
Abstract
Ischemia/reperfusion injury (IRI) can lead to cellular and, eventually, organ dysfunction, with the liver being one of the most frequently affected organs. Melatonin, a molecule that has notable antioxidant and anti-inflammatory properties, has been shown to protect against hepatic IRI. The purpose of this review is to summarize the protective effects of melatonin on hepatic IRI. The review initially summarizes the antioxidant properties of melatonin. We then discuss the protective effects of melatonin against endothelial and mitochondrial dysfunction. Thereafter, we introduce some information covering melatonin-related signaling pathways, including heme oxygenase-1 (HO-1), toll-like receptor (TLR), c-Jun N-terminal kinase (JNK), and so on. Furthermore, the clinical application of melatonin to hepatic diseases is considered. Finally, the safety of melatonin is evaluated. Taken together, the information compiled in this review will serve as a comprehensive reference regarding the pharmacological benefits of melatonin on hepatic IRI, aid in the design of future experimental research, and promote melatonin as a new therapeutic target.
Key messages
Summarizing the protective effects of melatonin in hepatic IRI and discussing the protective effects of melatonin against endothelial and mitochondrial dysfunction.
Introducing the melatonin-related signaling pathways in IRI.
Discussing the clinical application of melatonin in hepatic diseases.
Introduction
Ischemia/reperfusion injury (IRI) is tissue damage induced by blood deprivation (ischemia) followed by reperfusion, during which a large number of various mediators are released that can lead to cellular and, eventually, organ dysfunction. The liver is one of the most frequently affected organs. Effective drug therapy is the primary treatment strategy and research direction for hepatic IRI.
Melatonin, a molecule with notable antioxidant properties, protects against IRI in various organs, including the heart, brain, and kidney (Citation1–3). In 1996, Sewerynek and colleagues were the first to demonstrate that melatonin protects the liver against IRI (Citation4). Since then, many reports have confirmed this finding. Hernández reported that melatonin reduces liver damage and the associated inflammation of hepatic tissue after ischemia reperfusion (IR) (Citation5). Sener and colleagues compared the effects of melatonin with those of N-acetylcysteine and concluded that the former agent was superior to the latter glutathione precursor in reducing hepatic IRI (Citation6). Kang and co-workers demonstrated that melatonin treatment reduced the IR-induced pro-inflammatory and pro-apoptotic gene expression in the rat liver (Citation7). Finally, Liang and colleagues reported that melatonin was hepatoprotective, most likely via mechanisms involving inhibition of the IκB kinase (IKK) and c-Jun N-terminal kinase (JNK) pathways and regulation of cell proliferation (Citation8). The collective findings strongly suggest that melatonin is an attractive candidate for reducing hepatic IRI in humans.
In the current review, we initially overview the antioxidant properties of melatonin. We then discuss the protective effects of melatonin against endothelial and mitochondrial dysfunction, with a focus on the protective mechanisms germane to hepatic IRI. We also consider melatonin-related signaling pathways, such as heme oxygenase-1 (HO-1), toll-like receptor (TLR), JNK, and so on. Furthermore, we discuss the clinical application of melatonin in hepatic diseases. Finally, the safety of melatonin is evaluated. Taken together, the information compiled in this review will serve as a comprehensive reference regarding the pharmacological effects of melatonin in hepatic IRI.
Melatonin and antioxidation
Melatonin is a powerful antioxidant produced by the pineal gland. Melatonin and its metabolites have potent antioxidant/anti-inflammatory properties and have been proved to be highly effective in a variety of disorders linked to inflammation and oxidative stress (Citation9). Melatonin not only neutralizes reactive nitrogen species (RNS) and reactive oxygen species (ROS) but also stimulates several antioxidant enzymes, such as SOD, GRed, and GPx (Citation10), thereby stabilizing cell membranes (Citation11). Ahmadiasl and colleagues (Citation12) found that melatonin caused a reduction in malondialdehyde (MDA) production, which reflects the degree of lipid peroxidation, indicating a reduction in lipid peroxidation and cellular damage. This protective effect of melatonin may be in part due to the scavenging of the very reactive ONOO- and OH (Citation13). A study by Bharti and colleagues (Citation14) elucidated the ameliorative effects of buffalo epiphyseal proteins and melatonin against fluoride-induced oxidative stress. Further, this study revealed that exogenous administration of melatonin and buffalo epiphyseal proteins may have therapeutic potential for reducing fluoride-induced oxidative stress-mediated pathogenesis and cardiac, hepatic, and renal damage.
Melatonin has a particularly effective ability to neutralize free radicals (Citation15) and prevent tissue damage associated with oxidative stress. Thus, it exhibits both direct scavenging actions on free radicals and related products (Citation16) as well as indirect antioxidative actions via its ability to stimulate the cellular antioxidant defense system by increasing mRNA levels and the activities of several important antioxidant enzymes to promote the synthesis of another important intracellular antioxidant, glutathione (Citation17); to reduce the activity of the pro-oxidative enzyme nitric oxide synthase (Citation18); and to diminish free radical formation at the mitochondrial level by reducing the leakage of electrons from the electron transport chain (Citation19). Additionally, different studies have demonstrated the protective role of melatonin against oxidative damage induced by drugs, toxins, and different diseases (Citation20–23). This combination of actions makes melatonin an important agent in combating ischemic injury.
In the present review, we highlight the antioxidation of melatonin in two essential pathological developments, endothelial dysfunction and mitochondrial dysfunction. The protection of melatonin against oxidative stress in the endothelium and mitochondria is the basis of its protective effects against liver IRI.
Melatonin and endothelial dysfunction
Endothelial dysfunction is an important pathological change in hepatic IRI, causing vascular stress and remodeling, as well as the expression of pro-inflammatory and pro-apoptotic genes. Research has demonstrated that endothelial dysfunction is related to nitric oxide (NO) and endothelin-1 (ET-1) (Citation24). Risbano and Gladwin reported that the dysregulation of vasodilator systems largely involves the NO pathway, with almost every step being subject to impairments (Citation25). The dysregulation includes a reduction in endothelial NO synthase (eNOS) function, the enzymatic ‘uncoupling’ of eNOS, the increased coupling of NO with superoxide and cell-free hemoglobin, the elaboration of the endogenous competitive inhibitors of eNOS (asymmetric dimethylarginine, ADMA), and the oxidation of target soluble guanylyl cyclase, the molecular target of NO.
The pathophysiology of hepatic IR is related to the level of NO. NO is produced by two NOS isoforms—eNOS and inducible NO synthase (iNOS). Endothelial NOS is expressed constitutively and exclusively in sinusoidal endothelial cells; these cells release small amounts of NO for short intervals. In contrast, iNOS synthesizes large amounts of NO for sustained periods and is transcriptionally up-regulated in all liver cells, including hepatocytes, sinusoidal endothelial cells, Kupffer cells, and stellate cells, and this up-regulation typically occurs in response to inflammatory mediators (Citation26). Endothelial NOS is considered to be essential for endothelial function and is a major protective factor against vascular endothelium pathophysiology, and the up-regulation of eNOS contributes to the protective mechanism of the endothelium by increasing blood flow (Citation27,Citation28). In contrast, iNOS increases free radical formation, thus promoting ischemic injury (Citation29). An increase in eNOS and iNOS expression has been reported in the liver of young animals during IRI (Citation30). However, in that study, the adult rats suffering from ischemia and reperfusion exhibited increased expression of iNOS but not of eNOS. It is therefore possible that, in young animals, elevated NO production due to the up-regulation of eNOS might attenuate the hepatic microcirculatory disturbance after IRI through the vasodilatory actions of the enzyme.
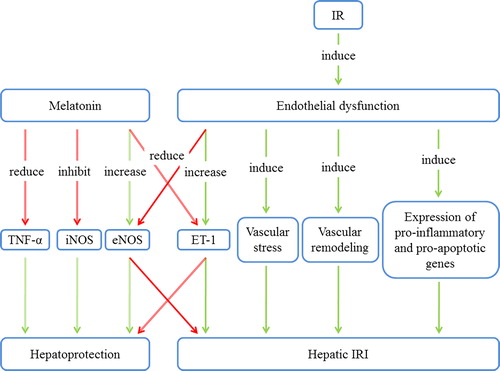
Numerous reports of an age-dependent decrease in endothelium-dependent relaxation, a process thought to be mediated by NO, have been published (Citation31). Furthermore, it has been suggested that the role of NO in the endothelium is reduced in older animals. This hypothesis is further supported by evidence of an age-associated decline in the levels of the NO precursors arginine, citrulline, and aspartic acid (Citation30). Conversely, the venular shear rates were significantly reduced in aged rats (before and after IR); lower shear rates are associated with a decreased endothelial production of NOS (Citation32). Rodriguez-Reynoso and colleagues reported that exogenous melatonin preserves functional and energetic status during IR, thereby reducing concentrations of tumor necrosis factor α (TNF-α) and inhibiting iNOS expression and NO production (Citation33). The mRNA levels of eNOS and iNOS were significantly increased after IR. Melatonin augmented the rise in eNOS mRNA levels, whereas it reduced the elevation of iNOS mRNA levels. Other studies also demonstrated that melatonin protected against ischemic injury through the inhibition of iNOS and the activation of eNOS (Citation34,Citation35). These results uniformly support the finding that melatonin pretreatment increases NO bioavailability and reduces endothelin expression, consequently playing a protective role in preserving both liver function and structure during hepatic IRI (Citation36). A schematic diagram of the relationship between melatonin and endothelial dysfunction is provided in .
Figure 1. The relationship between melatonin and endothelial dysfunction in hepatic IRI. Melatonin exerts hepatoprotective effects via inhibition of iNOS, stimulation of eNOS, and reduction of TNF-α and ET-1. Endothelial dysfunction leads to vascular stress and remodeling and to the expression of pro-inflammatory and pro-apoptotic genes, thus worsening hepatic IRI. eNOS = endothelial NO synthase; ET-1 = endothelin-1; iNOS = inducible NO synthase; IRI = ischemia/reperfusion injury; TNF-α = tumor necrosis factor α.
Melatonin and mitochondrial dysfunction
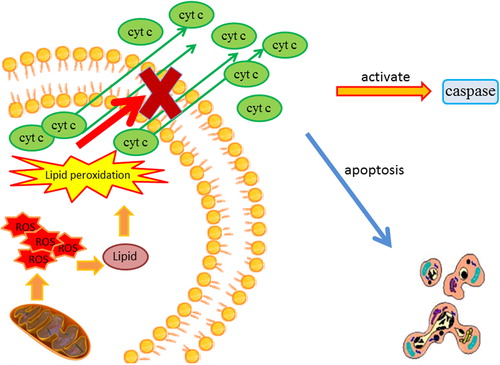
Recent studies have provided strong evidence for the involvement of mitochondria in the process of apoptotic cell death and cellular necrosis (Citation37). Mitochondria are also the primary source of intracellular ROS production, and ROS overproduction damages mitochondrial membranes and proteins and promotes lipid peroxidation. Lipid peroxidation increases mitochondrial membrane permeability, ultimately leading to a loss of mitochondrial integrity that results in the discharge of cytochrome c into the cytoplasm, subsequent activation of caspase activity, and initiation of apoptotic cell death (Citation38). The progress of this pathological change is summarized in .
Figure 2. The involvement of mitochondria in apoptotic cell death and cellular necrosis. The overproduction of ROS by the mitochondria causes lipid peroxidation. Lipid peroxidation increases the permeability of the mitochondrial membrane. The increased membrane permeability ultimately leads to a loss of mitochondrial integrity that results in the release of cytochrome c into the cytoplasm, the subsequent activation of caspase activity, and the initiation of apoptotic cell death. ROS = reactive oxygen species.
In particular, mitochondrial dysfunction is a key factor in IR-induced cell damage. Mitochondrial dysfunction worsened the effect of IRI in steatotic livers (Citation39). Frederiks and colleagues documented that increased serum levels of mitochondrial enzymes are indicative of cell necrosis in an ischemic liver (Citation40). Moreover, a large body of evidence suggests that a channel formed in mitochondrial membranes, which has been identified as the permeability transition pore, is involved in IR-associated cell damage and increases the permeability of the inner mitochondrial membrane to solutes (Citation38). Permeability transition pore opening is triggered by the association of calcium overload with an inducer, such as oxidative stress or high phosphate concentrations; both of these conditions are encountered during IR. The opening of this pore leads to loss of the mitochondrial membrane potential and mitochondrial swelling, which results in mitochondrial uncoupling and the inhibition of ATP synthesis (Citation41).
The mitochondria are believed to be a major target of pharmaceutical melatonin (Citation42). Interestingly, hepatic mitochondria contain concentrations of melatonin that exceed those in the serum (Citation43). Many investigations have provided significant evidence that melatonin protects against the IR-induced impairment of mitochondrial respiration, ATP synthesis, mitochondrial swelling, and lipid peroxidation (Citation41,Citation44). During IR, the availability of excess metabolic substrates in steatotic liver cells causes a sustained shift in the electrochemical potential of the mitochondria, resulting in the increased production of superoxide anions and H2O2 compared with that in lean livers (Citation45). The permeability transition pore is highly sensitive to mitochondrial redox states, and oxidative stress triggers permeability transition pore opening (Citation46). Long-term melatonin treatment protects against the age-related oxidative damage of lipids, protein, DNA, and mitochondrial respiratory function in the brains of senescence-accelerated mice (Citation41).
The ability of melatonin to preserve, at least in part, mitochondrial physiology under conditions of oxidative stress is consistent with the previously summarized mitochondrial actions (Citation19). Thus, the disruption of the mitochondrial respiratory cycle and ATP synthesis due to toxin exposure is restored by melatonin administration (Citation19,Citation46). Under these conditions, melatonin likely reduces electron leakage and free radical generation, thereby contributing to its ability to protect against molecular damage at the mitochondrial level, as summarized here and elsewhere (Citation47). However, it has been demonstrated that melatonin increases the expression of uncoupling protein (Citation48), and uncoupling protein is thought to prevent mitochondrial superoxide generation by increasing proton flow into the matrix, thus rendering electron flow through the respiratory complexes more efficient (Citation49). Therefore, melatonin may decrease free radical generation in mitochondria not only by reducing electron leakage from the mitochondrial permeability transition pore (MPTP) but also by promoting electron leakage via the uncoupling protein, which is the physiological pathway for decreasing free radical generation. The ATP depletion that occurs during ischemia leads to a loss of the mitochondrial transmembrane potential, which is associated with Ca2+ overload and causes the opening of the MPTP (Citation50). Mitochondrial glutamate dehydrogenase (GDH) activity, a marker of mitochondrial membrane integrity, is reduced in IR rats. Melatonin also stabilizes microsomal membranes, enabling them, in a concentration-dependent manner, to resist the rigidity induced by free radical attack (Citation51).
Once the MPTP is open, mitochondria undergo rapid swelling and rupture due to ion overload and a hyperosmotic effect. A study by Kim and Lee showed that the rate of mitochondrial swelling driven by the addition of succinate was significantly elevated in mitochondria isolated from livers 5 h after reperfusion. The mitochondria from the melatonin-pretreated rats showed reduced mitochondrial swelling. Additionally, along with the increased mitochondrial swelling, a rise was observed in the amount of mitochondrial cytochrome c released into the cytoplasm, which was suppressed by melatonin (Citation42).
Overall, we conclude that exogenous melatonin preserves mitochondrial function and the energy state of cells during hepatic IR. The effects of melatonin on experimental models subjected to hepatic IRI are summarized in .
Table I. The effects of melatonin on experimental models subjected to hepatic IRI.
Critical signaling pathways
Melatonin protects against hepatic IRI via numerous signaling pathways, including HO-1, TLR, and JNK. Here, we identify the pathways by which melatonin protects the liver during IRI; similarly, a diagram of the pathways regulated by melatonin is provided in .
Figure 3. The downstream pathways of melatonin in hepatic IRI. Melatonin increases HO-1 and inhibits JNK. HO-1 inhibits JAK2/STAT1, TRIF, IFN-β, CXCL-10, and MyD88. Bicyclol, NAC, neutrophils, and repressed MyD88 cause attenuation of TLR4. TLR4 repression leads to the suppression of NF-κB. These changes result in hepatoprotection. CXCL-10 = C-X-C motif ligand 10; HO-1 = heme oxygenase 1; IRI = ischemia/reperfusion injury; JAK2 = Janus kinase 2; JNK = c-Jun N-terminal kinase; MyD88 = myeloid differentiation factor 88; NAC = N-acetylcysteine; NF-κB = nuclear factor κB; STAT1 = signal transducer and activator of transcription 1; TLR-4 = toll-like receptor 4; TRIF = toll-receptor-associated activator of interferon.
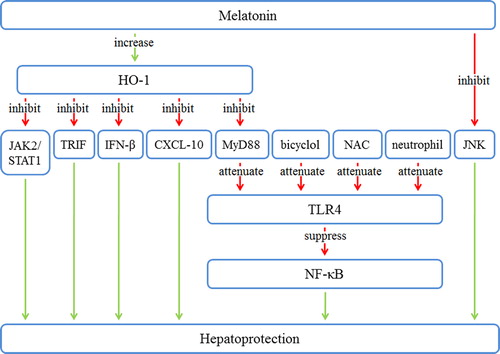
Heme oxygenase (HO) is the rate-limiting enzyme in heme degradation, which results in the formation of CO, biliverdin, and free iron (Citation52). HO-1 is the inducible form of the enzyme. It has been indicated that HO-1 induction plays a significant protective role against inflammatory processes and oxidative tissue injury (Citation53). HO-1 imparts a critical protective action that is activated during cellular stress and is thought to play a key role in hepatic IR via its antioxidative and anti-inflammatory functions (Citation54). The importance of the cross-talk between HO-1 and the TLR4 system is well known, with TLR4 being one of the putative HO-1 repressors in non-infectious hepatic IR (Citation55). Kang and Lee found that melatonin attenuated the IR-induced increase in serum alanine aminotransferase activity and that the HO-1 inhibitor zinc protoporphyrin (ZnPP) reversed this attenuation (Citation56). Melatonin augmented the levels of HO activity and of HO-1 protein and mRNA expression, and this enhancement was reversed by ZnPP. Melatonin enhanced NF-E2-related factor-2 (Nrf2) nuclear translocation, while ZnPP reversed this rise. Melatonin attenuated the IR-induced overexpression of TLR4 and its adaptor proteins, toll-receptor-associated activator of interferon (IFN) (TRIF) and myeloid differentiation factor 88 (MyD88); ZnPP reversed the effect of melatonin on TLR4 and TRIF expression. Melatonin suppressed the elevated interaction between TLR4/TRIF and TLR4/MyD88, which was prevented by ZnPP. Melatonin diminished the increased activation of Janus kinase 2 (JAK2), signal transducer and activator of transcription 1 (STAT1), and IFN-β, with ZnPP nullifying these inhibitory effects. Melatonin suppressed the levels of chemokine (C-X-C motif) ligand 10 (CXCL-10), and ZnPP reversed this inhibition. In addition, CoPP-induced HO-1 overexpression ameliorates hepatic IRI in type-1 IFN by down-regulating STAT1 phosphorylation downstream of TLR4, thus resulting in the reduction of CXCL-10 (Citation57). One study also demonstrated that HO-1 protein and mRNA expression increased 5 h after reperfusion, and melatonin augmented these increases (Citation7). The accumulated results suggest that melatonin ameliorates hepatic IRI by up-regulating HO-1 as an adaptive antioxidative and anti-inflammatory process.
TLRs are pattern-recognition receptors (PRRs) that recognize conserved pathogen-associated molecular patterns (PAMPs). Activation of the sentinel TLR system plays an important role in infectious and inflammatory disease states (Citation58). In particular, hepatic IRI is exacerbated by the activation of TLR4 by high-mobility group box 1 (HMGB1), a damage-associated molecular pattern (DAMP) protein that is released from dying cells (Citation59). MyD88-dependent signaling in the TLR4 activation pathway leads to direct nuclear factor κB (NF-κB) activation and to the induction of pro-inflammatory cytokines, whereas MyD88-independent signaling mediated by IFN regulatory factor 3 (IRF3) induces type I IFN (IFN-α/β) and IFN-inducible genes, such as CXCL-10 (Citation60). TLR3 regulates the amplification events of inflammation in experimental polymicrobial septic peritonitis and ischemic gut injury (Citation61). Recent studies have shown that TLR3 also plays critical roles in a variety of liver diseases, including fibrosis, viral hepatitis, and primary biliary cirrhosis (Citation62). Abundant evidence has demonstrated that IR, specific to the liver, promotes TLR4-dependent inflammation and organ injury (Citation59,Citation63). The study by Kang and colleagues indicated that TLR4 protein expression markedly increases 1 h after reperfusion and rises further after 5 h (Citation7). This pattern coincided with increased serum alanine aminotransferase (ALT) levels, indicating that TLR4 overexpression is significantly linked to liver damage and dysfunction. Melatonin markedly reduced the rise in TLR4 protein expression at 1 and 5 h after reperfusion, suggesting that a protective effect of melatonin in hepatic IR might be associated with its suppression of TLR4 overexpression. In addition, melatonin was reported to depress TLR3-mediated inflammatory factors and NF-κB activation in respiratory syncytial virus (RSV)-infected macrophages (Citation64). The cross-talk between melatonin and the TLR system in hepatic IR is pivotal.
JNKs are involved in the regulation of metabolism and inflammatory and immune responses (Citation65). Consequently, dysregulated JNK signaling has been suggested to be an important factor in the pathology of IRI (Citation66). Among several intracellular signaling pathways induced after IR, the JNK signaling pathway is thought to play a pivotal role in mediating IRI (Citation67). Studies have shown that melatonin abates hepatic IRI by inhibiting hepatic necrosis and apoptosis, improving the balance between NO and endothelin, and suppressing the JNK pathway (Citation8). Using TLR4 chimeric mice, Tsung and colleagues demonstrated the involvement of JNK activation in TLR4-mediated hepatic IRI (Citation63). It is well established that blood deprivation and reperfusion activates Kupffer cells (KCs), which subsequently generate ROS, which in turn activate JNK and enhance the production of pro-inflammatory cytokines, such as TNF-α (Citation68). KC-derived TNF-α binds to its receptor on the surface of hepatocytes and induces JNK and IKK signal transduction through a cascade of protein–protein interactions. While JNK promotes hepatocyte cell death, IKK activation further enhances the recruitment of leukocytes to the liver (Citation69). Selective pharmacological inhibitors of JNK and its substrate c-Jun decrease both pericentral hepatocyte necrosis and non-parenchymal cell death, and they improve survival in experimental models of hepatic warm IRI, resection, and transplantation; thus, JNK is a potential novel target for reducing hepatic IRI (Citation70). Here, melatonin elicited effects similar to those observed in the context of JNK and c-Jun inhibition, limiting hepatocyte necrosis and improving survival after warm IR and liver resection. Based on the summarized findings, melatonin exerts hepatoprotective effects in hepatic IRI via such mechanisms as JNK pathway inhibition.
From bench to bedside
While the current literature leaves little doubt that melatonin administration may mediate hepatoprotective actions, many questions remain as to how melatonin would be used for clinical treatment. Based on the available evidence, it appears possible that melatonin can exert important protective effects in hepatic diseases.
To examine the hypothesis that melatonin protects against IR-induced hepatic diseases, a pilot study by Schemmer and colleagues evaluated the use of melatonin in patients undergoing major liver resections (Citation71). This study opened the door to yet another important indication for melatonin in human liver surgery, namely as an adjunct to reduce IRI in liver transplantation. The researchers demonstrated that melatonin reduces cold ischemic injury in rat livers and suggested that melatonin may be useful in liver transplantation (Citation72). This idea was supported by Casillas-Ramírez and colleagues in their review of liver transplantation (Citation73). Thus, melatonin administration could be beneficial to patients not only by minimizing the damage to the transplant but also by serving as a protective agent for the attenuation of reperfusion injury.
In addition, melatonin is reported to exert protective effects in other hepatic diseases. Pashkov and colleagues observed that melatonin treatment might be useful for limiting drug-induced hepatitis; this finding was supported by the results of Popov and colleagues (Citation74,Citation75). Recently, it was found that 81.7% of patients with drug-induced hepatitis experienced improvements in their condition when using a combination treatment that included melatonin, compared with 66.5% of patients receiving standard therapy that included hepatoprotectors without melatonin (Citation76). These findings are consistent with the generally protective actions of melatonin against drug-mediated hepatic toxicity (Citation77). Moreover, Fourman and colleagues suggested that melatonin is involved in the pathogenesis of autoimmune hepatitis (Citation78). Cuesta and colleagues observed that the exogenous administration of melatonin reduces hepatic inflammation, which is in line with its well-known anti-inflammatory actions (Citation79).
Liver transplantation-associated IRI plays an important role in the induction of graft injury. Prolonged cold storage remains a risk factor for liver graft outcome, especially when steatosis is present. Steatotic livers exhibit exacerbated endoplasmic reticulum (ER) stress, which occurs in response to cold IRI. In addition, defective liver autophagy correlates with liver damage. Zaouali and colleagues evaluated the combined effect of melatonin and trimetazidine as additives to the Institute George Lopez (IGL)-1 solution on the modulation of ER stress and autophagy in steatotic liver grafts through the activation of adenosine monophosphate activated protein (AMP)-activated protein kinase (AMPK) (Citation80). Their data confirm the close relationship between AMPK activation and ER stress and autophagy after cold IRI. The addition of melatonin and temozolomide (TMZ) to the IGL-1 solution improved steatotic liver graft preservation through AMPK activation, which reduces ER stress and increases autophagy. Furthermore, it is important to remember that there is an ongoing clinical trial in Europe titled ‘Impact of melatonin in the pretreatment of organ donor and the influence in the evolution of liver transplant’ (http://ClinicalTrials.gov/show/NCT01860716). The research by the Aragon Institute of Health Sciences is believed to provide data supporting the use of melatonin in liver transplants.
Non-alcoholic fatty liver disease is the most common chronic liver disease, and non-alcoholic steatohepatitis (NASH) is its advanced form. Oxidative stress and hepatocyte apoptosis are considered to be involved in NASH pathogenesis and, particularly, in the progression of NASH to liver fibrosis and cirrhosis, which is initiated by inflammation that promotes disease progression. Cichoz-Lach and colleagues evaluated the effects of melatonin and L-tryptophan on selected biochemical blood parameters in patients with NASH (Citation81). The outcome suggests that melatonin and tryptophan significantly reduce plasma levels of pro-inflammatory cytokines and may be useful in the treatment of NASH. In addition, Gonciarz and colleagues demonstrated that treatment with melatonin for 3 months significantly improved plasma liver enzymes in NASH patients without any side effects (Citation82). Throughout the course of treatment, patient plasma melatonin levels persisted above baseline. These findings show that treatment with melatonin significantly improves plasma liver enzymes in NASH patients, but studies involving larger cohort trials and longer melatonin treatments are required before this indole can be accepted for use as a treatment for NASH. Additionally, the results of another study, which documented the beneficial effects of melatonin on liver enzymes in NASH patients, encourages further controlled trials of melatonin administered over a longer period of time using liver histology as an end-point (Citation83).
Publications have indicated that melatonin has a role in reducing liver cirrhosis and hepatic encephalopathy. Celinski and colleagues reported that patients with liver cirrhosis and portal hypertension exhibited significantly elevated fasting and postprandial plasma melatonin levels compared with healthy subjects and that the significant alteration of plasma melatonin in liver cirrhosis patients with portal hypertension compared with healthy controls is possibly due to portal systemic shunting and decreased liver degradation in cirrhotic patients (Citation84). Montagnese and colleagues indicated that plasma melatonin profile abnormalities were observed in patients with mild to moderately decompensated cirrhosis and that the abnormalities are substantially unrelated to the sleep disturbances prevalent in this population (Citation85).
Numerous studies indicate that unusual melatonin levels are associated with hepatic encephalopathy. It has been indicated that abnormal pituitary hormone and melatonin circadian patterns are present in cirrhosis in advance of the development of hepatic encephalopathy (Citation86). A recent study also showed that elevated blood melatonin levels during both the night and day may account for some of the clinical manifestations of hepatic encephalopathy (Citation87). These results suggest that melatonin may have some roles in hepatic encephalopathy and that disturbances in melatonin rhythm may be a new target for treating these diseases.
Abundant evidence indicates that melatonin is involved in preventing tumor initiation, promotion, and progression (Citation88). In animal model studies, melatonin exhibits preventative actions against nitrosodiethylamine (NDEA)-induced liver cancer (Citation89). In addition to its direct oncostatic action, melatonin protects hematopoietic precursors from the toxic effects of anticancer chemotherapeutic drugs. In hepatomas, melatonin inhibits linoleic acid uptake through its activation of MT1 and MT2 receptors, thereby preventing the formation of the mitogenic metabolite 1,3-hydroxyoctadecadienoic acid (Citation90). To assess the clinical efficacy of transcatheter arterial chemoembolization (TACE) and TACE+ melatonin on inoperable advanced primary hepatocellular carcinoma, Yan and colleagues studied 100 patients with inoperable advanced primary hepatocellular carcinoma (Citation91). The results were significantly better in the TACE+ melatonin group than in the TACE group. Melatonin protected liver function from the damage caused by TACE. The IL-2 levels of all patients significantly increased, whereas the expression of the soluble IL-2 receptor decreased after treatment with TACE+ melatonin compared with the TACE group. These results suggest that melatonin enhances the immunological activities of patients given melatonin, which exhibited protective actions against the damaging effects of TACE on liver function. Melatonin may also improve the effects of TACE by enhancing the survival and resection rates after the two-stage operation. In a preliminary study that investigated the influence of melatonin on angiogenesis, Lissoni and colleagues evaluated the effects of melatonin therapy on vascular endothelial growth factor (VEGF) blood levels in advanced cancer patients (Citation92). This study showed that the melatonin-induced control of neoplastic growth is associated with a decline in VEGF secretion, suggesting that the indoleamine may control tumor growth at least in part by acting as a natural anti-angiogenic molecule that inhibits angiogenesis-dependent cancer proliferation (Citation93). These results document that melatonin may be a valuable target in liver cancer treatment.
Future studies will demonstrate whether melatonin meets expectations not only in experimental cell/animal studies but also for patients. However, the available evidence suggests that melatonin will prove efficacious as a powerful hepatoprotective agent in humans. The clinical evidence in support of this hypothesis is summarized in .
Table II. Clinical evidence supporting the protective effects of melatonin in liver diseases.
Safety analysis
Several characteristics make melatonin a devoted defender to reduce IRI. One of the most important characteristics is that melatonin is endogenously produced and has a low toxicity profile (Citation94). Numerous studies have confirmed that melatonin is well tolerated and non-toxic (Citation95,Citation96). In a pharmacokinetic and safety study (Citation97), melatonin in propylene glycol was evaluated in adult male Sprague-Dawley rats. Following a single intravenous injection of 5 or 15 mg/kg, the melatonin plasma concentrations increased to 39 and 199 million pg/mL, respectively, at 2 min and 128,000 and 772,000 pg/mL, respectively, at 120 min. Within 60 min of injection, the blood pressure, heart rate and body temperature remained unaffected. A melatonin dose of 5 mg/kg did not influence the complete blood count results at 60 min, but 15 mg/kg melatonin slightly affected the differential white cell and platelet counts. A melatonin dose of 5 or 15 mg/kg caused a slight elevation in some liver enzymes 60 min after injection, and higher doses of melatonin also increased plasma creatinine and lactate dehydrogenase levels. A 5.5% reduction in body weight was observed 24 h after the completion of six daily injections of melatonin. Gross post-mortem and histological examinations of the brain, kidney, liver, and spleen did not reveal any evidence of toxicity. In conclusion, melatonin in propylene glycol markedly elevates the plasma levels of indoleamine with no serious toxicity. This preparation should be further evaluated in human patients.
One study indicated that melatonin might, under some circumstances, have inconsistent effects. Morera and colleagues (Citation98) reviewed the adverse effects of melatonin consumption in humans. From the 307 articles reviewed, 9 reported adverse effects. The melatonin doses associated with unusual side effects ranged from 1 to 36 mg. The reported reactions included one patient with autoimmune hepatitis, one case of confusion due to a melatonin overdose, one case of optic neuropathy, four cases with fragmented sleep, one case with a psychotic episode, one case of nystagmus, four cases of seizures, one case of headache, and two cases of skin eruptions. It was not always clear what other drugs were being taken by the patients who received melatonin. Additionally, in the 307 studies reviewed, the total number of patients was not specified, and the percentage of adverse effects reported in these studies seemed lower than that which would be expected in the placebo group.
Conclusion
Currently, melatonin is of significant interest to investigators studying hepatic IRI treatment, and its multifarious activities and involvement with signaling pathways provide many avenues for researchers to explore. Increasing lines of evidence strongly suggest that melatonin protects the liver from IRI. Melatonin appears to be a promising drug for reducing the damage and molecular changes associated with IR. However, the biological functions of melatonin remain only partially characterized. New frontiers to be explored in subsequent melatonin studies should include: 1) assessing melatonin in older animals and humans to identify a method of strategic intervention that will eventually increase the number and types of liver donors used, thus addressing the current organ shortage, and 2) investigating the effect of immune modulation by melatonin on hepatic IRI through inhibition of TLR signaling. Further investigations into the targets and functions of melatonin will aid in the development of new strategies for its use to protect against and recover from hepatic IRI and related diseases.
Acknowledgements
Yue Li, Yang Yang, and Yingtong Feng contributed equally to this work.
Declaration of interest: This study was supported by grants from the National Natural Science Foundation of China (81070951, 81222015, 81000938, 81170213, 81102091), the Excellent Doctoral Support Project of the Fourth Military Medical University (2013D01), and the New Century Talent Supporting Project by Chinese education ministry (NCET-12-1004), the Chinese Changjiang Scholars and Innovative Research Team in University (IRT1053). The authors report no conflicts of interest.
References
- Yang Y, Duan W, Jin Z, Yi W, Yan J, Zhang S, et al. JAK2/STAT3 activation by melatonin attenuates the mitochondrial oxidative damage induced by myocardial ischemia/reperfusion injury. J Pineal Res. 2013;55:275–86.
- Koh PO. Melatonin regulates the calcium-buffering proteins, parvalbumin and hippocalcin, in ischemic brain injury. J Pineal Res. 2012;53: 358–65.
- Sezgin G, Oztürk G, Güney S, Sinanoğlu O, Tunçdemir M. Protective effect of melatonin and 1,25-dihydroxyvitamin D3 on renal ischemia-reperfusion injury in rats. Ren Fail. 2013;35:374–9.
- Sewerynek E, Reiter RJ, Melchiorri D, Ortiz GG, Lewinski A. Oxidative damage in the liver induced by ischemia/reperfusion: protection by melatonin. Hepatogastroenterology. 1996;43:898–905.
- Hernández JA. [Effect of pretreatment with melatonin on the oxidative and inflammatory damage induced by hepatic ischemia/reperfusion in Zucker rats]. An R Acad Nac Med (Madr). 2011;128:391–415. [In Spanish]
- Sener G, Tosun O, Sehirli AO, Kacmaz A, Arbak S, Ersoy Y, et al. Melatonin and N-acetylcysteine have beneficial effects during hepatic ischemia and reperfusion. Life Sci. 2003;72:2707–18.
- Kang JW, Koh EJ, Lee SM. Melatonin protects liver against ischemia and reperfusion injury through inhibition of toll-like receptor signaling pathway. J Pineal Res. 2011;50:403–11.
- Liang R, Nickkholgh A, Hoffmann K, Kern M, Schneider H, Sobirey M, et al. Melatonin protects from hepatic reperfusion injury through inhibition of IKK and JNK pathways and modification of cell proliferation. J Pineal Res. 2009;46:8–14.
- Mayo JC, Sainz RM, Tan DX, Hardeland R, Leon J, Rodriguez C, et al. Anti-inflammatory actions of melatonin and its metabolites, N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK) and N1-acetyl-5-methoxykynuramine (AMK), in macrophages. J Neuroimmunol. 2005;165:139–49.
- Reiter RJ, Tan DX, Osuna C, Gitto E. Actions of melatonin in the reduction of oxidative stress: a review. J Biomed Sci. 2000;7:444–58.
- Rodriguez C, Mayo JC, Sainz RM, Antolin I, Herrera F, Martin V, et al. Regulation of antioxidant enzymes: a significant role for melatonin. J Pineal Res. 2004;36:1–9.
- Ahmadiasl N, Banaei S, Alihemmati A, Baradaran B, Azimian E. The anti-inflammatory effect of erythropoietin and melatonin on renal ischemia reperfusion injury in male rats. Adv Pharm Bull. 2014; 4:49–54.
- Reiter RJ, Oh CS, Fujimori O. Melatonin its intracellular and genomic actions. Trends Endocrinol Metab. 1996;7:22–7.
- Bharti VK, Srivastava RS, Kumar H, Bag S, Majumdar AC, Singh G, et al. Effects of melatonin and epiphyseal proteins on fluoride-induced adverse changes in antioxidant status of heart, liver, and kidney of rats. Adv Pharmacol Sci. 2014;2014:532969.
- Tan DX, Chen LD, Poeggeler B, Manchester LC, Reiter RJ. Melatonin: a potent, endogenous hydroxyl radical scavenger. J Endocrinol. 1993; 1:57–60.
- Rodriguez AB, Nogales G, Marchena JM, Ortega E, Barriga C. Suppression of both basal and antigen-induced lipid peroxidation in ring dove heterophils by melatonin. Biochem Pharmacol. 1999;58:1301–6.
- Winiarska K, Fraczyk T, Malinska D, Drozak J, Bryla J. Melatonin attenuates diabetes-induced oxidative stress in rabbits. J Pineal Res. 2006;40:168–76.
- Pozo D, Reiter RJ, Calvo JR, Guerrero JM. Physiological concentrations of melatonin inhibit nitric oxide synthase in rat cerebellum. Life Sci. 1994;55:PL455–60.
- León J, Acuña-Castroviejo D, Escames G, Tan DX, Reiter RJ. Melatonin mitigates mitochondrial malfunction. J Pineal Res. 2005;38:1–9.
- Espino J, Bejarano I, Paredes SD, González D, Barriga C, Reiter RJ, et al. Melatonin counteracts alterations in oxidative metabolism and cell viability induced by intracellular calcium overload in human leucocytes: changes with age. Basic Clin Pharmacol Toxicol. 2010; 107:590–7.
- Leon J, Acuña-Castroviejo D, Sainz RM, Mayo JC, Tan DX, Reiter RJ. Melatonin and mitochondrial function. Life Sci. 2004;75:765–90.
- Espino J, Bejarano I, Redondo PC, Rosado JA, Barriga C, Reiter RJ, et al. Melatonin reduces apoptosis induced by calcium signaling in human leukocytes: evidence for the involvement of mitochondria and bax activation. J Membr Biol. 2010;233:105–18.
- Espino J, Bejarano I, Paredes SD, Barriga C, Rodríguez AB, Pariente JA. Protective effect of melatonin against human leukocyte apoptosis induced by intracellular calcium overload: relation with its antioxidant actions. J Pineal Res. 2011;51:195–206.
- Javeshghani D, Barhoumi T, Idris-Khodja N, Paradis P, Schiffrin EL. Reduced macrophage-dependent inflammation improves endothelin-1-induced vascular injury. Hypertension. 2013;62:112–17.
- Risbano MG, Gladwin MT. Therapeutics targeting of dysregulated redox equilibrium and endothelial dysfunction. Handb Exp Pharmacol. 2013;218:315–49.
- Shah V, Kamath PS. Nitric oxide in liver transplantation: pathobiology and clinical implications. Liver Transpl. 2003;9:1–11.
- Albrecht EW, Stegeman CA, Heeringa P, Henning RH, van Goor H. Protective role of endothelial nitric oxide synthase. J Pathol. 2003;199: 8–17.
- Marletta MA. Nitric oxide synthase: aspects concerning structure and catalysis. Cell. 1994;78:927–30.
- Dalkara T, Endres M, Moskowitz MA. Mechanisms of NO neurotoxicity. Prog Brain Res. 1998;118:231–9.
- Strolin Benedetti M, Dostert P, Marrari P, Cini M. Effect of ageing on tissue levels of amino acids involved in the nitric oxide pathway in rat brain. J Neural Transm Gen Sect. 1993;94:21–30.
- Soltis EE. Effect of age on blood pressure and membrane dependent vascular responses in the rat. Circ Res. 1987;61:889–97.
- Ranjan V, Xiao Z, Diamond SL. Constitutive NOS expression in cultured endothelial cells is elevated by fluid shear stress. Am J Physiol. 1995;269:H550–5.
- Rodriguez-Reynoso S, Leal C, Portilla E, Olivares N, Muniz J. Effect of exogenous melatonin on hepatic energetic status during ischemia/reperfusion: possible role of tumor necrosis factor-α and nitric oxide. J Surg Res. 2001;100:141–9.
- Kilic U, Kilic E, Reiter RJ, Bassetti CL, Hermann DM. Signal transduction pathways involved in melatonin-induced neuroprotection after focal cerebral ischemia in mice. J Pineal Res. 2005;38:67–71.
- Wang WZ, Fang XH, Stephenson LL, Baynosa RC, Khiabani KT, Zamboni WA. Microcirculatory effects of melatonin in rat skeletal muscle after prolonged ischemia. J Pineal Res. 2005;39:57–65.
- Zhang WH, Li JY, Zhou Y. Melatonin abates liver ischemia/reperfusion injury by improving the balance between nitric oxide and endothelin. Hepatobiliary Pancreat Dis Int. 2006;5:574–9.
- Zamzami N, Hirsch T, Dallaporta B, Petit PX, Kroemer G. Mitochondrial implication in accidental and programmed cell death: apoptosis and necrosis. J Bioenerg Biomembr. 1997;29:185–93.
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–49.
- Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology. 2004;40:46–54.
- Frederiks WM, Vogels IM, Fronik GM. Plasma ornithine carbamyl transferase level as an indicator of ischaemic injury of rat liver. Cell Biochem Funct. 1984;2:217–20.
- Okatani Y, Wakatsuki A, Reiter RJ, Enzan H, Miyahara Y. Protective effect of melatonin against mitochondrial injury induced by ischemia and reperfusion of rat liver. Eur J Pharmacol. 2003;469:145–52.
- Kim SH, Lee SM. Cytoprotective effects of melatonin against necrosis and apoptosis induced by ischemia/reperfusion injury in rat liver. J Pineal Res. 2008;44:165–71.
- Venegas C, García JA, Escames G, Ortiz F, López A, Doerrier C, et al. Extrapineal melatonin: analysis of its subcellular distribution and daily fluctuations. J Pineal Res. 2012;52:217–27.
- Kireev R, Bitoun S, Cuesta S, Tejerina A, Ibarrola C, Moreno E, et al. Melatonin treatment protects liver of Zucker rats after ischemia/ reperfusion by diminishing oxidative stress and apoptosis. Eur J Pharmacol. 2013;701:185–93.
- Yang S, Zhu H, Li Y, Lin H, Gabrielson K, Trush MA, et al. Mitochondrial adaptations to obesity related oxidant stress. Arch Biochem Biophys. 2000;378:259–68.
- Jou MJ, Peng TI, Yu PZ, Jou SB, Reiter RJ, Chen JY, et al. Melatonin protects against common deletion of mitochondrial DNA-augmented mitochondrial oxidative stress and apoptosis. J Pineal Res. 2007;43: 389–403.
- Pappolla MA, Chyan YJ, Poeggeler B, Bozner P, Ghiso J, Le Daux SP, et al. Alzheimer h protein mediated oxidative damage to mitochondrial DNA: prevention by melatonin. J Pineal Res. 1999;27:226–9.
- Jiménez-Aranda A, Fernández-Vázquez G, Campos D, Tassi M, Velasco-Perez L, Tan DX, et al. Melatonin induces browning of inguinal white adipose tissue in Zucker diabetic fatty rats. J Pineal Res. 2013;55:416–23.
- Mattiasson G, Sullivan PG. The emerging functions of UCP2 in health, disease, and therapeutics. Antioxid Redox Signal. 2006;8:1–38.
- Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans. 2006;34:232–7.
- Garcia JJ, Reiter RJ, Guerrero JM, Escames G, Yu BP, Oh CS, et al. Melatonin prevents changes in microsomal membrane fluidity during induced lipid peroxidation. FEBS Lett. 1997;408:297–300.
- Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–54.
- Zhu X, Fan WG, Li DP, Kung H, Lin MC. Heme oxygenase-1 system and gastrointestinal inflammation: a short review. World J Gastroenterol. 2011;17:4283–8.
- Shen XD, Ke B, Zhai Y, Gao F, Anselmo D, Lassman CR, et al. Stat4 and Stat6 signaling in hepatic ischemia/reperfusion injury in mice: HO-1 dependence of Stat4 disruption-mediated cytoprotection. Hepatology. 2003;37:296–303.
- Shen XD, Ke B, Zhai Y, Gao F, Busuttil RW, Cheng G, et al. Toll- like receptor and heme oxygenase-1 signaling in hepatic ischemia/reperfusion injury. Am J Transplant. 2005;5:1793–800.
- Kang JW, Lee SM. Melatonin inhibits type 1 interferon signaling of toll-like receptor 4 via heme oxygenase-1 induction in hepatic ischemia/reperfusion. J Pineal Res. 2012;53:67–76.
- Tsuchihashi S, Zhai Y, Bo Q, Busuttil RW, Kupiec-Weglinski JW. Heme oxygenase-1 mediated cytoprotection against liver ischemia and reperfusion injury: inhibition of type-1 interferon signaling. Transplantation. 2007;83:1628–34.
- O'neill LA. Therapeutic targeting of Toll-like receptors for inflammatory and infectious diseases. Curr Opin Pharmacol. 2003;3:396–403.
- Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201:1135–43.
- Hertzog PJ, O'neill LA, Hamilton JA. The interferon in TLR signaling: more than just antiviral. Trends Immunol. 2003;24:534–9.
- Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med. 2008;205:2609–21.
- Xiao X, Zhao P, Rodriguez-Pinto D, Qi D, Henegariu O, Alexopoulou L, et al. Inflammatory regulation by TLR3 in acute hepatitis. J Immunol. 2009;183:3712–19.
- Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, et al. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol. 2005;175:7661–8.
- Huang SH, Cao XJ, Wei W. Melatonin decreases TLR3-mediated inflammatory factor expression via inhibition of NF-kappa B activation in respiratory syncytial virus-infected RAW264.7 macrophages. J Pineal Res. 2008;45:93–100.
- Huang G, Shi LZ, Chi H. Regulation of JNK and p38 MAPK in the immune system: signal integration, propagation and termination. Cytokine. 2009;48:161–9.
- Johnson GL, Nakamura K. The c-Jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim Biophys Acta. 2007;1773:1341–8.
- Schwabe RF, Brenner DA. Mechanisms of liver injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290:583–9.
- Schemmer P, Schoonhoven R, Swenberg JA, Bunzendahl H, Thurman RG. Gentle in situ liver manipulation during organ harvest decreases survival after rat liver transplantation: role of Kupffer cells. Transplantation. 1998;65:1015–20.
- Fan C, Li Q, Zhang Y, Liu X, Luo M, Abbott D, et al. IkappaBalpha and IkappaBbeta possess injury context-specific functions that uniquely influence hepatic NF-kappaB induction and inflammation. J Clin Invest. 2004;113:746–55.
- Uehara T, Bennett B, Sakata ST, Satoh Y, Bilter GK, Westwick JK, et al. NK mediates hepatic ischemia reperfusion injury. J Hepatol. 2005;42: 850–9.
- Schemmer P, Nickkholgh A, Schneider H, Sobirey M, Weigand M, Koch M, et al. PORTAL: pilot study on the safety and tolerance of preoperative melatonin application in patients undergoing major liver resection: a double-blind randomized placebo-controlled trial. BMC Surg. 2008;8:2.
- Freitas I, Bertone V, Guarnaschelli C, Ferrigno A, Boncompagni E, Rizzo V, et al. In situ demonstration of improvement of liver mitochondria function by melatonin after cold ischemia. In Vivo. 2006;20: 229–37.
- Casillas-Ramírez A, Mosbah IB, Ramalho F, Roselló-Catafau J, Peralta C. Past and future approaches to ischemia-reperfusion lesion associated with liver transplantation. Life Sci. 2006;79:1881–94.
- Pashkov AN, Popov SS, Semenikhina AV, Rakhmanova TI. Glutathione system and activity of NADPH-generating enzymes in the liver of intact rats and animals with toxic hepatitis receiving melatonin. Bull Exp Biol Med. 2005;139:565–8.
- Popov SS, Shul’gin KK, Pashkov AN, Zoloedov VI, Shvedov GI. [Intensity of free radical oxidation and superoxide dismutase activity in patients with drug hepatitis in combination therapy with melaxen or epifamin]. Eksp Klin Gastroenterol. 2011;9:36–40. [In Russian]
- Popov SS, Pashkov AN, Zoloedov VI, Shvedov GI. The use of melatonin in combined therapy of drug-induced hepatitis. Klin Med (Mosk). 2013;91:50–3.
- Reiter RJ, Tan DX, Sainz RM, Mayo JC, Lopez-Burillo S. Melatonin: reducing the toxicity and increasing the efficacy of drugs. J Pharm Pharmacol. 2002;54:1299–321.
- Fourman LT, Robert Meyer B. Autoimmune hepatitis in association with ramelteon. J Clin Gastroenterol. 2013;47:651–4.
- Cuesta S, Kireev R, Forman K, García C, Escames G, Ariznavarreta C, et al. Melatonin improves inflammation processes in liver of senescence-accelerated prone male mice (SAMP8). Exp Gerontol. 2010; 45:950–6.
- Zaouali MA, Boncompagni E, Reiter RJ, Bejaoui M, Freitas I, Pantazi E, et al. AMPK involvement in endoplasmic reticulum stress and autophagy modulation after fatty liver graft preservation: a role for melatonin and trimetazidine cocktail. J Pineal Res. 2013;55:65–78.
- Cichoz-Lach H, Celinski K, Konturek PC, Konturek SJ, Slomka M. The effects of L-tryptophan and melatonin on selected biochemical parameters in patients with steatohepatitis. J Physiol Pharmacol. 2010; 61:577–80.
- Gonciarz M, Gonciarz Z, Bielanski W, Mularczyk A, Konturek PC, Brzozowski T, et al. The pilot study of 3-month course of melatonin treatment of patients with nonalcoholic steatohepatitis: effect on plasma levels of liver enzymes, lipids and melatonin. J Physiol Pharmacol. 2010;61:705–10.
- Gonciarz M, Gonciarz Z, Bielanski W, Mularczyk A, Konturek PC, Brzozowski T, et al. The effects of long-term melatonin treatment on plasma liver enzymes levels and plasma concentrations of lipids and melatonin in patients with nonalcoholic steatohepatitis: a pilot study. J Physiol Pharmacol. 2012;63:35–40.
- Celinski K, Konturek PC, Slomka M, Cichoz-Lach H, Gonciarz M, Bielanski W, et al. Altered basal and postprandial plasma melatonin, gastrin, ghrelin, leptin and insulin in patients with liver cirrhosis and portal hypertension without and with oral administration of melatonin or tryptophan. J Pineal Res. 2009;46:408–14.
- Montagnese S, Middleton B, Mani AR, Skene DJ, Morgan MY. On the origin and the consequences of circadian abnormalities in patients with cirrhosis. Am J Gastroenterol. 2010;105:1773–81.
- Chojnacki C, Walecka-Kapica E, Klupińska G, Wachowska-Kelly P, Żylińska K, Winczyk K, et al. Serotonin and melatonin secretion and metabolism in patients with liver cirrhosis. Pol Arch Med Wewn. 2012;122:392–7.
- Chojnacki C, Wachowska-Kelly P, Błasiak J, Reiter RJ, Chojnacki J. Melatonin secretion and metabolism in patients with hepatic encephalopathy. J Gastroenterol Hepatol. 2013;28:342–7.
- Alvarez-García V, González A, Alonso-González C, Martínez-Campa C, Cos S. Melatonin interferes in the desmoplastic reaction in breast cancer by regulating cytokine production. J Pineal Res. 2012;52:282–90.
- Subramanian P, Mirunalini S, Dakshayani KB, Pandi-Perumal SR, Trakht I, Cardinali DP. Prevention by melatonin of hepatocarcinogenesis in rats injected with N-nitrosodiethylamine. J Pineal Res. 2007; 43:305–12.
- Srinivasan V, Spence DW, Pandi-Perumal SR, Trakht I, Cardinali DP. Therapeutic actions of melatonin in cancer: possible mechanisms. Integr Cancer Ther. 2008;7:189–203.
- Yan JJ, Shen F, Wang K, Wu MC. Patients with advanced primary hepatocellular carcinoma treated by melatonin and transcatheter arterial chemoembolization: a prospective study. Hepatobiliary Pancreat Dis Int. 2002;1:183–6.
- Lissoni P, Rovelli F, Malugani F, Bucovec R, Conti A, Maestroni GJ. Anti-angiogenic activity of melatonin in advanced cancer patients. Neuro Endocrinol Lett. 2001;2245–7.
- Kim KJ, Choi JS, Kang I, Kim KW, Jeong CH, Jeong JW. Melatonin suppresses tumor progression by reducing angiogenesis stimulated by HIF-1 in a mouse tumor model. J Pineal Res. 2013;54:264–70.
- Buscemi N, Vandermeer B, Hooton N, Pandya R, Tjosvold L, Hartling L, et al. Efficacy and safety of exogenous melatonin for secondary sleep disorders and sleep disorders accompanying sleep restriction: metaanalysis. BMJ. 2006;332:385–93.
- Manda K, Ueno M, Anzai K. AFMK, a melatonin metabolite, attenuates X-ray-induced oxidative damage to DNA, proteins and lipids in mice. J Pineal Res. 2007;42:386–93.
- Tan DX, Manchester LC, Terron MP, Flores LJ, Reiter RJ. One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species? J Pineal Res. 2007;42:28–42.
- Cheung RT, Tipoe GL, Tam S, Ma ES, Zou LY, Chan PS. Preclinical evaluation of pharmacokinetics and safety of melatonin in propylene glycol for intravenous administration. J Pineal Res. 2006;41:337–43.
- Morera AL, Henry M, de La Varga M. Safety in melatonin use. Actas Esp Psiquiatr. 2001;29:334–37.