Abstract
Dietary salt intake is the most important factor contributing to hypertension, but the salt susceptibility of blood pressure (BP) is different in individual subjects. Although the pathogenesis of salt-sensitive hypertension is heterogeneous, it is mainly attributable to an impaired renal capacity to excrete sodium (Na+ ). We recently identified two novel mechanisms that impair renal Na+ -excreting function and result in an increase in BP. First, mineralocorticoid receptor (MR) activation in the kidney, which facilitates distal Na+ reabsorption through epithelial Na+ channel activation, causes salt-sensitive hypertension. This mechanism exists not only in models of high-aldosterone hypertension as seen in conditions of obesity or metabolic syndrome, but also in normal- or low-aldosterone type of salt-sensitive hypertension. In the latter, Rac1 activation by salt excess causes MR stimulation. Second, renospecific sympathoactivation may cause an increase in BP under conditions of salt excess. Renal beta2 adrenoceptor stimulation in the kidney leads to decreased transcription of the gene encoding WNK4, a negative regulator of Na+ reabsorption through Na+ -Cl − cotransporter in the distal convoluted tubules, resulting in salt-dependent hypertension. Abnormalities identified in these two pathways of Na+ reabsorption in the distal nephron may present therapeutic targets for the treatment of salt-sensitive hypertension.
| Abbreviations | ||
| Ang II | = | angiotensin II |
| AT1 | = | angiotensin II type 1 |
| BP | = | blood pressure |
| CTRP | = | complement C1q tumor necrosis factor-related protein |
| DCT | = | distal convoluted tubules |
| EKODE | = | 12,13-epoxy-9-oxo-10(trans)-octadecanoic acid |
| ENaC | = | epithelial Na+ channel |
| GAP | = | GTPase-activating protein |
| GDI | = | GDP-dissociation inhibitor |
| GEF | = | GDP/GTP exchange factor |
| GR | = | glucocorticoid receptor |
| HDAC8 | = | histone deacetylase-8 |
| ICV | = | intracerebroventricular |
| MR | = | mineralocorticoid receptor |
| Na+ | = | sodium |
| NADPH | = | reduced nicotinamide-adenine dinucleotide phosphate |
| NCC | = | Na+ -Cl− cotransporter |
| nGRE | = | negative glucocorticoid response element |
| PAC | = | plasma aldosterone concentration |
| ROS | = | reactive oxygen species |
| Sgk-1 | = | serum and glucocorticoid-regulated kinase-1 |
| SHR | = | spontaneously hypertensive rats |
| Tiam1 | = | T cell lymphoma invasion and metastasis1 |
Key messages
The salt sensitivity of blood pressure is modulated solely by abnormal function of Na+ excretion in the kidney.
The aberrant Rac1-mineralocorticoid receptor pathway and the beta2 adrenoceptor-WNK4 pathway in the kidney may cause increased Na+ reabsorption in the distal nephron, leading to the development of salt-sensitive hypertension.
Introduction
Essential hypertension is caused by both genetic and environmental etiologies. Dietary intake of excess salt is a major environmental factor leading to the development of hypertension (Citation1). However, it is well known that the degree of blood pressure (BP) sensitivity to salt intake differs among patients with essential hypertension (Citation2). The causative factors of salt-sensitive essential hypertension vary and may interact with each other (Citation3,Citation4). Moreover, aging (Citation2), changes in body weight (Citation5), and other attributes alter the salt sensitivity of BP. In contrast, secondary salt-sensitive hypertension is caused by a specific, identifiable etiology. Recent investigations have begun to clarify etiologic factors in the development of primary salt-sensitive hypertension. We propose two novel mechanisms by which excess salt may cause the elevation of BP in salt-sensitive hypertension.
Role of kidney in salt-sensitive hypertension
In spite of the complex pathophysiology of salt-sensitive hypertension, the increase in salt-sensitivity is solely attributable to abnormal function of sodium (Na+) excretion in the kidney (Citation6). Described simply, BP is a product of cardiac output and peripheral vascular resistance. In salt-sensitive hypertension, excess salt induces Na+ retention, resulting in a rise in BP due to an increase in cardiac output. These acute hemodynamic changes are chronically altered, possibly due to whole body autoregulation, resulting in increased vascular resistance (Citation6). However, initial retention of Na+ is critical for the salt-induced increase in BP. According to Guyton's model, the relationship between arterial pressure and renal Na+ excretion is shifted to the right in patients with salt-sensitive hypertension, and its slope is less steep than that of normotensive subjects (Citation7). This implies that during high salt intake, salt-sensitive hypertensive subjects require higher BP to excrete Na+ sufficiently under conditions of abnormal kidney function.
In Dahl salt-sensitive and -resistant rats, when inter-strain renal transplants were performed, the change in BP in response to salt loading was largely determined by the genotype of the donor kidney (Citation8); Dahl salt-resistant rats transplanted with the kidney of salt-sensitive rats showed salt-induced increases in BP, whereas salt-sensitive rats transplanted with the kidney of salt-resistant rats did not. Interestingly, compared to that of wild-type mice, the increase in BP induced by angiotensin II (Ang II) infusion was less in mice lacking Ang II type 1 (AT1) receptor in the kidney, but was almost the same as that in mice lacking cardiac and vascular AT1 receptor (Citation9). Because Ang II stimulates Na+ reabsorption in proximal tubules (Citation10,Citation11), these results suggest that salt retention may play an important role in Ang II-induced hypertension. It was also reported that the Ang II-induced increase in BP was attenuated in mice lacking the AT1 receptor in renal proximal tubules (Citation12). Thus, abnormal control of renal Na + reabsorption may play an essential role in the hypertensive effects of Ang II. An abnormal BP set point for kidney Na+ excretion can be observed in the development of hypertension by treatment with vasoconstrictor substances such as Ang II, aldosterone, and norepinephrine, components known to participate in renal sympathetic drive. Based on these observations, we have recently suggested novel mechanisms for salt-induced BP rise (Citation13–15).
Aldosterone overproduction in salt-sensitive hypertension with obesity
Aldosterone causes the retention of Na+ by stimulation of epithelial Na+ channel (ENaC) absorption in the distal nephron and possibly in the proximal tubules (Citation16–18). Primary aldosteronism, in which the plasma aldosterone concentration (PAC) is increased due to adrenal aldosterone overproduction, is a well-known cause of secondary salt-sensitive hypertension. In addition, overproduction of aldosterone is also observed in some types of hypertension in the absence of primary aldosteronism.
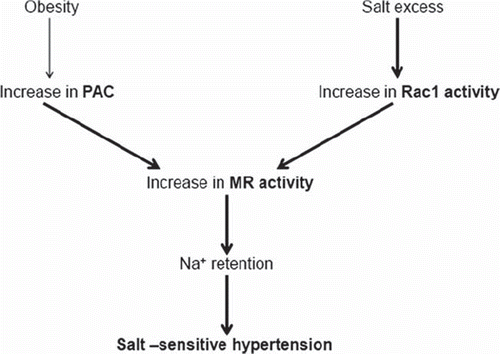
It was reported that PAC (Citation19) or urinary aldosterone levels (Citation20) are increased in obese subjects, and that body weight loss causes a decrease in PAC (Citation19). The presence of aldosterone-releasing factors that elevate PAC in adipose tissue (Citation21) was proposed, because plasma from obese, spontaneously hypertensive rats (SHR), termed SHR/NDmcr-cps, stimulated aldosterone production in cultured adrenal cells (Citation22). Investigators posited the involvement of aldosterone-releasing factor candidates such as 12,13-epoxy-9-oxo-10(trans)-octadecanoic acid (EKODE) (Citation23) or complement C1q tumor necrosis factor-related protein (CTRP) (Citation24). In addition, under conditions of hyperreactivity of the renin-angiotensin system and hyperinsulinemia, increases in PAC in subjects with obesity or metabolic syndrome were observed (Citation21). In fact, Rocchini et al. demonstrated that obese hypertensive adolescents experienced decreased BP with salt restriction to a greater extent than those that were non-obese (Citation25), suggesting an increased salt-sensitivity of BP under conditions of obesity. Moreover, successful body weight loss negated the decrease in BP brought about by salt restriction, but unsuccessful body weight loss did not. In addition, SHR/NDmcr-cps were susceptible to salt-induced organ damage possibly through an inadequate suppression of PAC, which could have been caused by a failure in the negative regulation of PAC by salt-loading (Citation26,Citation27). Since the mineralocorticoid receptor (MR) blocker normalized salt-induced cardiac and renal abnormalities in SHR/NDmcr-cps, both salt excess and hyperaldosteronism are thought to activate MR, leading to organ damage. Thus, aldosterone overproduction may contribute to the important public health concern of obesity by causing increased salt sensitivity of BP as well as organ damage ().
Figure 1. Mineralocorticoid receptor (MR) activation in salt-sensitive hypertension. Plasma aldosterone concentration (PAC) is increased in obese hypertensive patients even in the absence of primary aldosteronism. MR is also stimulated in normal- or low-aldosterone type of salt-sensitive hypertension, potentially due to Rac1 activation. As a result, sodium (Na+) is retained and hypertension develops. See text for a description of detailed mechanisms of aldosterone overproduction in obese hypertension and of Rac1 activation in normal- or low-aldosterone types of hypertension.
Rac1 and MR activation in low-aldosterone-type salt-sensitive hypertension
In Dahl salt-sensitive hypertensive rats, PAC is decreased under conditions of salt loading, yet salt excess elevated BP associated with renal MR activation (Citation13,Citation14). Thus, some other factor, other than increased PAC, may enhance aldosterone-induced MR activation in Dahl salt-sensitive rats. Previous studies revealed cross-talk between steroid receptors and intracellular signaling factors. For example, Rho family members and their regulatory proteins were involved in the transactivation of several steroid receptors (Citation28,Citation29). Rac1, which is a member of the Rho family GTPases, regulates diverse biological processes including cell cycle, cell migration, cell adhesion, and activation of a reduced nicotinamide-adenine dinucleotide phosphate (NADPH) oxidase (Citation30) and was recently suggested to serve as a key regulator of nuclear transcription factor (Citation31). Shibata et al. recently postulated that the interaction between MR and Rac1 represents an alternative pathway to potentiate MR activity (Citation14). Arhgdia−/− mice, in which Rac1 is activated specifically in the kidney, showed renal injury and MR activation, which was ameliorated by not only an MR blocker but also by a Rac1 inhibitor (but not by Rho-kinase inhibitor) (Citation14). Transient transfection assays in HEK293 cells showed that constitutively active Rac1 enhanced MR activity associated with nuclear translocation of MR. Thus, it was suggested that the signaling interaction between Rac1 and MR is a novel pathway that leads to salt-induced kidney injury. MR is located mainly in the distal nephron; therefore, MR activation can induce Na+reabsorption, leading to Na+retention and resultant hypertension. Accordingly, salt loading increased BP in Arhgdia−/− mice (Citation13). Interestingly, high salt intake increased Rac1 levels (but not Rho levels) in the kidneys of Dahl salt-sensitive rats, but decreased Rac1 levels in those of Dahl salt-resistant rats (Citation13). Treatment with renal serum and glucocorticoid-regulated kinase-1 (Sgk-1), a downstream signaling molecule of MR, revealed similar changes in Rac1 under conditions of salt loading. Despite a reduction in PAC, salt-loaded Dahl salt-sensitive rats showed increased MR signaling in the kidney. Moreover, in Dahl salt-sensitive rats, treatment with Rac inhibitor as well as MR blocker inhibited salt-induced BP increase and reduced renal injury. Thus, Rac1 activation by salt excess may stimulate MR activity in the kidney of salt-sensitive hypertension, resulting in increased BP, despite lowering PAC ().
Mechanism of Rac1 activation in salt-sensitive hypertension
Rac1 activity is regulated by three groups of molecules: GDP/GTP exchange factor (GEF), GTPase-activating protein (GAP), and GDP-dissociation inhibitor (GDI) (Citation30). Interestingly, the gene encoding human GDI alpha (ARHGDIA) is located in quantitative trait loci that influence BP, as shown by several genome-wide linkage analyses of human hypertension (Citation32,Citation33). Because GDI alpha binds to the GDP-bound, inactive form of Rac1 and inhibits both basal and GEF-stimulated dissociation of GDP from the inactive Rac1 (Citation30), deletion of GDI alpha causes an exaggerated BP response to salt through the up-regulation of Rac1. In fact, Arhgdia−/− mice which lack GDI alpha and activated Rac1 show increased salt sensitivity of BP, as mentioned above (Citation14). Thus, increased Rac1 activity resulting from GEF-stimulated GDP dissociation may contribute to salt-induced increases in BP in salt-sensitive hypertension. Notably, Rac-specific, GEF T cell lymphoma invasion and metastasis 1 (Tiam1) was increased during salt-loading in Dahl salt-sensitive rats but not in Dahl salt-resistant rats (Citation13). Thus, we postulate that Rac1 may play a causative role in increasing the salt sensitivity of BP and kidney damage via MR activation, although further experiments are required to elucidate the mechanism of Rac1 activation by salt excess. We further demonstrated that Rac1 and aldosterone interdependently caused MR over-activity in salt-sensitive hypertension (Citation13). Aldosterone infusion caused hypertension and renal Rac1 activation in salt-loaded rats but not in non-salt-loaded rats (Citation13). Moreover, Rac1 and Tiam1 activities were decreased by adrenalectomy in salt-loaded Dahl salt-sensitive rats, and salt-induced Rac1 activation was recovered with aldosterone infusion in adrenalectomized Dahl salt-sensitive rats. Therefore, salt-sensitive hypertension may develop due to a pathological response of Rac1 to salt excess and aldosterone.
Salt sensitivity in young salt-sensitive hypertension
In the experiments mentioned above, young (4-week-old) Dahl salt-sensitive rats were selected for use, because earlier reports showed that salt-dependent forms of experimental hypertension were more severe when induced in immature animals (Citation34). We confirmed that in Dahl salt-sensitive rats, salt loading of young rats caused severe renal injury associated with a greater BP increase than salt loading of adult-aged rats, which did not cause renal damage (Citation35). Also, salt loading increased BP and induced renal injury in young heminephrectomized Sprague-Dawley rats, but not in adult Sprague-Dawley rats (Citation36). Because PAC is higher in younger-aged rats (Citation37), cross-talk between aldosterone and Rac1 is thought to facilitate salt-induced BP increase and the progression of renal injury in salt-sensitive hypertension. In fact, PAC was significantly higher in salt-loaded young heminephrectomized rats than that in adults, despite suppression of PAC by salt loading (Citation36). Also, treatment with aldosterone synthase inhibitor and adrenalectomy ameliorated salt-induced hypertension and renal injury in young Dahl salt-sensitive and young heminephrectomized rats (Citation35,Citation36). These data are compatible with data from the Framingham Offspring Study which showed that normotensive subjects with higher PAC, despite having relatively greater salt intake, were prone to the onset of hypertension after 4 years even when PAC was within normal limits (Citation38). Thus, inappropriate suppression of PAC by salt loading may lead to hypertension, possibly through cross-talk between Rac1 and MR. Progression of renal damage caused by high salt intake during young ages may critically impact health outcomes at mature ages, because it enhances susceptibility to salt-induced increases in BP, which occur probably as a result of an abnormality in renal Na+-excreting function. This hypothesis may explain previous human data that showed salt restriction during the first 6 months of life decreased BP in subjects 15 years of age (Citation39). It is noteworthy that the International Study of Salt and Blood Pressure (INTERSALT) (Citation40) showed that age-related increases in BP are observed in developed societies that are characterized by high salt intake but are not seen in populations that are known to have a very low salt intake.
Central sympathetic drive in salt-sensitive hypertension
In an early study, Fujita T. and Bartter et al. demonstrated that suppression of plasma norepinephrine under conditions of salt loading is an inadequate treatment for salt-sensitive hypertensive subjects as compared to non-salt-sensitive subjects (Citation1). Also, several human and animal studies showed the role of the sympathetic nervous system in salt-induced hypertension. Renal sympathoactivation is thought to be important for the development of hypertension because renal denervation prevented a salt-induced increase in BP in salt-sensitive hypertensive models (Citation41). Moreover, recent human studies indicated that catheter-based renal denervation induced a substantial and sustained decrease in BP in patients with resistant hypertension (Citation42).
Data collected from a study of human subjects showed that salt loading caused skeletal muscle (forearm) vasodilation, which was associated with an increase in cardiac output and visceral (renal and hepatic) vasoconstriction in salt-sensitive hypertensive patients (Citation43). Thus, we proposed a role for kidney-specific sympathoactivation in salt-sensitive hypertension. The observed redistribution of blood flow (Citation44) resembles the hemodynamic pattern associated with a ‘fight-or-flight’ response triggered by mental stress (Citation44). Thus, central sympathoexcitation, which induces stimulation of sympathetic nerve activity on a selective basis, may be a key factor in salt-sensitive hypertension. In fact, salt-induced renal sympathoactivation mediated by the dysfunction of the central nervous system has been suggested by studies that showed an impaired central property in the baroreceptor reflex of renal sympathetic nerve activity in salt-sensitive young SHR, when electrical stimulation of the aortic depressor nerve was employed (Citation45,Citation46).
Sympathoactivation by brain oxidative stress in salt-sensitive hypertension
Several investigators, including us, reported that reactive oxygen species (ROS) production is up-regulated in several organs of salt-sensitive hypertension animal models (Citation47–49). In addition, a stimulating effect of ROS on the sympathetic nervous system was suggested in several studies (Citation50,Citation51). Thus, we hypothesized that in salt-sensitive hypertension, increased brain ROS may stimulate sympathetic nerve activity, resulting in an increase in BP.
Salt loading increased NADPH-induced ROS overproduction in the hypothalamus of Dahl salt-sensitive rats (Citation52) and in young heminephrectomized rats (Citation53). To clarify whether brain ROS overproduction increases BP via sympathoactivation, Fujita M. et al. examined changes in BP and renal sympathetic nerve activity in response to tempol infusion into the lateral cerebral ventricle. Under conditions of salt loading, intracerebroventricular (ICV) antioxidants decreased BP and renal sympathetic nerve activity to a greater extent in Dahl salt-sensitive rats that in Dahl salt-resistant rats (Citation52). Salt-loaded young heminephrectomized rats showed similar changes in response to ICV antioxidants (Citation53). Moreover, chronic ICV antioxidant treatment of salt-loaded heminephrectomized rats prevented a BP increase and the resultant progression of renal injury (Citation53). Thus, it was suggested that ROS overproduction in the brain causes an increase in BP through renal sympathetic over-activity in salt-sensitive hypertension (Citation5).
Renal sympathoactivation and WNK4 suppression in salt-sensitive hypertension
It is well known that the WNK family of serine-threonine kinases plays a key role in renal Na+ reabsorption (Citation54). WNK4 decreases distal Na + reabsorption through the inhibition of the Na+-Cl− cotransporter (NCC) (Citation55). Loss-of-function mutations in WNK4 caused pseudohypoaldosteronism II, a secondary salt-sensitive hypertension, through activation of NCC in distal convoluted tubules (DCT) (Citation56,Citation57). In addition, expression of a transgene encoding the dominant-negative mutant WNK4 induced hypertension, which was associated with increased NCC expression in the DCT of mice, whereas expression of a transgene for wild-type WNK4 showed the opposite effect (Citation58). In normal rats, the renal expression of WNK4 was decreased by low salt intake but was increased by high salt intake (Citation59). Thus, changes in NCC activity via WNK4 activity are thought to contribute to renal adaptive modulation in response to changes in dietary salt intake, a process crucial for maintaining Na+balance and BP homeostasis.
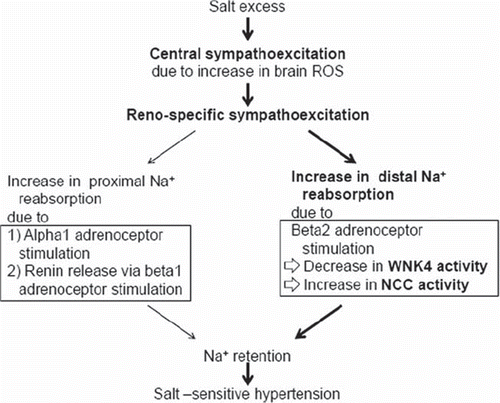
It is well established that renal sympathoactivation enhances proximal Na+reabsorption through Ang II overproduction that is mediated by increased renin release following beta1 adrenoceptor stimulation and by direct alpha1 adrenoceptor activation. We recently suggested that NCC and ENaC are involved in sympathetic control of Na+reabsorption. In rats with sensory nerve degeneration, high salt intake increased the NCC activity associated with renal sympathoactivation (Citation60). Renal beta2 adrenoceptor activation and cAMP production promotes Na+reabsorption through ENaC (Citation61). Because ENaC is downstream of WNK4 signaling, NCC regulation of WNK4 may also be mediated through beta2 adrenoceptor activity. Mu et al. recently suggested a mechanism in which renal beta2 adrenoceptor stimulation accelerates tubular Na+ reabsorption caused by NCC activation in DCT. In mice, norepinephrine infusion caused a salt-induced increase in BP that was associated with NCC activation mediated by WNK4 down-regulation (Citation15). Because the salt-induced BP rise was abolished by the beta-blocker propranolol, we examined the effect of the beta agonist isoproterenol on salt sensitivity of BP in beta1 and beta2 adrenoceptor-deficient mice. An isoproterenol-induced increase in salt sensitivity was observed in beta1 adrenoceptor-deficient mice but not in beta2 adrenoceptor-deficient mice. Moreover, in the same mouse model isoproterenol treatment did not induce a salt-induced increase in BP upon treatment with the NCC blocker hydrochlorothiazide. Because WNK4 is a negative regulator of NCC activity, we reason that WNK4 suppression by beta2 adrenoceptor activation induces increased Na+reabsorption through NCC activation in DCT.
Distal nephron-specific glucocorticoid receptor (GR)-deficient mice showed no beta agonist-induced WNK4 inhibition or BP increase (Citation15). Accordingly, we found a negative glucocorticoid response element (nGRE) in the promoter region of WNK4. Interestingly, our data (Citation15) suggest that beta2 adrenoceptor stimulation results in cyclic AMP-dependent inhibition of histone deacetylase-8 (HDAC8) activity and increased histone acetylation; in turn, histone acetylation leads to binding of the GR to nGRE in the promoter region of WNK4, resulting in the inhibition of WNK4 transcription. These findings implicate the epigenetic modulation of WNK4 transcription in the development of salt-sensitive hypertension through NCC activation. Thus, renal beta2 adrenoceptor stimulation may cause a salt-induced increase in BP through WNK4 suppression due to epigenetic modulation of WNK4 ().
Figure 2. Renospecific sympathoexcitation and WNK4 suppression in salt-sensitive hypertension. Salt excess causes central sympathoexcitation, possibly as a result of brain reactive oxygen species (ROS) overproduction. The resultant renospecific sympathostimulation increases sodium (Na+) reabsorption and blood pressure. It is well known that renal sympathoactivation enhances proximal Na + reabsorption through Ang II overproduction via increased renin release by beta1 adrenoceptor stimulation and through direct alpha1 adrenoceptor activation (left in the figure). We recently suggested an additional pathway of sympathoactivation-induced increase in Na + reabsorption (right in the figure). Beta2 adrenoceptor stimulation in distal convoluted tubules decreased WNK4 activity, resulting in activation of the Na+ -Cl− cotransporter (NCC). The latter mechanism may be important in some types of salt-sensitive hypertension as mentioned in the text.
Conclusion
Recently two novel mechanisms of salt-sensitive hypertension were identified: one involves the Rac1-MR-ENaC activation pathway, and the other involves the renal sympathetic (beta2 adrenoceptor) activation-WNK4 pathway. The important role of MR and the renal sympathetic nervous system as regulators of Na + reabsorption and contributors to the development of salt-induced hypertension has been established previously. However, data presented here clarify the precise mechanisms of abnormal Na + reabsorption in the distal nephron and identify two potential therapeutic strategies. The Rac1 inhibitor and HDAC8 agonist may be useful for the treatment of salt-sensitive hypertension.
Declaration of interest: The authors report no conflicts of interest.
References
- Fujita T, Henry WL, Bartter FC, Lake CR, Delea CS. Factors influencing blood pressure in salt-sensitive patients with hypertension. Am J Med. 1980;69:334–44.
- Luft FC, Weinberger MH. Heterogeneous responses to changes in dietary salt intake: the salt-sensitivity paradigm. Am J Clin Nutr. 1997;65(Suppl):612S–7S.
- Fujita T, Noda H, Ando K. Sodium susceptibility and potassium effects in young patients with borderline hypertension. Circulation. 1984;69:468–76.
- Fujita T, Ando K. Hemodynamic and endocrine changes associated with potassium supplementation in sodium-loaded hypertensives. Hypertension. 1984;6:184–92.
- Ando K, Fujita M. Reactive oxygen species and the central nervous system in salt-sensitive hypertension: possible relationship to obesity-induced hypertension. Clin Exp Pharmacol Physiol. 2012;39:111–6.
- Guyton AC. Kidneys and fluids in pressure regulation. Small volume but large pressure changes. Hypertension. 1992;19 (Suppl 1):l2–8.
- Hall JE, Brands MW, Henegar JR. Angiotensin II and long-term arterial pressure regulation: the overriding dominance of the kidney. J Am Soc Nephrol. 1999;10 (Suppl 12):S258–65.
- Dahl LK, Heine M, Thompson K. Genetic influence of the kidneys on blood pressure. Evidence from chronic renal homografts in rats with opposite predispositions to hypertension. Circ Res. 1974;40:94–101.
- Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, . Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103:17985–90.
- Li Y, Yamada H, Kita Y, Kunimi M, Horita S, Suzuki M, . Roles of ERK and cPLA2 in the angiotensin II-mediated biphasic regulation of Na+-HCO3− transport. J Am Soc Nephrol. 2008;19:252–9.
- Horita S, Zheng Y, Hara C, Yamada H, Kunimi M, Taniguchi S, . Biphasic regulation of Na+ -HCO3− cotransporter by angiotensin II type 1A receptor. Hypertension. 2002;40:707–12.
- Gurley SB, Riquier-Brison AD, Schnermann J, Sparks MA, Allen AM, Haase VH, . AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab. 2011;13:469–75.
- Shibata S, Mu SY, Kawarazaki H, Muraoka K, Ishizawa K, Yoshida S, . Rac1 GTPase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor-dependent pathway. J Clin Invest. 2011;121:3233–43.
- Shibata S, Nagase M, Yoshida S, Kawarazaki W, Kurihara H, Tanaka H, . Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med. 2008;14:1370–6.
- Mu SY, Shimosawa T, Ogura S, Wang H, Uetake U, Kawakami-Mori F, . Epigenetic modulation of the renal β-adrenergic–WNK4 pathway in salt-sensitive hypertension. Nat Med. 2011;17:573–80.
- Xu G, Liu A, Liu X. Aldosterone induces collagen synthesis via activation of extracellular signal-regulated kinase 1 and 2 in renal proximal tubules. Nephrology. 2008;13:694–701.
- Pinto V, Pinho MJ, Hopfer U, Jose PA, Soares-da-Silva P. Oxidative stress and the genomic regulation of aldosteronestimulated NHE1 activity in SHR renal proximal tubular cells. Mol Cell Biochem. 2008;310:191–201.
- Drumm K, Kress TR, Gassner B, Krug AW, Gekle M. Aldosterone stimulates activity and surface expression of NHE3 in human primary renal proximal tubule epithelial cells (RPTEC). Cell Physiol Biochem. 2006;17:21–8.
- Engeli S, Böhnke J, Gorzelniak K, Janke J, Schling P, Bader M, . Weight loss and the renin-angiotensin-aldosterone system. Hypertension. 2005;45:356–62.
- Bentley-Lewis R, Adler GK, Perlstein T, Seely EW, Hopkins PN, Williams GH, . Body mass index predicts aldosterone production in normotensive adults on a high-salt diet. J Clin Endocrinol Metab. 2007;92:4472–5.
- Briet M, Schiffrin EL. The role of aldosterone in the metabolic syndrome. Curr Hypertens Rep. 2011;13:163–72.
- Nagase M, Yoshida S, Shibata S, Nagase T, Gotoda T, Ando K, . Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol. 2006;17:3438–46.
- Goodfriend TL, Ball DL, Egan BM, Campbell WB, Nithipatikom K. Epoxy-keto derivative of linoleic acid stimulates aldosterone secretion. Hypertension. 2004;43:358–63.
- Wong GW, Wang J, Hug C, Tsao TS, Lodish HF. A family of Acrp30/adiponectin structural and functional paralogs. Proc Natl Acad Sci U S A. 2004;101:10302–7.
- Rocchini AP, Key J, Bondie D, Chico R, Moorehead C, Katch V, . The effect of weight loss on the sensitivity of blood pressure to sodium in obese adolescents. N Engl J Med. 1989;321:580–5.
- Nagase M, Matsui H, Shibata S, Gotoda T, Fujita T. Salt-induced nephropathy in obese spontaneously hypertensive rats via paradoxical activation of the mineralocorticoid receptor: role of oxidative stress. Hypertension. 2007;50: 877–83.
- Matsui H, Ando K, Kawarazaki H, Nagae A, Fujita M, Shimosawa T, . Salt excess causes left ventricular diastolic dysfunction in rats with metabolic disorder. Hypertension. 2008;52:287–94.
- Rubino D, Driggers P, Arbit D, Kemp L, Miller B, Coso O, . Characterization of Brx, a novel Dbl family member that modulates estrogen receptor action. Oncogene. 1998; 16:2513–26.
- Su LF, Knoblauch R, Garabedian MJ. Rho GTPases as modulators of the estrogen receptor transcriptional response. J Biol Chem. 2001;276:3231–7.
- Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81:153–208.
- Wu X, Tu X, Joeng KS, Hilton MJ, Williams DA, Long F. Rac1 activation controls nuclear localization of beta-catenin during canonical Wnt signaling. Cell. 2008;133:340–53.
- Franceschini N, MacCluer JW, Göring HH, Cole SA, Rose KM, Almasy L, . A quantitative trait loci-specific gene-by-sex interaction on systolic blood pressure among American Indians: the Strong Heart Family Study. Hypertension. 2006;48:266–70.
- Wilk JB, Djousse L, Arnett DK, Hunt SC, Province MA, Heiss G, . Genome-wide linkage analyses for age at diagnosis of hypertension and early-onset hypertension in the HyperGEN study. Am J Hypertens. 2004;17:839–44.
- Zicha J, Kunes J, Jelínek J. Experimental hypertension in young and adult animals. Hypertension. 1986;8:1096–104.
- Kawarazaki H, Ando K, Nagae A, Fujita M, Matsui H, Fujita T. Mineralocorticoid receptor activation contributes to salt-induced hypertension and renal injury in prepubertal Dahl salt-sensitive rats. Nephrol Dial Transplant. 2010;25:2879–89.
- Kawarazaki H, Ando K, Fujita M, Matsui H, Nagae A, Muraoka K, . Mineralocorticoid receptor activation: a major contributor to salt-induced renal injury and hypertension in young rats. Am J Physiol Renal Physiol. 2011;300: F1402–9.
- Fiselier TJ, Lijnen P, Monnens L, van Munster P, Jansen M, Peer P. Levels of renin, angiotensin I and II, angiotensin-converting enzyme and aldosterone in infancy and childhood. Eur J Pediatr. 1983;141:3–7.
- Vasan RS, Evans JC, Larson MG, Wilson PW, Meigs JB, Rifai N, . Serum aldosterone and the incidence of hypertension in nonhypertensive persons. N Engl J Med. 2004;351:33–41.
- Geleijnse JM, Hofman A, Witteman JC, Hazebroek AA, Valkenburg HA, Grobbee DE. Long-term effects of neonatal sodium restriction on blood pressure. Hypertension. 1997; 29:913–7.
- Stamler J, Rose G, Elliott P, Dyer A, Marmot M, Kesteloot H, . Findings of the International Cooperative INTERSALT Study. Hypertension. 1991;17(Suppl):I9–15.
- Fujita T, Sato Y. Role of hypothalamic-renal noradrenergic systems in hypotensive action of potassium. Hypertension. 1992;20:466–72.
- Krum H, Schlaich M, Whitbourn R, Sobotka PA, Sadowski J, Bartus K, . Catheter-based renal sympathetic denervation for resistant hypertension: a multicentre safety and proof-of-principle cohort study. Lancet. 2009;373: 1275–81.
- Fujita T, Ando K, Ogata E. Systemic and regional hemodynamics in patients with salt-sensitive hypertension. Hypertension. 1990;16:235–44.
- Brod J, Fencl V, Hejl Z, Jirka J. Circulatory changes underlying blood pressure elevation during acute emotional stress (mental arithmetic) in normotensive and hypertensive subjects. Clin Sci. 1959;18:269–79.
- Ono A, Kuwaki T, Cao WH, Kumada M, Fujita T. High calcium diet prevents baroreflex impairment in salt-loaded spontaneously hypertensive rats. Hypertension. 1994;24:83–90.
- Ono A, Kuwaki T, Kumada M, Fujita T. Differential central modulation of the baroreflex by salt loading in normotensive and spontaneously hypertensive rats. Hypertension. 1997; 29:808–14.
- Matsui H, Shimosawa T, Uetake Y, Wang H, Ogura S, Kaneko T, . Protective effect of potassium against the hypertensive cardiac dysfunction. Hypertension. 2006;48:225–31.
- Kido M, Ando K, Oba S, Fujita T. Renoprotective effect of pravastatin in salt-loaded Dahl salt-sensitive rats. Hypertens Res. 2005;28:1009–15.
- Kido M, Ando K, Onozato ML, Tojo A, Yoshikawa M, Ogita T, . Protective effect of dietary potassium against the vascular injury in salt-sensitive hypertension. Hypertension. 2008;51:225–31.
- Xu H, Fink GD, Galligan JJ. Nitric oxide-independent effects of tempol on sympathetic nerve activity and blood pressure in DOCA-salt rats. Am J Physiol Heart Circ Physiol. 2002;283:H885–92.
- Shokoji T, Nishiyama A, Fujisawa Y, Hitomi H, Kiyomoto H, Takahashi N, . Renal sympathetic nerve responses to tempol in spontaneously hypertensive rats. Hypertension. 2003;41:266–73.
- Fujita M, Kuwaki T, Ando K, Fujita T. Sympatho-inhibitory action of endogenous adrenomedullin through inhibition of oxidative stress in the brain. Hypertension. 2005;45: 1165–72.
- Fujita M, Ando K, Kawarazaki H, Kawarasaki C, Muraoka K, Ohtsu H, . Sympathoexcitation by brain oxidative stress mediates arterial pressure elevation in salt-induced chronic kidney disease. Hypertension. 2012;59:105–12.
- Hoorn EJ, Nelson JH, McCormick JA, Ellison DH. The WNK kinase network regulating sodium, potassium, and blood pressure. J Am Soc Nephrol. 2011;22:605–14.
- Kahle KT, Wilson FH, Leng O, Lalioti MD, O'Connell AD, Dong K, . WNK4 regulates the balance between renal NaCl reabsorption and K+secretion. Nat Genet. 2003;35:372–6.
- Yang CL, Zhu X, Ellison DH. The thiazide-sensitive Na-Cl cotransporter is regulated by a WNK kinase signaling complex. J Clin Invest. 2007;117:3403–11.
- Ring AM, Cheng SX, Leng Q, Kahle KT, Rinehart J, Lalioti MD, . WNK4 regulates activity of the epithelial Na+ channel in vitro and in vivo. Proc Natl Acad Sci U S A. 2007;104:4020–4.
- Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, . Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet. 2006;38:1124–32.
- O'Reilly M, Marshall E, Macgillivray T, Mittal M, Xue W, Kenyon CJ, . Dietary electrolyte-driven responses in the renal WNK kinase pathway in vivo. J Am Soc Nephrol. 2006;17:2402–13.
- Li J, Wang D. Function and regulation of epithelial sodium transporters in the kidney of a salt-sensitive hypertensive rat model. J Hypertens. 2007;25:1065–72.
- Vasquez MM, Castro R, Seidner SR, Henson BM, Ashton DJ, Mustafa SB. Induction of serum- and glucocorticoid-induced kinase-1(SGK1) by cAMP regulates increases in α-ENaC. J Cell Physiol. 2008;217:632–42.