designed specifically to address the role of ROS in the development of hypertension. With a better understanding of processes regulating ROS metabolism and identification of factors that promote oxidative excess in humans, it should be possible to target therapies more effectively so that damaging actions of oxygen free radicals can be reduced. Such therapies could have potential in the management of redox-sensitive diseases associated with cardiovascular and renal damage, including hypertension (Citation46,165).
Declaration of interest: Work from the author's laboratory was supported by grants 44018 and 57886, both from the Canadian Institutes of Health Research (CIHR). R.M.T. is supported through a Canada Research Chair/Canadian Foundation for Innovation award and A.C.M. by a fellowship from the CIHR. The authors report no conflicts of interest.
References
- Narayan KM, Ali MK, Koplan JP. Global noncommunicable diseases—where worlds meet. N Engl J Med. 2010;363:1196–8.
- Harrison DG, Vinh A, Lob H, Madhur MS. Role of the adaptive immune system in hypertension. Curr Opin Pharmacol. 2010;10:203–7.
- Schiffrin EL. T lymphocytes: a role in hypertension? Curr Opin Nephrol Hypertens. 2010;19:181–6.
- Touyz RM. Molecular and cellular mechanisms in vascular injury in hypertension: role of angiotensin II. Curr Opin Nephrol Hypertens. 2005;14:125–131.
- Harris DM, Cohn HI, Pesant S, Eckhart AD. GPCR signalling in hypertension: role of GRKs. Clin Sci (Lond). 2008;115:79–89.
- Vaziri ND, Rodriguez-Iturbe B. Mechanisms of disease: oxidative stress and inflammation in the pathogenesis of hypertension. Nat Clin Pract Nephrol. 2006;2:582–59.
- Landmesser U, Harrison DG, Drexler H. Oxidant stress-a major cause of reduced endothelial nitric oxide availability in cardiovascular disease. Eur J Clin Pharmacol. 2006;62: 13–19.
- Touyz RM, Schiffrin EL. Reactive oxygen species in vascular biology: implications in hypertension. Histochem Cell Biol. 2004;122:339–352.
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95.
- Ushio-Fukai M, Alexander RW, Akers M, Griendling KK. p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J Biol Chem. 1998;273:15022–15029.
- Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–2183.
- Touyz RM, Tabet F, Schiffrin EL. Redox-dependent signalling by angiotensin II and vascular remodelling in hypertension. Clin Exp Pharmacol Physiol. 2003;30:860–866.
- Millar TM, Phan V, Tibbles LA. ROS generation in endothelial hypoxia and reoxygenation stimulates MAP kinase signaling and kinase-dependent neutrophil recruitment. Free Radic Biol Med. 2007;42:1165–1677.
- Tabet F, Savoia C, Schiffrin EL, Touyz RM. Differential calcium regulation by hydrogen peroxide and superoxide in vascular smooth muscle cells from spontaneously hypertensive rats. J Cardiovasc Pharmacol. 2004;44:200–208.
- Romanowski A, Murray IR, Huston MJ. Effects of hydrogen peroxide on normal and hypertensive rats. Pharm Acta Helv. 1960;35:354–357.
- Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NAD(P)H oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923.
- Zalba G, Beaumont FJ, San Jose G, Fortuno A, Fortuno MA, Etayo JC. Vascular NADH/NAD(P)H oxidase is involved in enhanced superoxide production in spontaneously hypertensive rats. Hypertension. 2000;35:1055–1061.
- Jung O, Schreiber JG, Geiger H, Pedrazzini T, Busse R, Brandes RP. gp91phox-containing NADPH oxidase mediates endothelial dysfunction in renovascular hypertension. Circulation. 2004;109:1795–801.
- Kagota S, Tada Y, Kubota Y, Nejime N, Yamaguchi Y, Nakamura K, Kunitomo M, Shinozuka K. Peroxynitrite is Involved in the dysfunction of vasorelaxation in SHR/NDmcr-cp rats, spontaneously hypertensive obese rats. J Cardiovasc Pharmacol. 2007;50:677–685.
- Klanke B, Cordasic N, Hartner A, Schmieder RE, Veelken R, Hilgers KF. Blood pressure versus direct mineralocorticoid effects on kidney inflammation and fibrosis in DOCA-salt hypertension. Nephrol Dial Transplant. 2008;23: 3456–3463.
- Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, Holland SM, Harrison DG. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40:511–515.
- Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M, Takai S, Yamanishi K, Miyazaki M, Matsubara H, Yabe-Nishimura C. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation. 2005;112:2677–85.
- Lavi S, Yang EH, Prasad A, Mathew V, Barsness GW, Rihal CS, Lerman LO, Lerman A. The interaction between coronary endothelial dysfunction, local oxidative stress, and endogenous nitric oxide in humans. Hypertension. 2008; 51:127–133.
- Cottone S, Mulè G, Guarneri M, Palermo A, Lorito MC, Riccobene R, Arsena R, Vaccaro F, Vadalà A, Nardi E, Cusimano P, Cerasola G. Endothelin-1 and F2-isoprostane relate to and predict renal dysfunction in hypertensive patients. Nephrol Dial Transplant. 2009;24:497–503.
- Duffy SJ, Gokce N, Holbrook M, Huang A, Frei B, Keaney JF Jr, Vita JA. Treatment of hypertension with ascorbic acid. Lancet. 1999;354:2048–9.
- Kurl S, Tuomainen TP, Laukkanen JA, Nyyssönen K, Lakka T, Sivenius J, Salonen JT. Plasma vitamin C modifies the association between hypertension and risk of stroke. Stroke. 2002;33:1568–1573.
- Kamgar M, Zaldivar F, Vaziri ND, Pahl MV. Antioxidant therapy does not ameliorate oxidative stress and inflammation in patients with end-stage renal disease. J Natl Med Assoc. 2009;101:336–44.
- Touyz RM, Briones AM. Reactive oxygen species and vascular biology: implications in human hypertension. Hypertens Res. 2011;34:5–14.
- Rodrigo R, Prat H, Passalacqua W, Araya J, Bächler JP. Decrease in oxidative stress through supplementation of vitamins C and E is associated with a reduction in blood pressure in patients with essential hypertension. Clin Sci (Lond). 2008;114:625–34.
- Darko D, Dornhorst A, Kelly FJ, Ritter JM, Chowienczyk PJ. Lack of effect of oral vitamin C on blood pressure, oxidative stress and endothelial function in Type II diabetes. Clin Sci (Lond). 2002;103:339–344.
- Schiffrin EL. Antioxidants in hypertension and cardiovascular disease. Mol Interv. 2010;10:354–62.
- Fridovich I. Superoxide anion radical (O2-.), superoxide dismutases, and related matters. J Biol Chem. 1997; 272:18515–18517.
- Giustarini D, Dalle-Donne I, Tsikas D, Rossi R. Oxidative stress and human diseases: Origin, link, measurement, mechanisms, and biomarkers. Crit Rev Clin Lab Sci. 2009;46:241–81.
- Nishino T, Okamoto K, Eger BT, Pai EF, Nishino T. Mammalian xanthine oxidoreductase - mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008;275:3278–3289.
- Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res. 2002;91:406–413.
- Adlam D, Bendall JK, De Bono JP, Alp NJ, Khoo J, Nicoli T, Yokoyama M, Kawashima S, Channon KM. Relationships between nitric oxide-mediated endothelial function, eNOS coupling and blood pressure revealed by eNOS-GTP cyclohydrolase 1 double transgenic mice. Exp Physiol. 2007;92:119–126
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–9.
- Geiszt M. NAD(P)H oxidases: New kids on the block. Cardiovasc Res 2006;71:289–299.
- Touyz RM, Briones AM, Sedeek M, Burger D, Montezano AC. NOX Isoforms and Reactive Oxygen Species in Vascular Health. Mol Interv. 2011;11:27–35.
- Bokoch GM, Zhao T. Regulation of the phagocyte NAD(P)H oxidase by Rac GTPase. Antioxid Redox Signal. 2006;8:1533–1548.
- Guzik TJ, Chen W, Gongora MC, Guzik B, Lob HE, Mangalat D, Hoch N, Dikalov S, Rudzinski P, Kapelak B, Sadowski J, Harrison DG. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J Am Coll Cardiol. 2008;52:1803–1809.
- Leto TL, Morand S, Hurt D, Ueyama T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid Redox Signal. 2009; 11:260.
- Petry A, Weitnauer M, Görlach A. Receptor activation of NADPH oxidases. Antioxid Redox Signal. 2010;13:467.
- Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–297.
- Briones AM, Tabet F, Callera GE, Montezano AC, Yogi A, He Y, Quinn MT, Salaices M, Touyz RM. Differential regulation of Nox1, Nox2 and Nox4 in vascular smooth muscle cells from WKY and SHR. J Am Soc Hypertens. 2011; 5:137–153.
- Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–471.
- Nisimoto Y, Tsubouchi R, Diebold BA, Qiao S, Ogawa H, Ohara T, Tamura M. Activation of NAD(P)H oxidase 1 in tumour colon epithelial cells. Biochem J. 2008;415:57–65.
- Gianni D, Dermardirossian C, Bokoch GM. Direct interaction between Tks proteins and the N-terminal proline-rich region (PRR) of NoxA1 mediates Nox1-dependent ROS generation. Eur J Cell Biol. 2011;90:164–71.
- Dutta S, Rittinger K. Regulation of NOXO1 activity through reversible interactions with p22 and NOXA1. PLoS One. 2010;5:e10478.
- Fernandes DC, Manoel AHO, Wosniak J, Laurindo FR. Protein disulfide isomerise overexpression in vascular smooth muscle cells induces spontaneous preemptive NAD(P)H oxidase activation nad Nox1 mRNA expression: effects of nitrosothiol exposure. Arch Biochem Biophys 2009;484:197–204.
- Lee MY, San Martin A, Mehta PK, Dikalova AE, Garrido AM, Datla SR, Lyons E, Krause KH, Banfi B, Lambeth JD, Lassègue B, Griendling KK. Mechanisms of vascular smooth muscle NAD(P)H oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol. 2009;29:480–487.
- Manea A, Tanase LI, Raicu M, Simionescu M. Transcriptional regulation of NADPH oxidase isoforms Nox1 and Nox4, by nuclear factor-kappaB in human aortic smooth muscle cells, Biochem. Biophys. Res. Commun. 2010;396: 901–907.
- Tabet F, Schiffrin EL, Callera GE, He Y, Yao G, Ostman A, Kappert K, Tonks NK, Touyz RM. Redox-sensitive signaling by angiotensin II involves oxidative inactivation and blunted phosphorylation of protein tyrosine phosphatase SHP-2 in vascular smooth muscle cells from SHR. Circ Res. 2008; 103:149–154.
- Rathore R, Zheng YM, Niu CF, Liu QH, Korde A, Ho YS, Wang YX. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2 + ]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic Biol Med. 2008;45:1223–31.
- Dikalova AE, Góngora MC, Harrison DG, Lambeth JD, Dikalov S, Griendling KK. Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via eNOS uncoupling. Am J Physiol Heart Circ Physiol. 2010;299:H673–9.
- Yogi A, Mercure C, Touyz J, Callera GE, Montezano AC, Aranha AB, Tostes RC, Reudelhuber T, Touyz RM. Renal redox-sensitive signaling, but not blood pressure, is attenuated by Nox1 knockout in angiotensin II-dependent chronic hypertension. Hypertension. 2008;51:500–506.
- Basset O, Deffert C, Foti M, Bedard K, Jaquet V, Ogier-Denis E, Krause KH. NADPH oxidase 1 deficiency alters caveolin phosphorylation and angiotensin II-receptor localization in vascular smooth muscle. Antioxid Redox Signal. 2009;11:2371–84.
- Niu XL, Madamanchi NR, Vendrov AE, Tchivilev I, Rojas M, Madamanchi C, Brandes RP, Krause KH, Humphries J, Smith A, Burnand KG, Runge MS. Nox activator 1: a potential target for modulation of vascular reactive oxygen species in atherosclerotic arteries. Circulation. 2010;121: 549–59.
- Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, Pagano PJ, Schiffrin EL. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circ Res. 2002;90:1205–1213.
- Gupte SA, Kaminski PM, George S, Kouznestova L, Olson SC, Mathew R, Hintze TH, Wolin MS. Peroxide generation by p47phox-Src activation of Nox2 has a key role in protein kinase C-induced arterial smooth muscle contraction. Am J Physiol Heart Circ Physiol. 2009;296:H1048–1057.
- Han W, Li H, Villar VA, Pascua AM, Dajani MI, Wang X, Natarajan A, Quinn MT, Felder RA, Jose PA, Yu P. Lipid rafts keep NADPH oxidase in the inactive state in human renal proximal tubule cells. Hypertension. 2008;51:481–485.
- Touyz RM, Mercure C, He Y, Javeshghani D, Yao G, Callera GE, Yogi A, Lochard N, Reudelhuber TL. Angiotensin II-dependent chronic hypertension and cardiac hypertrophy are unaffected by gp91phox-containing NAD(P)H oxidase. Hypertension. 2005;45:530–537.
- Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab. 2009;29:1262–72.
- Violi F, Sanguigni V, Carnevale R, Plebani A, Rossi P, Finocchi A, Pignata C, De Mattia D, Martire B, Pietrogrande MC, Martino S, Gambineri E, Soresina AR, Pignatelli P, Martino F, Basili S, Loffredo L. Hereditary deficiency of gp91(phox) is associated with enhanced arterial dilatation: results of a multicenter study. Circulation. 2009;120: 1616–22.
- Loukogeorgakis SP, van den Berg MJ, Sofat R, Nitsch D, Charakida M, Haiyee B, de Groot E, MacAllister RJ, Kuijpers TW, Deanfield JE. Role of NADPH oxidase in endothelial ischemia/reperfusion injury in humans. Circulation. 2010;121:2310–6.
- Geiszt M, Kopp JB, Várnai P, Leto TL. Identification of renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci U S A. 2000 ;97:8010–4.
- Sedeek M, Callera G, Montezano A, Gutsol A, Heitz F, Szyndralewiez C, Page P, Kennedy CR, Burns KD, Touyz RM, Hébert RL. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am J Physiol Renal Physiol. 2010;299:F1348–58.
- Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:677–83.
- Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, Papaharalambus C, Lassègue B, Griendling KK. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res. 2009;105:249–59.
- Tong X, Hou X, Jourd'heuil D, Weisbrod RM, Cohen RA. Upregulation of Nox4 by TGF{beta}1 oxidizes SERCA and inhibits NO in arterial smooth muscle of the prediabetic Zucker rat. Circ Res. 2010;107:975–83.
- Wu RF, Ma Z, Liu Z, Terada LS. Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Mol Cell Biol. 2010;30:3553–68.
- Manea A, Tanase LI, Raicu M, Simionescu M. Jak/STAT signaling pathway regulates nox1 and nox4-based NADPH oxidase in human aortic smooth muscle cells. Arterioscler Thromb Vasc Biol. 2010;30:105–12.
- Ismail, A. Sturrock, P. Wu, B. Cahill, K. Norman, T. Huecksteadt, K. Sanders, T. Kennedy and J. Hoidal, NOX4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: the role of autocrine production of transforming growth factor-{beta}1 and insulin-like growth factor binding protein-3. Am. J. Physiol. Lung Cell Mol. Physiol. 2009;296:489–499.
- Schröder K, Wandzioch K, Helmcke I, Brandes RP. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler Thromb Vasc Biol. 2009;29: 239–45.
- Zhang M, Brewer AC, Schröder K, Santos CX, Grieve DJ, Wang M, Anilkumar N, Yu B, Dong X, Walker SJ, Brandes RP, Shah AM. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U S A. 2010;107:18121–6.
- Ray R, Murdoch CE, Wang M, Santos CX, Zhang M, Alom-Ruiz S, Anilkumar N, Ouattara A, Cave AC, Walker SJ, Grieve DJ, Charles RL, Eaton P, Brewer AC, Shah AM. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler Thromb Vasc Biol. 2011;31:1368–76.
- Serrander L, Jaquet V, Bedard K, Plastre O, Hartley O, Arnaudeau S, Demaurex N, Schlegel W, Krause KH. NOX5 is expressed at the plasma membrane and generates superoxide in response to protein kinase C activation. Biochimie. 2007;89:1159–1167.
- Fulton DJ. Nox5 and the regulation of cellular function. Antioxid Redox Signal. 2009;11:2443–52.
- Jay DB, Papaharalambus CA, Seidel-Rogol B, Dikalova AE, Lassègue B, Griendling KK. Nox5 mediates PDGF-induced proliferation in human aortic smooth muscle cells. Free Radic Biol Med. 2008;45:329–335.
- Montezano AC, Paravicini TM, Chignalia AZ, Yusuf H, Almasri M, He Y, He G, Callera GE, Krause K-H, Lambeth D, Touyz RM. Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 5 (Nox5) regulation by Angiotensin II and Endothelin-1 is Mediated via Calcium/Calmodulin-dependent Pathways in Human Endothelial Cells. Circ Res. 2010;106:1363–1373.
- Wosniak J Jr, Santos CX, Kowaltowski AJ, Laurindo FR. Cross-talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid Redox Signal. 2009;11:1265–1269.
- Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–96.
- Daiber A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta. 2010;1797:897–906.
- Graham KA, Kulawiec M, Owens KM, Li X, Desouki MM, Chandra D, Singh KK. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol Ther. 2010;10:223–31.
- Gongora MC, Qin Z, Laude K, Kim HW, McCann L, Folz JR, Dikalov S, Fukai T, Harrison DG. Role of extracellular superoxide dismutase in hypertension. Hypertension. 2006; 48:473–481.
- Tajima M, Kurashima Y, Sugiyama K, Ogura T, Sakagami H. The redox state of glutathione regulates the hypoxic induction of HIF-1. Eur J Pharmacol. 2009;606:45–49.
- Sui H, Wang W, Wang PH, Liu LS. Effect of glutathione peroxidase mimic ebselen (PZ51) on endothelium and vascular structure of stroke-prone spontaneously hypertensive rats. Blood Press. 2005;14:366–372.
- Welch WJ, Chabrashvili T, Solis G, Chen Y, Gill PS, Aslam S, Wang X, Ji H, Sandberg K, Jose P, Wilcox CS. Role of extracellular superoxide dismutase in the mouse angiotensin slow pressor response. Hypertension. 2006 ;48:934–941.
- Simic DV, Mimic-Oka J, Pljesa-Ercegovac M, Savic-Radojevic A, Opacic M, Matic D, Ivanovic B, Simic T. Byproducts of oxidative protein damage and antioxidant enzyme activities in plasma of patients with different degrees of essential hypertension. J Hum Hypertens. 2006;20:149–55.
- Godin N, Liu F, Lau GJ, Brezniceanu ML, Chénier I, Filep JG, Ingelfinger JR, Zhang SL, Chan JS. Catalase overexpression prevents hypertension and tubular apoptosis in angiotensinogen transgenic mice. Kidney Int. 2010;77: 1086–1097.
- Collins AR, Lyon CJ, Xia X, Liu JZ, Tangirala RK, Yin F, Boyadjian R, Bikineyeva A, Praticò D, Harrison DG, Hsueh WA. Age-accelerated atherosclerosis correlates with failure to upregulate antioxidant genes. Circ Res. 2009;104:e42–54.
- Feissner RF, Skalska J, Gaum WE, Sheu SS. Crosstalk signaling between mitochondrial Ca2 + and ROS. Front Biosci. 2009;14:1197–218.
- Monteiro HP, Arai RJ, Travassos LR. Protein tyrosine phosphorylation and protein tyrosine nitration in redox signaling. Antioxid Redox Signal. 2008;10:843–89.
- Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res. 2004;95:210–216.
- Hirooka Y. Role of reactive oxygen species in brainstem in neural mechanisms of hypertension. Auton Neurosci. 2008;142:20–24.
- Wilcox CS. Oxidative stress and nitric oxide deficiency in the kidney: a critical link to hypertension? Am J Physiol Regul Integr Comp Physiol. 2005 Oct;289:R913–935.
- Mori T, Cowley AW Jr, Ito S. Molecular mechanisms and therapeutic strategies of chronic renal injury: physiological role of angiotensin II-induced oxidative stress in renal medulla. J Pharmacol Sci. 2006;100:2–8.
- Johns EJ, O'Shaughnessy B, O'Neill S, Lane B, Healy V. Impact of elevated dietary sodium intake on NAD(P)H oxidase and SOD in the cortex and medulla of the rat kidney. Am J Physiol Regul Integr Comp Physiol. 2010; 299:R234–40.
- Hayden MR, Whaley-Connell A, Sowers JR. Renal redox stress and remodeling in metabolic syndrome, type 2 diabetes mellitus, and diabetic nephropathy: paying homage to the podocyte. Am J Nephrol. 2005;25:553–69.
- Manning RD Jr, Tian N, Meng S. Oxidative stress and antioxidant treatment in hypertension and the associated renal damage. Am J Nephrol. 2005;25:311–7.
- Hisaki R, Fujita H, Saito F, Kushiro T. Tempol attenuates the development of hypertensive renal injury in Dahl salt-sensitive rats. Am J Hypertens. 2005;18:707–13.
- Blendea MC, Jacobs D, Stump CS, McFarlane SI, Ogrin C, Bahtyiar G, Stas S, Kumar P, Sha Q, Ferrario CM, Sowers JR. Abrogation of oxidative stress improves insulin sensitivity in the Ren-2 rat model of tissue angiotensin II overexpression. Am J Physiol Endocrinol Metab. 2005; 288:E353–9.
- Chabrashvili T, Tojo A, Onozato ML, Kitiyakara C, Quinn MT, Fujita T, Welch WJ, Wilcox CS. Expression and cellular localization of classic NADPH oxidase subunits in the spontaneously hypertensive rat kidney. Hypertension. 2002;39:269–74.
- Montezano AC, Burger D, Ceravolo GS, Yusuf H, Montero M, Touyz RM. Novel Nox homologues in the vasculature: focusing on Nox4 and Nox5. Clin Sci (Lond). 2011; 120:131–41.
- Schneider R, Iscovitz H, Ilan Z, Bernstein K, Gros M, Iaina A. Oxygen free radical scavenger system intermediates in essential hypertensive patients before and immediately after sublingual captopril administration. Isr J Med Sci. 1990;26:491–5.
- Nakazono K, Watanabe N, Matsuno K, Sasaki J, Sato T, Inoue M. Does superoxide underlie the pathogenesis of hypertension? Proc Natl Acad Sci U S A. 1991; 88:10045–10048.
- Nuyt AM. Mechanisms underlying developmental programming of elevated blood pressure and vascular dysfunction: evidence from human studies and experimental animal models. Clin Sci (Lond). 2008;114:1–17.
- Török J. Participation of nitric oxide in different models of experimental hypertension. Physiol Res. 2008;57: 813–825.
- Viel EC, Benkirane K, Javeshghani D, Touyz RM, Schiffrin EL. Xanthine oxidase and mitochondria contribute to vascular superoxide anion generation in DOCA-salt hypertensive rats. Am J Physiol Heart Circ Physiol. 2008; 295:H281–288.
- Fukai T, Ishizaka N, Rajagopalan S, Laursen JB, Capers QT, Taylor WR, Harrison DG, de Leon H, Wilcox JN, Griendling KK. p22phox mRNA expression and NAD(P)H oxidase activity are increased in aortas from hypertensive rats. Circ Res. 1997;80:45–51.
- Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–8.
- Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, Cave AC, Shah AM. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res. 2003;93:802–5.
- Modlinger P, Chabrashvili T, Gill PS, Mendonca M, Harrison DG, Griendling KK, Li M, Raggio J, Wellstein A, Chen Y, Welch WJ, Wilcox CS. RNA silencing in vivo reveals role of p22phox in rat angiotensin slow pressor response. Hypertension. 2006;47:238–44.
- Laude K, Cai H, Fink B, Hoch N, Weber DS, McCann L, Kojda G, Fukai T, Schmidt HH, Dikalov S, Ramasamy S, Gamez G, Griendling KK, Harrison DG. Hemodynamic and biochemical adaptations to vascular smooth muscle overexpression of p22phox in mice. Am J Physiol Heart Circ Physiol. 2005;288:H7–12.
- Virdis A, Neves MF, Amiri F, Touyz RM, Schiffrin EL. Role of NAD(P)H oxidase on vascular alterations in angiotensin II-infused mice. J Hypertens. 2004;22:535–542.
- Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ Res. 2001;89:408–414.
- Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, Krause KH. Decreased bloodpressure in NOX1-deficient mice. FEBS Lett. 2006;580:497–504.
- Dikalova A, Clempus R, Lassegue B, Cheng G, McCoy J, Dikalov S, San Martin A, Lyle A, Weber DS, Weiss D, Taylor WR, Schmidt HH, Owens GK, Lambeth JD, Griendling KK. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112:2668–2676.
- de Cavanagh EM, Ferder LF, Ferder MD, Stella IY, Toblli JE, Inserra F. Vascular structure and oxidative stress in salt-loaded spontaneously hypertensive rats: effects of losartan and atenolol. Am J Hypertens. 2010; 23:1318–25.
- Lob HE, Marvar PJ, Guzik TJ, Sharma S, McCann LA, Weyand C, Gordon FJ, Harrison DG. Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension. 2010;55:277–283.
- Zimmerman MC, Lazartigues E, Lang JA, Sinnayah P, Ahmad IM, Spitz DR, Davisson RL. Superoxide mediates the actions of angiotensin II in the central nervous system. Circ Res. 2002;91:1038–1045.
- Davisson RL, Zimmerman MC. Angiotensin II, oxidant signaling, and hypertension: down to a T? Hypertension. 2010;55:228–30.
- Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, Weyand CM, Harrison DG, Guzik TJ. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation. 2010; 122:2529–37.
- Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, Paradis P, Schiffrin EL. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension. 2011;57:469–76.
- Yamamoto E, Tamamaki N, Nakamura T, Kataoka K, Tokutomi Y, Dong YF, Fukuda M, Matsuba S, Ogawa H, Kim-Mitsuyama S. Excess salt causes cerebral neuronal apoptosis and inflammation in stroke-prone hypertensive rats through angiotensin II-induced NADPH oxidase activation. Stroke. 2008;39:3049–56.
- Callera GE, Touyz RM, Teixeira SA, Muscara MN, Carvalho MH, Fortes ZB, Nigro D, Schiffrin EL, Tostes RC. ETA receptor blockade decreases vascular superoxide generation in DOCA-salt hypertension. Hypertension. 2003; 42:811–817.
- Chen X, Touyz RM, Park JB, Schiffrin EL. Antioxidant effects of vitamins C and E are associated with altered activation of vascular NADPH oxidase and superoxide dismutase in stroke-prone SHR. Hypertension. 2001;38:606–610.
- Lin JY, Moens AL. Tetrahydrobiopterin and hypertension: more than an emigrating story. Am J Physiol Heart Circ Physiol. 2011;300:H715.
- Antihypertensive therapy increases tetrahydrobiopterin levels and NO/cGMP signaling in small arteries of angiotensin II-infused hypertensive rats. Am J Physiol Heart Circ Physiol. 2011;300:H718–24.
- Caimi G, Mulè G, Hopps E, Carollo C, Lo Presti R. Nitric oxide metabolites and oxidative stress in mild essential hypertension. Clin Hemorheol Microcirc. 2010;46:321–5.
- Lacy F, Kailasam MT, O'Connor DT, Schmid-Schonbein GW, Parmer RJ. Plasma hydrogen peroxide production in human essential hypertension: role of heredity, gender, and ethnicity. Hypertension. 2000;36:878–884.
- Yasunari K, Maeda K, Nakamura M, Yoshikawa J. Oxidative stress in leukocytes is a possible link between blood pressure, blood glucose, and C-reacting protein. Hypertension. 2002;39:777–780.
- Lacy F, O'Connor DT, Schmid-Schönbein GW. Plasma hydrogen peroxide production in hypertensives and normotensive subjects at genetic risk of hypertension. J Hypertens. 1998;16:291–303.
- Feairheller DL, Park JY, Sturgeon KM, Williamson ST, Diaz KM, Veerabhadrappa P, Brown MD. Racial differences in oxidative stress and inflammation: in vitro and in vivo. Clin Transl Sci. 2011;4:32–7.
- Kruger R, Schutte R, Huisman HW, Van Rooyen JM, Malan NT, Fourie CM, Louw R, van der Westhuizen FH, van Deventer CA, Malan L, Schutte AE. Associations between reactive oxygen species, blood pressure and arterial stiffness in black South Africans: the SABPA study. J Hum Hypertens. 2011 Jan 27. doi:10.1038/jhh.2010.134.
- Touyz RM, Schiffrin EL. Increased generation of superoxide by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients: role of phospholipase D-dependent NAD(P)H oxidase-sensitive pathways. J Hypertens. 2001;19:1245–1254.
- Touyz RM, Yao G, Quinn MT, Pagano PJ, Schiffrin EL. p47phox associates with the cytoskeleton through cortactin in human vascular smooth muscle cells: role in NAD(P)H oxidase regulation by angiotensin II. Arterioscler Thromb Vasc Biol. 2005;25:512–518.
- Zalba G, San Jose G, Moreno MU, Fortuno A, Diez J. NADPH oxidase-mediated oxidative stress: genetic studies of the p22(phox) gene in hypertension. Antioxid Redox Signal. 2005;7:1327–1336.
- Moreno MU, Jose GS, Fortuno A, Beloqui O, Diez J, Zalba G. The C242T CYBA polymorphism of NADPH oxidase is associated with essential hypertension. J Hypertens. 2006;24:1299–306.
- Pacher P, Nivorozhkin A, Szabó C. Therapeutic Effects of Xanthine Oxidase Inhibitors: Renaissance Half a Century after the Discovery of Allopurinol Pharmacol Rev 2006;58:87–114.
- Saez GT, Tormos C, Giner V, Chaves J, Lozano JV, Iradi A, Redon J. Factors related to the impact of antihypertensive treatment in antioxidant activities and oxidative stress by-products in human hypertension. Am J Hypertens. 2004;17:809–816.
- Mullan BA, Young IS, Fee H, McCance DR. Ascorbic acid reduces blood pressure and arterial stiffness in type 2 diabetes. Hypertension. 2002;40:804–9.
- Zureik M, Galan P, Bertrais S, Mennen L, Czernichow S, Blacher J, Ducimetière P, Hercberg S. Effects of long-term daily low-dose supplementation with antioxidant vitamins and minerals on structure and function of large arteries. Arterioscler Thromb Vasc Biol. 2004;24:1485–1491.
- Caner M, Karter Y, Uzun H, Curgunlu A, Vehid S, Balci H, Yucel R, Güner I, Kutlu A, Yaldiran A, Oztürk E. Oxidative stress in human sustained and white coat hypertension. Int J Clin Pract. 2006;60:1565–1571.
- Hasnain BI, Mooradian AD. Recent trials of antioxidant therapy: what should we be telling our patients? Cleve Clin J Med. 2004;71:327–334.
- McCance DR, Holmes VA, Maresh MJ, Patterson CC, Walker JD, Pearson DW, Young IS; Diabetes and Pre-eclampsia Intervention Trial (DAPIT) Study Group. Vitamins C and E for prevention of pre-eclampsia in women with type 1 diabetes (DAPIT): a randomised placebo-controlled trial. Lancet. 2010;376:259–66.
- Roberts JM, Myatt L, Spong CY, Thom EA, Hauth JC, Leveno KJ, Pearson GD, Wapner RJ, Varner MW, Thorp JM Jr, Mercer BM, Peaceman AM, Ramin SM, Carpenter MW, Samuels P, Sciscione A, Harper M, Smith WJ, Saade G, Sorokin Y, Anderson GB; Eunice Kennedy Shriver National Institute of Child Health and Human Development Maternal-Fetal Medicine Units Network. Vitamins C and E to prevent complications of pregnancy-associated hypertension. N Engl J Med. 2010;362:1282–91.
- Al-Solaiman Y, Jesri A, Zhao Y, Morrow JD, Egan BM. Low-Sodium DASH reduces oxidative stress and improves vascular function in salt-sensitive humans. J Hum Hypertens. 2009;23:826–35.
- Chen S, Ge Y, Si J, Rifai A, Dworkin LD, Gong R. Candesartan suppresses chronic renal inflammation by a novel antioxidant action independent of AT1R blockade. Kidney Int. 2008;74:1128–1138.
- Wray DW, Uberoi A, Lawrenson L, Bailey DM, Richardson RS. Oral antioxidants and cardiovascular health in the exercise-trained and untrained elderly: a radically different outcome. Clin Sci (Lond). 2009;116:433–41.
- Greig D, Alcaino H, Castro PF, Garcia L, Verdejo HE, Navarro M, López R, Mellado R, Tapia F, Gabrielli LA, Nogerol C, Chiong M, Godoy I, Lavandero S. Xanthine-oxidase inhibitors and statins in chronic heart failure: effects on vascular and functional parameters. J Heart Lung Transplant. 2011;30:408–13.
- Robbins IM, Hemnes AR, Gibbs JS, Christman BW, Howard L, Meehan S, Cabrita I, Gonzalez R, Oyler T, Zhao L, Du RH, Mendes LA, Wilkins MR. Safety of sapropterin dihydrochloride (6r-bh4) in patients with pulmonary hypertension. Exp Lung Res. 2011;37:26–34.
- Renke M, Tylicki L, Rutkowski P, Larczynski W, Neuwelt A, Aleksandrowicz E, Łysiak-Szydłowska W, Rutkowski B. The effect of N-acetylcysteine on blood pressure and markers of cardiovascular risk in non-diabetic patients with chronic kidney disease: a placebo-controlled, randomized, cross-over study. Med Sci Monit. 2010;16:PI13–8.
- Oliveira PJ, Goncalves L, Monteiro P, Providencia LA, Moreno AJ. Are the antioxidant properties of carvedilol important for the protection of cardiac mitochondria? Curr Vasc Pharmacol. 2005;3:147–158.
- Fortuño A, Bidegain J, Robador PA, Hermida J, López-Sagaseta J, Beloqui O, Díez J, Zalba G. Losartan metabolite EXP3179 blocks NADPH oxidase-mediated superoxide production by inhibiting protein kinase C: potential clinical implications in hypertension. Hypertension. 2009;54:744–50.
- Silva PS, Fontana V, Palei AC, Sertório JT, Biagi C, Tanus-Santos JE. Antihypertensive effects exerted by enalapril in mild to moderate hypertension are not associated with changes in the circulating levels of nitric oxide-related markers. Eur J Clin Pharmacol. 2011;67:365–70.
- Kuklińska AM, Mroczko B, Musiał WJ, Sawicki R, Kozieradzka A, Usowicz-Szaryńska M, Kamiński K, Knapp M, Szmitkowski M. Hypotensive effect of atorvastatin is not related to changes in inflammation and oxidative stress. Pharmacol Rep. 2010;62:883–90.
- Kim JA, Neupane GP, Lee ES, Jeong BS, Park BC, Thapa P. NADPH oxidase inhibitors: a patent review. Expert Opin Ther Pat. 2011;21:1147–1158.