Abstract
High-ceiling diuretics (HCD), known potent inhibitors of housekeeping Na+,K+,2Cl cotransporter (NKCC1) and renal-specific NKCC2, decrease [Cl−]i, hyperpolarize vascular smooth muscle cells (VSMC), and suppress contractions evoked by modest depolarization, phenylephrine, angiotensin II, and UTP. These actions are absent in nkcc1 / knock-out mice, indicating that HCD interact with NKCC1 rather than with other potential targets. These findings also suggest that VSMC-specific inhibitors of NKCC1 may be considered potential pharmacological therapeutic tools in treatment of hypertension. It should be underlined that side by side with attenuation of peripheral resistance and systemic blood pressure, HCD blocked myogenic tone (MT) in renal afferent arterioles. Keeping this in mind, attenuation of MT might be a mechanism underlying the prevalence of end-stage renal disease documented in hypertensive African-Americans with decreased NKCC1 activity and in hypertensive patients subjected to chronic HCD treatment. The role of NKCC1-mediated MT in protection of the brain, heart, and other encapsulated organs deserves further investigation.
| Abbreviations | ||
| AII | = | angiotensin II |
| BP | = | blood pressure |
| Em | = | |
| = | electrical membrane potential | |
| HCD | = | high-ceiling diuretics |
| KCC | = | K+,Cl cotransporter |
| MHS and MNS | = | Milan hypertensive and normotensive strains |
| MT | = | myogenic tone |
| NCC | = | Na + ,Cl cotransporter |
| NKCC | = | Na + ,K + ,2Cl cotransporter |
| PE | = | phenylephrine |
| SHR | = | spontaneously hypertensive rats |
| SPAK | = | 20/SPS1-related proline/alanine-rich kinase |
| VSMC | = | vascular smooth muscle cells |
| WNK | = | ‘with no lysine’ (K) kinases |
Key messages
Na+,K+,2Cl cotransport isoform 1 (NKCC1) is involved in regulation of vascular smooth muscle cell (VSMC) contractions via [Cl−]i increment, membrane depolarization, and activation of voltage-gated Ca2+ channels that, in turn, leads to elevation of peripheral resistance.
NKCC1 also plays a key role in regulation of myogenic tone (MT) in renal afferent arteriole.
Thus, systems providing cell type-specific regulation of NKCC1 in resistant vessels with low MT may be considered potential pharmacological targets in hypertension, whereas attenuation of NKCC1-dependent MT might be a mechanism underlying the prevalence of end-stage renal disease documented in hypertensive with decreased NKCC1 activity and/or subjected to chronic treatment with high-ceiling diuretics.
The hypothesis of involvement of Na+ ,K + ,2Cl cotransporter (NKCC) in the pathogenesis of hypertension is based on two initial observations. First, in the mid-1980s, Bianchi and co-workers obtained F1 hybrids of the Milan hypertensive and normotensive strains (MHS and MNS, respectively) and subjected them to X-ray irradiation and bone-marrow transplantation. They discerned that NKCC activity in erythrocytes from F1 MHS bone-marrow recipients is elevated to a similar extent as in donor strains compared to MNS bone-marrow recipients (Citation1). Second, in erythrocytes from F2 hybrids of MHSxMNS (Citation1) as well as spontaneously hypertensive rats developed by K. Okamoto (SHR) and normotensive WKY rats (Citation2), NKCC activity correlates positively with blood pressure (BP) (for more details, see (Citation3,Citation4)).
Two NKCC isoforms providing electroneutral symport of monovalent ions with stoichiometry of 1Na + :1K + :2Cl have been cloned from vertebrate cDNA libraries. NKCC2, expressed exclusively in apical membranes of renal epithelial cells, plays a major role in renal handling of salt and osmotically obliged water. In contrast, NKCC1 is expressed in all types of cells studied so far, including vascular smooth muscle cells (VSMC) and erythrocytes (Citation5,Citation6). Both isoforms are inhibited by high-ceiling diuretics (HCD), such as furosemide and bumetanide (Citation7). The renal mechanisms implicating NKCC1 and NKCC2 in BP regulation have been considered in recent publications (Citation8,Citation9). This mini-review focuses on the role of NKCC1 in the maintenance of systemic and local BP via regulation of VSMC contractions and myogenic responses.
Molecular mechanisms involved during NKCC1 regulation of vasoconstriction and hypertension
Under baseline conditions ([Na+]o >>[Na +]i and [Cl−]o2 >>[Cl−]i2), NKCC1 provides inwardly directed net ion flux and maintenance of [Cl−]i above values predicted by the Nernst equilibrium potential (Citation7). Importantly, unlike the dominant contribution of K + permeability (PK) to resting membrane potential (Em) in skeletal and cardiac muscles, PK and PCl values in VSMC are somewhat similar (Citation10), suggesting the involvement of NKCC1-mediated [Cl−]i modulation in the regulation of Em and Em-dependent VSMC contractions. Indeed, HCD decrease [Cl−]i (Citation11,Citation12), hyperpolarize VSMC (Citation11), and attenuate the activation of voltage-gated L-type Ca2+ channels (Citation12). These studies disclose the mechanism underlying the inhibitory action of HCD on the baseline tone of smooth muscles (Citation13–15) as well as their contraction triggered by modest K+ -induced depolarization (Citation12), electrical stimulation (Citation16), histamine (Citation17), angiotensin II (AII) (Citation18), thromboxane A2 (Citation19,Citation20), oxytocin (Citation21,Citation22), the α-adrenergic agonist phenylephrine (PE) (Citation12,Citation23–25), and agonists of P2Y purinergic receptors (Citation26). We have reported that the inhibitory action of bumetanide on VSMC contractions triggered by modest depolarization, α-adrenergic stimulation, and UTP is completely abolished in nkcc1-null mice (Citation27). These results show that HCD inhibit VSMC contractions via their interactions with NKCC1 rather than with other potential targets.
Meyer and co-workers observed that attenuation of systolic BP was accompanied by decreased left ventricular pressure without any changes in myocardial contraction parameters (Citation28). Garg and co-workers examined the role of attenuated tone in resistance arteries under BP declines evoked by bumetanide. They found that contractile responses in PE-treated, isolated, third-order mesenteric arteries were associated with transient NKCC activation and were suppressed by bumetanide (Citation24). Recently, the augmented inhibitory action of bumetanide on PE-induced contractions of mesenteric arteries and aortae from SHR was demonstrated by Lee and co-workers (Citation29). The bumetanide-sensitive component of PE-induced contractions was also sharply decreased in mice lacking SPAK (20/SPS1-related proline/alanine-rich kinase) (Citation30), playing a key role in phosphorylation and activation of NKCC1 (Citation31). Viewed collectively, these data indicate that augmented NKCC1 activity may contribute to the pathogenesis of hypertension via its involvement in the regulation of VSMC contractions. Indeed, systolic BP measured by tail cuff of femoral artery catheter was decreased by 15–20 mmHg in nkcc1 / mice compared to wild-type animals (Citation28,Citation32,Citation33). More recently, Kim and co-workers applied radiotelemetry for mean arterial BP measurement. This investigation did not detect any differences between nkcc1 / and wild-type mice (Citation34). Additional studies should be performed to clarify the nature of this discrepancy.
The mechanisms underlying altered NKCC1 activity in hypertension are still a controversial matter that probably reflects its diverse regulatory pathways and the mosaic origin of hypertension. Thus, in VSMC, [Ca2+]i and aldosterone elevations increase NKCC1 activity, whereas cAMP inhibits this carrier without alteration of its expression (Citation23,Citation35,Citation36). Numerous early investigations have demonstrated abnormal [Ca2 +]i handling and cAMP-mediated signaling in primary hypertension (for review, see (Citation37,Citation38)). Side by side with [Ca2 +]i- and cAMP-mediated signaling, the role of ‘with no lysine’ (K) kinases (WNK) in the regulation of NKCC1 and other Cl -coupled ion carriers has been well documented in numerous in-vitro studies (Citation39–41). Recently, Bergaya and co-workers reported that Wnk+/ mice exhibit a pronounced decrease in NKCC1 phosphorylation and BP responses following α-adrenergic activation (Citation42). However, data on mutations of genes encoding these enzymes are limited to rare monogenic (Mendelian) forms of hypertension, such as Gordon's disease and familial hypertension of pseudohypoaldosteronism type II (Citation43). It should also be noted that mutations of WNK1 and WNK4, seen in monogenic hypertension, contribute to BP elevation via their involvement in the abnormal activity of renal Na + ,Cl and K + ,Cl cotransporters (NCC and KCC, respectively) rather than via NKCC1 (Citation44).
Sterile 20/SPS1-related proline/alanine-rich kinase (SPAK) is a downstream substrate of WNK1 and WNK4 and an upstream regulator of NCC and NKCC (Citation31). NKCC1 expression is increased, whereas phosphorylated NKCC1 is decreased in the aortic tissues of SPAK-deficient mice (Citation30). Rafigi and co-workers generated knock-in mice in which SPAK cannot be activated by WNKs (Citation45). These animals display reduced BP that was accompanied by attenuated expression in the kidney of NCC and NKCC2 protein. To the best of our knowledge, there are no reports of altered SPAK expression and/or activity in hypertension.
Recently, Lee and co-workers reported that increased nkcc1 mRNA and NKCC1 protein content of the aorta and heart of SHR is accompanied by hypomethylation of the nkcc1 gene promoter (Citation29). Importantly, methylation of nkcc1 promoter in normotensive WKY rats was increased with age, whereas in SHR it remained hypomethylated after development of hypertension (Citation46). Both increased nkcc1 expression and inhibitory action of bumetanide on mesenteric artery contractions were increased with age in SHR but not in WKY rats. They also found that at 18 weeks of age the activity of DNA methyltransferase 3B (DNTB3B) in the aorta of WKY was 3-fold higher than that of age-matched SHR (Citation46). Viewed collectively, these data strongly suggest that the maintenance of hypomethylation in nkcc1 promoter due to the decreased DNTB3B activity underlies age-dependent development of hypertension in SHR via augmented expression of this carrier in VSMC that, in turn, leads to depolarization and contraction of resistance arteries.
NKCC1 as a regulator of myogenic tone (MT)
Myogenic tone (MT) is the intrinsic ability of small arteries to constrict in response to increases in intraluminal pressure in the absence of blood flow. In all types of blood vessels studied so far, МT is accompanied by depolarization of the sarcolemma that, in turn, culminates in activation of voltage-gated Ca2 + channels and elevation of [Ca2 +]i (Citation47,Citation48); in several resistance vessels, MT is caused by heightened myosin light chain kinase sensitivity to Ca2+ or by С2+ -independent modification of actin microfilaments (Citation49). Considering this, it is noteworthy that both the magnitude and kinetics of MT are different in different blood vessels. Thus, in renal afferent arterioles, intraluminal pressure elevation from 90 to 160 mmHg results in almost 2-fold attenuation of inner diameter within the first 5–10 s (Citation50). In brain arteries, MT developing in 30 s of intraluminal pressure elevation from 20 to 80 mmHg leads to complete normalization of vessel diameter (Citation51), whereas in mesenteric arteries the partial normalization of the diameter occurs in 3–5 min of pressure elevation (Citation27). This diversity is probably elicited by a VSMC-specific set of signaling systems involved in myogenic responses. In contrast to the ubiquitous role of depolarization and voltage-gated Ca2+ channels, these upstream signaling systems provide specific mechanisms of adaptation to an altered supply of oxygen and blood-derived fuels to tissues, triggered by BP and blood flow modulation.
Numerous experiments have demonstrated that furosemide and other HCD suppress the autoregulation of renal blood flow via their interaction with NKCC2 that, in turn, leads to inhibition of tubuloglomerular feedback (Citation8). Wang and co-workers were the first to report that these compounds reversibly inhibit MT in renal afferent arterioles (Citation20). In contrast to renal afferent arterioles, bumetanide diminished MT in mesenteric arteries by 50%–60% (Citation27). This discrepancy might be explained by the actions of HCD on targets distinct from NKCC1. Indeed, in VSMC, furosemide partially inhibited KCC (Citation52) and thromboxane A2 receptor-mediated signaling (Citation19) and activated cyclic nucleotide phosphodiesterase (Citation22). Keeping this in mind, we investigated MT regulation in mesenteric arteries isolated from nkcc1 / mice. We discovered that the inhibitory action of bumetanide on MT is completely abolished in nkcc1-null mice (Citation27). These results disclosed, for the first time, that HCD suppress MT via their interactions with NKCC1.
Myogenic tone as a drug target
Because blood flow resistance varies inversely to the fourth power of vessel diameter (Citation53), the involvement of abnormal MT responses in protecting against hypertension-induced organ damage was the subject of numerous investigations (Citation54). Importantly, sustained suppression of MT evokes hypertrophic vascular remodeling that, in turn, decreases the sensitivity of MT to BP variations. As a result of this vicious circle, systemic BP elevation is transferred to the microcirculation and high BP-induced damage of encapsulated organs, such as the brain, heart, retina, and kidneys (Citation55,Citation56). Because of this, the impact of antihypertensive drugs and other therapeutics on MT needs careful examination.
Calcium channel blockers, such as nifedipine, amlodipine, and diltiazem, which target L-type voltage-gated channels, exhibit potent and long-lasting antihypertensive actions, ranking second in the pharmaceutical market for hypertension treatment. It is generally accepted that the blood–brain barrier protects the brain through abundant voltage-gated Ca2 + channels from the side-effects of these drugs. However, such is not the case with other encapsulated target organs, including the kidneys. A growing body of evidence points to a problem associated with the development of kidney insufficiency in patients treated with calcium channel blockers compared to other antihypertensive drugs, such as thiazides and renin-angiotensin-aldosterone system inhibitors (Citation56–59). An analysis of publications has demonstrated an increased incidence of heart failure, side by side with renal complications, in hypertensive patients treated with calcium channel blockers (Citation60). Both in-vivo and in-vitro studies strongly indicate that problems associated with the chronic administration of calcium channel blockers to hypertensive patients are caused by MT inhibition in afferent arterioles that leads to pressure elevation in the local microcirculation despite decreased systemic BP. Indeed, given in vivo, these compounds cause a greater increase in the glomerular filtration rate than in renal plasma flow. Such observations were confirmed in isolated, perfused, intact, and hydronephrotic kidneys and by direct visualization of the nephron circulation (for review, see (Citation61–63)). The preferential afferent arteriole actions of calcium antagonists indicate a greater abundance of L-type channels compared to efferent arterioles. Indeed, Hansen and co-workers demonstrated that Cav1.2 L-type channel subunits in rabbit kidneys are expressed in afferent but not in efferent arterioles (Citation64).
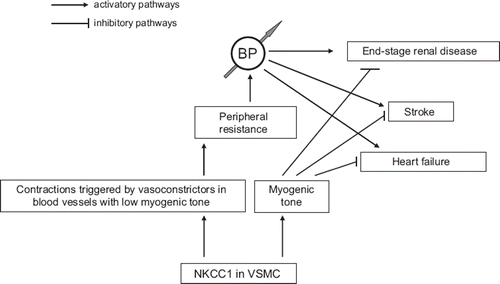
Loutzenhiser and co-workers developed a model of isolated perfused hydronephrotic kidneys that allows direct observation of the renal microvasculature in the absence of its regulation via tubuloglomerular feedback. With this model, they demonstrated that low doses of bumetanide and furosemide dose-dependently inhibited contractions of rat afferent arterioles (Citation20). Their results as well as the absence of HCD actions on MT in mesenteric arteries isolated from nkcc1 / mice (Citation27) prompted us to propose that augmented NKCC1 activity protects the kidneys from high BP-induced damage, whereas chronic HCD usage accelerates the development of proteinuria and end-stage kidney disease (Citation9,Citation65). Indeed, highly active NKCC1 keeps unchanged renal blood flow via its manifestation in VSMC of afferent arteriole possessing sharp MT in spite of elevation of systolic BP caused by augmented NKCC1 in mesenteric arteries and other blood vessels with low NKCC1-sensitive MT (). Considering this, HCD and other inhibitors of NKCC1 abolish this protective mechanism via inhibition of MT in the renal afferent arteriole. This hypothesis is consistent with decreased by more than 2-fold NKCC1 activity in erythrocytes from normotensive and hypertensive blacks compared to their Caucasian counterparts (for more details, see (Citation6,Citation65)) and with ∼4-fold greater prevalence of end-stage renal disease documented in hypertensive African-Americans compared to hypertensive whites (Citation66,Citation67). Further investigations should clarify the relative impact of NKCC1 in the regulation of [Cl−]i, Em, and contractions of VSMC from afferent and efferent arterioles as well as the spectrum of MT-mediated side-effects of chronic HCD administration that is widespread in the treatment of heart failure and other volume-expanded disorders.
Figure 1. Mechanisms of NKCC1 involvement in vascular smooth muscle cell (VSMC)-mediated blood pressure (BP) regulation and target organ damage: a working hypothesis.
Conclusions and unresolved issues
We have drawn three major conclusions from the data considered in the mini-review:
First, NKCC1 is involved in BP elevation via [Cl−]i increment, membrane depolarization, and voltage-gated Ca2+ channel activation that, in turn, leads to augmented MT and contractions evoked by electrical stimuli and vasoconstrictors ().
Second, cell-type specific inhibition of NKCC1 in mesenteric arteries and other resistant blood with low-active MT may be considered a potential pharmacological approach for treatment of hypertension.
Third, attenuation of NKCC1-dependent MT might be a mechanism underlying the prevalence of end-stage renal disease documented in hypertensive African-Americans with decreased NKCC1 activity and in hypertensive patients subjected to chronic HCD treatment (). The role of NKCC1-mediated MT in protection of the brain, heart, and other encapsulated organs deserves further investigation.
Keeping in mind the dual mechanism of NKCC1 in the pathogenesis of hypertension and its complications, several questions should be answered and alternative pharmacological tools should be tested to achieve the therapeutic targeting of this carrier.
NKCC in cultured VSMC and isolated vessels is well documented to be activated by vasoconstrictors, such as PE and AII, and inhibited by cAMP-elevating compounds and other vasodilators (Citation23,Citation35). Does reciprocal regulation of NKCC1 by vasoconstrictors and vasodilators contribute to vascular tone control?
Side by side with vasoactive compounds, NKCC1 is reciprocally affected by cell shrinkage and swelling (Citation68). Our studies have revealed that VSMC contractions might be evoked by anisosmotic media (Citation12,Citation69). Does cell volume modulation have an impact on NKCC-mediated VSMC contractions?
Endothelium-denuded vessels in an overwhelming number of investigations have demonstrated the vasodilatory actions of HCD. More recently, we reported that treatment with the nitric oxide synthase blocker L-NAME did not affect the inhibitory action of bumetanide on MT and contractions of mesenteric arteries evoked by modest depolarization and agonists of α-adrenergic and P2Y-receptors (Citation27). In contrast, Wang and co-workers noted that L-NAME slowed the dilating action of bumetanide on the MT of renal afferent arterioles (Citation20). This observation raises questions about the molecular origin of HCD-sensitive endothelial elements involved in the regulation of VSMC contractions.
Should we consider [Cl−]i-dependent modulation of Em to be the only physiologically relevant consequence of NKCC1 regulation? In other words, does [Cl−]i affect cellular functions via its interaction with intracellular sensor(s), as shown for other monovalent ions, such as Na+ and HCO32 (Citation70)?
Autoregulation of blood flow in kidney and other target organs display remarkable differences between human and experimental rodents. Thus, the novel approaches for in-vivo measurement of the actions of HCD and other drugs on MT in humans should be developed in forthcoming investigations.
Currently available inhibitors, such as bumetanide and furosemide, exhibit similar affinity for NKCC1 and NKCC2. Importantly, the apparent affinity of NKCC for HCD is sharply increased by diverse activators of these carriers (Citation71). Thus, NKCC1 should have reduced HCD sensitivity under baseline conditions compared to permanently active NKCC2. Because of this, vasodilatation through NKCC1 inhibition will be subjugated by massive diuresis caused by NKCC2 inhibition. Moreover, the lack of function of NKCC1 in the inner ear epithelium causes profound deafness in mice and humans (Citation72,Citation73). These data suggest that cell type-specific inhibitors of SPAK and other upstream NKCC1 activators are probably the preferable challenge in antihypertensive therapy.
Acknowledgements
The editorial help of Ovid Da Silva is appreciated.
Declaration of interest: This study was supported by grants from the Canadian Institutes of Health Research (MOP-81392), the Heart and Stroke Foundation of Canada, the Kidney Foundation of Canada, the Russian Foundation for Fundamental Research (09-0073/04), and the Federal Aim Program 2009–2013 ‘Research and Teaching Resources of the Innovating Russia’.
Related Research Data
References
- Bianchi G, Ferrari P, Trizio P, Ferrandi M, Torielli L, Barber BR, . Red blood cell abnormalities and spontaneous hypertension in rats. A genetically determined link. Hypertension. 1985;7:319–25.
- Kotelevtsev IuV, Orlov SN, Pokudin NI, Agnaev VM, Postnov IuV. [Genetic analysis of inheritance of Na+,K+ cotransport, calcium level in erythrocytes and blood pressure in F2 hybrids of spontaneously hypertensive and normotensive rats]. Bull Exp Biol Med. 1987;103:456–8.
- Orlov SN, Adragna N, Adarichev VA, Hamet P. Genetic and biochemical determinants of abnormal monovalent ion transport in primary hypertension. Am J Physiol. 1999;276: C511–36.
- Garay RP, Alda O. What can we learn from erythrocyte Na-K-Cl cotransporter NKCC1 in human hypertension. Pathophysiology. 2007;14:167–70.
- Jiang G, Akar F, Cobbs SL, Lomashvilli K, Lakkis R, Gordon FJ, . Blood pressure regulates the activity and function of Na-K-2Cl cotransporter in vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2004;286:H1552–7.
- Orlov SN, Gossard F, Pausova Z, Akimova OA, Tremblay J, Grim CE, . Decreased NKCC1 activity in erythrocytes from African-Americans with hypertension and dyslipidemia. Am J Hypertens. 2010;23:321–6.
- Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev. 2005;85:423–93.
- Orlov SN, Mongin AA. Salt sensing mechanisms in blood pressure regulation and hypertension. Am J Physiol Heart Circ Physiol. 2007;293:H2039–53.
- Orlov SN, Tremblay J, Hamet P. NKCC1 and hypertension: a novel therapeutic target involved in regulation of vascular tone and renal function. Curr Opin Nephrol Hypertens. 2010;19:163–8.
- Chipperfield AR, Harper AA. Chloride in smooth muscle. Prog Biophys Mol Biol. 2001;74:175–221.
- Davis JPL, Chipperfield AR, Harper AA. Accumulation of intracellular chloride by (Na-K-Cl) cotransport in rat arterial smooth muscle is enhanced in deoxycorticosterone acetate (DOCA)/salt hypertension. J Mol Cell Cardiol. 1993;25: 233–7.
- Anfinogenova YJ, Baskakov MB, Kovalev IV, Kilin AA, Dulin NO, Orlov SN. Cell-volume-dependent vascular smooth muscle contraction: role of Na+, K+, 2Cl cotransport, intracellular Cl and L-type Ca2+ channels. Pflügers Archiv. 2004;449: 42–55.
- Barthelmebs M, Stephan D, Fontaine C, Grima M, Imbs JL. Vascular effects of loop diuretics: an in vivo and in vitro study in the rat. Naunyn-Schmiedebergs Arch Pharmacol. 1994;349: 209–16.
- Lavallee SL, Iwamoto LM, Claybaugh JR, Dressel MV, Sato AK, Nakamura KT. Furosemide-induced airway relaxation in guinea pigs: relation to Na-K-2Cl cotransporter function. Am J Physiol. 1997;273:L211–6.
- Tian R, Aalkjaer C, Andreasen F. Mechanisms behind the relaxing effect of furosemide on the isolated rabbit ear artery. Pharmacol Toxicol. 1990;67:406–10.
- Kovalev IV, Baskakov MB, Anfinogenova YJ, Borodin YL, Kilin AA, Minochenko IL, . Effect of Na +, K+, 2Cl cotransport inhibitor bumetanide on electrical and contractile activity of smooth muscle cells in guinea pig ureter. Bull Exp Biol Med. 2003;136:145–9.
- Kovalev IV, Baskakov MB, Medvedev MA, Minochenko IL, Kilin AA, Anfinogenova YJ, . Na + ,K + ,2Cl cotransport and chloride permeability of the cell membrane in mezaton and histamine regulation of electrical and contractile activity in smooth muscle cells from the guinea pig ureter. Russian Physiol J. 2008;93:306–17.
- Stanke F, Devillier P, Breant D, Chavanon O, Sessa C, Bricca G, . Furosemide inhibits angiotensin II-induced contraction on human vascular smooth muscle. Br J Clin Pharmacol. 1998;46:571–5.
- Stanke-Labesque F, Craciwski JL, Bedouch P, Chavanon O, Magne JL, Bessard G, . Furosemide inhibits thrombaxane A2-induced contraction in isolated human internal artery and saphenous vein. J Cardiovasc Pharmacol. 2000;35: 531–7.
- Wang X, Breaks J, Loutzenhiser K, Loutzenhiser R. Effects of inhibition of the Na + /K + /2Cl- cotransporter on myogenic and angiotensin II responses of the rat afferent arteriole. Am J Physiol Renal Physiol. 2007;292:F999–1006.
- Mozhayeva MG, Bagrov YY. The inhibitory effects of furosemide on Ca2+ influx pathways associated with oxytocin-induced contractions of rat myometrium. Gen Physiol Biophys. 1995;14:427–36.
- Mozhayeva MG, Bagrov YY, Ostretsova IB, Gillespie JI. The effect of furosemide on oxytocin-induced contractions of the rat myometrium. Exp Physiol. 1994;79:661–7.
- Akar F, Skinner E, Klein JD, Jena M, Paul RJ, O'Neill WC. Vasoconstrictors and nitrovasodilators reciprocally regulate the Na + -K + -2Cl cotransporter in rat aorta. Am J Physiol. 1999;276:C1383–90.
- Garg P, Martin C, Elms SC, Gordon FJ, Wall SM, Garland CJ, . Effect of the Na-K-2Cl cotransporter NKCC1 on systematic blood pressure and smooth muscle tone. Am J Physiol Heart Circ Physiol. 2007;292:H2100–5.
- Palacios J, Espinoza F, Munita C, Cifuentes F, Michea L. Na + -K + -2Cl cotransporter is implicated in gender differences in the response of the rat aorta to phenylephrine. Br J Pharmacol. 2006;148:964–72.
- Koltsova SV, Maximov GV, Kotelevtsev SV, Lavoie JL, Tremblay J, Grygorczyk R, . Myogenic tone in mouse mesenteric arteries: evidence for P2Y receptor-mediated, Na + ,K + ,2Cl cotransport-dependent signaling. Purinergic Signal. 2009;5:343–9.
- Koltsova SV, Kotelevtsev SV, Tremblay J, Hamet P, Orlov SN. Excitation-contraction coupling in resistant mesenteric arteries: evidence for NKCC1-mediated pathway. Biochem Biophys Res Commun. 2009;379:1080–3.
- Meyer JW, Flagella M, Sutliff RL, Lorenz JN, Nieman ML, Weber GS, . Decreased blood pressure and vascular smooth muscle tone in mice lacking basolateral Na + -K + -2Cl cotransporter. Am J Physiol. 2002;283:H1846–55.
- Lee H-A, Baek I, Seok YM, Yang E, Cho H-M, Lee D-Y, . Promoter hypomethylation upregulates Na + -K + -2Cl cotransporter 1 in spontaneously hypertensive rats. Biochem Biophys Res Commun. 2010;396:252–7.
- Yang S-S, Lo Y-F, Wu C-C, Lin S-W, Yeh C-J, Chu P, . SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol. 2010;21:1868–77.
- Vitari AC, Thastrup J, Rafigi FH, Deak M, Morrice NA, Karlsson HK, . Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem J. 2006;397:223–31.
- Flagella M, Clarke LL, Miller ML, Erway LC, Giannella RA, Andriga A, . Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J Biol Chem. 1999;274:26946–55.
- Wall SM, Knepper MA, Hassel KA, Fischer MP, Shodeinde A, Shin W, . Hypotension in NKCC1 null mice: role of the kidney. Am J Physiol Renal Physiol. 2006;290:F409–16.
- Kim SM, Eisner C, Faulhaber-Walter R, Mizel D, Wall SM, Briggs JP, . Salt sensitivity of blood pressure in NKCC1-deficient mice. Am J Physiol Renal Physiol. 2008;295: F1230–8.
- Orlov SN, Resink TJ, Bernhardt J, Buhler FR. Na + -K + pump and Na + -K + co-transport in cultured vascular smooth muscle cells from spontaneously hypertensive rats: baseline activity and regulation. J Hypertens. 1992;10:733–40.
- Jiang G, Cobbs S, Klein JD, O'Neill WC. Aldosterone regulates the Na-K-Cl cotransporter in vascular smooth muscle. Hypertension. 2003;41:1131–5.
- Orlov SN, Li J-M, Tremblay J, Hamet P. Genes of intracellular calcium metabolism and blood pressure control in primary hypertension. Semin Nephrol. 1995;15:569–92.
- Hamet P, Orlov SN, Tremblay J. Intracellular signalling mechanisms in hypertension. In: Laragh JH, Brenner BM, editors. Hypertension: pathophysiology, diagnosis, and treatment. New York: Raven Press; 1995. p. 575–608.
- Gagnon F, Delpire E. Multiple pathways for protein phosphatase 1 (PP1) regulation of Na-K-2Cl cotransporter (NKCC1) function: the N-terminal tail of the Na-K-2Cl cotransporter serves as a regulator scaffold for the Ste20-related proline/alanine-rich kinase (SPAK) and PP1. J Biol Chem. 2010;285:14115–21.
- Sid B, Miranda L, Vertommen D, Viollet B, Rider MH. Stimulation of human and mouse erythrocyte Na + -K + -2Cl- cotransport by osmotic shrinkage does not involve AMP-activated protein kinase, but is associated with STE20/SPS1-related proline/alanine-rick kinase activation. J Physiol. 2010;588:2315–28.
- Hannemann A, Flatman PW. Phosphorylation and transport in the Na-K-2Cl cotransporters, NKCC1 and NKCC2A, compared in HEK-293 cells. PLoS One. 2011;25:e17992.
- Bergaya S, Faure S, Baudrie V, Rio M, Escoubet B, Bonnin P, . WNK1 regulates vasoconstriction and blood pressure response to α1-adrenergic stimulation in mice. Hypertension. 2011;58:439–45.
- Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, . Human hypertension caused by mutations in WNK kinases. Science. 2001;293: 1107–12.
- San-Cristobal P, de los Heros P, Ponce-Coria J, Moreno T, Gamba G. WNK kinases, renal ion transport and hypertension. Am J Nephrol. 2008;28:860–70.
- Rafigi FH, Zuber AM, Glover M, Richardson C, Fleming S, Jovanovic A, . Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol Med. 2010;2:63–75.
- Cho H-M, Lee H-A, Kim HY, Han HS, Kim IK. Expression of Na + ,K + -2Cl cotransporter is epigenetically regulated during postnatal development of hypertension. Am J Hypertens. 2011;24:1286–93.
- Schubert R, Mulvany MJ. The myogenic response: established facts and attractive hypothesis. Clin Sci. 1999;96:313–26.
- Hill MA, Davis MJ, Meininger GA, Potocnik SJ, Murphy TV. Arteriolar myogenic signaling mechanisms: implications for local vascular functions. Clin Hemorheol Microcirc. 2006;34:67–79.
- Schubert R, Lidington D, Bolz S-S. The emerging role of Ca2 + sensitivity regulation in promoting myogenic vasoconstriction. Cardiovasc Res. 2008;77:8–18.
- Loutzenhiser R, Bidani AK, Wang X. Systolic pressure and the myogenis response of the afferent arteriole. Acta Physiol Scand. 2004;181:407–13.
- Lagaud G, Gaudreault N, Moore EDW, Van Breemen C, Laher I. Pressure-dependent myogenic constriction of cefrebral arteries occurs independently of voltage-dependent activation. Am J Physiol Heart Circ Physiol. 2002;283: H2187–95.
- Adragna N, White RE, Orlov SN, Lauf PK. K-Cl cotransport in vascular smooth muscle and erythrocytes: possible implication in vasodilation. Am J Physiol. 2000;278:C381–90.
- Folkow B. Cardiovascular “remodeling” in rat and human: time axis, extent, and in vivo relevance. Physiology (Bethesda). 2010;25:264–5.
- Loutzenhiser R, Griffin K, Williamson G, Bidani A. Renal autoregulation: new perspectives regarding the protective and regulatory roles of the underlying mechanisms. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1153–67.
- Liu Y, Gutterman DD. Vascular control in humans: focus on the coronary micocirculation. Basic Res Cardiol. 2009;104: 211–27.
- Bidani A, Griffin KA, Williamson G, Wang X, Loutzenhiser R. Protective importance of the myogenic response in the renal circulation. Hypertension. 2009;54:393–8.
- Nathan S, Pepine CJ, Bakris GL. Calcium antagonists: effects on cardio-renal risk in hypertensive patients. Hypertension. 2005;46:637–42.
- Griffin KA, Picken MM, Bakris GL, Bidani AK. Class differences in the effects of calcium channel blockers in the rat remnant kidney model. Kidney Int. 1999;55:1849–60.
- Bakris GL, Toto RD, McCullough PA, Rocha R, Purkayastha D, Davis P, . Effect of different ACE inhibitor combinations on albuminuria: results of the GUARD study. Kidney Int. 2008;73:1303–9.
- Shibata MC, Leon H, Chatterley T, Dorgan M, Vandermeer B. Do calcium channel blockers increase the diagnosis of heart failure with hypertension? Am J Cardiol. 2010;106: 228–35.
- Loutzenhiser R, Epstein M. The renal hemodynamic effects of calcium antagonists. In: Epstein M, Loutzenhiser R, editors. Calcium antagonists and the kidney. Philadelphia: Hanley & Belfus, Inc.; 1990. p. 33–74.
- Hayashi K, Homma K, Wakino S, Tokuyama H, Sugano N, Saruta T, . T-type channel blockage as a determinant of kidney protection. Keio J Med. 2010;59:84–95.
- Inscho EW, Cook AK, Imig JD, Vial C, Evans RJ. Physiological role for P2X1 receptors in renal microvascular autoregulatory behavior. J Clin Invest. 2003;112:1895–905.
- Hansen PB, Jensen BL, Andreasen D, Scott O. Differential expression of T- and L-type voltage-dependent calcium channels in renal resistance vessels. Circ Res. 2001;89: 630–8.
- Orlov SN. Decreased Na + ,K + ,Cl cotransport and salt retention in Blacks: a provocative hypothesis. J Hypertens. 2005;23:1929–30.
- Boone CA. End-stage renal disease in African-Americans. Nephrol Nurs J. 2000;27:597–600.
- Kotchen TA, Piering AW, Cowley AW, Grim CE, Gaudet D, Hamet P, . Glomerular hyperfiltration in hypertensive African Americans. Hypertension. 2000;35:822–6.
- Orlov SN, Resink TJ, Bernhardt J, Buhler FR. Volume-dependent regulation of sodium and potassium fluxes in cultured vascular smooth muscle cells: dependence on medium osmolality and regulation by signalling systems. J Membrane Biol. 1992;126:199–210.
- Koltsova SV, Gusakova SV, Anfinogenova YJ, Baskakov MB, Orlov SN. Vascular smooth muscle contraction evoked by cell volume modulation: role of cytoskeleton network. Cell Physiol Biochem. 2008;21:29–36.
- Orlov SN, Hamet P. Intracellular monovalent ions as second messengers. J Membr Biol. 2006;210:161–72.
- Hannaert P, Alvarez-Guerra M, Pirot D, Nazaret C, Garay RP. Rat NKCC2/NKCC1 cotransport selectivity for loop diuretic drugs. Naunyn-Schmiedebergs Arch Pharmacol. 2002;365:193–9.
- Delpire E, Lu J, England R, Dull C, Thorne T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat Genet. 1999;22: 192–5.
- Lang F, Vallon V, Knipper M, Wangemann P. Functional significance of channels and transporters expressed in the inner ear and kidney. Am J Physiol Cell Physiol. 2007;293: C1187–208.