
Abstract
Uncontrolled generation of bradykinin (BK) due to insufficient levels of protease inhibitors controlling contact phase (CP) activation, increased activity of CP proteins, and/or inadequate degradation of BK into inactive peptides increases vascular permeability via BK-receptor 2 (BKR2) and results in subcutaneous and submucosal edema formation. Hereditary and acquired angioedema due to C1-inhibitor deficiency (C1-INH-HAE and -AAE) are diseases characterized by serious and potentially fatal attacks of subcutaneous and submucosal edemas of upper airways, facial structures, abdomen, and extremities, due to inadequate control of BK generation. A decreased activity of C1-inhibitor is the hallmark of C1-INH-HAE (types 1 and 2) due to a mutation in the C1-inhibitor gene, whereas the deficiency in C1-inhibitor in C1-INH-AAE is the result of autoimmune phenomena. In HAE with normal C1-inhibitor, a significant percentage of patients have an increased activity of factor XIIa due to a FXII mutation (FXII-HAE). Treatment of C1-inhibitor-dependent angioedema focuses on restoring control of BK generation by inhibition of CP proteases by correcting the balance between CP inhibitors and BK breakdown or by inhibition of BK-mediated effects at the BKR2 on endothelial cells. This review will address the pathophysiology, clinical picture, diagnosis and available treatment in C1-inhibitor-dependent angioedema focusing on BK-release and its regulation.
Inadequate control of bradykinin formation results in the formation of characteristic subcutaneous and submucosal edemas of the skin, upper airways, facial structures, abdomen and extremities as seen in hereditary and acquired C1-inhibitor-dependent angioedema.
Diagnosis of hereditary and acquired C1-inhibitor-dependent angioedema may be troublesome as illustrated by the fact that there is a significant delay in diagnosis; a certain grade of suspicion is therefore crucial for quick diagnosis.
Submucosal edema formation in hereditary and acquired C1-inhibitor-dependent angioedema is potentially life threatening and can occur at any age.
To date effective therapies for acute and prophylactic treatment are available.
Key Messages
Introduction
Angioedema is characterized by attacks of subcutaneous and submucosal swelling caused by an increase in vascular permeability. Given the broad range of clinical presentation and different pathophysiological mechanisms leading to this increased vasopermeability the different types of angioedema are difficult to identify. A recent valuable attempt of the European Academy of Allergy and Clinical Immunology resulted in a clinical useful classification of angioedema. The authors identified four individual types of acquired angioedema (AAE), which are idiopathic histaminergic AAE (IH-AAE), idiopathic non-histaminergic AAE (InH-AAE), AAE related to angiotensin-converting enzyme inhibitors (ACEI-AAE) and AAE with C1-inhibitor deficiency (C1-INH-AAE). Idiopathic histamine-mediated increase of vasopermeability (IH-AAE) is the most frequent form accounting for more than 85% of the AAE (Citation1). In addition, they proposed three different types of hereditary angioedema (HAE): C1-inh deficiency due to genetic C1-inh deficiency (C1-INH-HAE) and HAE with normal C1-inh with a factor XII (FXII) mutation (FXII-HAE) or without an identified cause (U-HAE), respectively (Citation1). Bradykinin (BK) seems to be responsible to increase vasopermeability in at least C1-inh-dependent angioedema; in the other types of angioedema the role of BK is less clear.
Excessive generation of BK as a result of inadequate control of contact phase (CP) activation, due to increased activity of enzymes of this system and/or impaired capacity to degrade BK may result in increased vasopermeability and the development of angioedema. The serine protease inhibitor, C1-inhibitor (C1-Inh), is a main regulator of enzymes involved in the generation of BK. The present article will review the pathophysiology, the clinical picture and emerging therapies available to treat C1-inh-dependent hereditary and AAE.
Clinical picture
C1-inh-dependent angioedema is characterized by relapsing episodic swellings affecting any tissue, including face, upper airways, limbs, or mucous membranes. In C1-INH-HAE prodromal symptoms preceding an angioedema attack are experienced by more than 50% of the patients and may include erythema marginatum, fatigue, muscle aches and/or local discomfort (Citation2,Citation3). It is important to realize that erythema marginatum may easily be misdiagnosed as urticaria pointing falsely to an allergic genesis of angioedema. However, the slow onset and development of edema as seen in C1-INH-HAE is less characteristic for allergic edema (Citation2). The episodes are self-limiting lasting for 1–5 days involving skin, upper airways and gastrointestinal tract (Citation2). In 15–30% of patients multiple anatomical unrelated localizations are involved (Citation2,Citation4). Zanichelli et al. report 46%, 33% and 6.4% of the attacks to affect the extremities, the gastrointestinal tract and upper airways, respectively (Citation5). In a Danish cohort 94.8% of the patients suffered from skin swelling, 59.7% from facial swelling and 96.1% from abdominal (mesenteric or intestinal) edemas, of which 16.9% resulted in abdominal complications (Citation2). Symptomatic systemic hypotension most probably due to abdominal fluid displacement has been reported in 8.6% of these patients (Citation2,Citation5). In C1-INH-AAE swelling pattern is comparable with C1-INH-HAE, but abdominal edema seems to occur less frequent (Citation1,Citation6). Abdominal edema may have a dramatic presentation with severe pain, vomiting and diarrhea. Diagnosis of abdominal attacks is troublesome and since abdominal edema attacks closely mimic acute abdominal diseases such as appendicitis, patients often undergo unnecessary surgical intervention due to improper diagnosis. The difficulty to identify abdominal attacks may also explain – in part – the discrepancy in the percentage of abdominal attacks reported in the literature (Citation2,Citation5). Edema of the upper airways is the most dreaded clinical presentation as this may lead to airway obstruction and even asphyxia. Laryngeal complications have been reported to occur in 20% in a smaller patient cohort up to 49% in larger cohorts of C1-INH-HAE (Citation2,Citation7). In a large cohort investigating 123 patients with C1-INH-HAE, 61 (49%) experienced laryngeal edema episodes, 4 of which needed cricothyrotomy and 2 needed intubation, respectively (Citation7). One patient died from fatal asphyxia. In another cohort, mortality from asphyxia was 1.4% in patients with diagnosed C1-INH-HAE, but increased to 34% in patients with unrecognized C1-INH-HAE (Citation8). It is important to stress on the fact that the location and the severity of an experienced attack do not predict the severity and location of a future attack (Citation9). Laryngeal attacks can occur at any time, also as a first manifestation of the disease as illustrated by the 9-year-old patient described by Bork et al. who died from asphyxia due to laryngeal attack as a first clinical presentation of C1-INH-HAE (Citation7). C1-inh-dependent angioedema attacks can be triggered by trauma, infection or emotional stress (Citation2,Citation5). However, often no obvious trigger can be identified. The pathogenetic mechanism on how such events can trigger an attack is still not fully understood. There is a broad variability in severity and the frequency of attacks between different patients but also in one particular patient from time-to-time. In a cohort of 103 patients with C1-INH-HAE, 22% of the patients suffered from more than 12 attacks/year, 23% from 6 to12 attacks/year, 37% from 1 to 5 attacks/year, 12.6% from less than 1 attack/year and 4.9% without any attacks, respectively (Citation5). The median duration of an attack was 1.58 days with a range from 1 to 8 days (Citation5). The recurrent angioedema attacks are associated with significant reduction in the quality of life, since they are associated with substantial disability (Citation2). Although the disability is reversible, the recurrent character of the symptoms leads to a significant limitation of daily activities and to an increased rate of absenteeism at work and school of both patients and caregivers (Citation10). The fear for evoking angioedema attacks or the unpredictable course of the disease may also have a strong negative impact on the quality of life. Also, the dependence of caregivers for administration of (intravenous) treatment may be bothersome for the patient, although self-management programs could partially overcome this issue (Citation11). To date no clinical or laboratory tools are available to predict the frequency and the severity of attacks. These tools would greatly facilitate the initiation of proper and timely (prophylactic) treatment and to design patient tailored therapy in order to minimize burden of illness.
BK as a central mediator in the pathogenesis of subcutaneous and submucosal edema formation in C1-inhibitor-dependent angioedema
The nonapeptide BK and the decapeptide Lys-bradykinin (Lys-BK, kallidin), the latter in circulation rapidly processed to Lys-desArg9-BK by carboxypeptidases, are the most important members of the kinin family. These kinins induce smooth muscle cell relaxation and increase vasopermeability resulting in the characteristic and submucosal swellings as seen in hereditary and acquired C1-INH dependent angioedema (Citation12). The importance of BK in the pathogenesis of the subcutaneous and submucosal swellings is illustrated by the following findings: First, increased levels of BK can be measured in patients suffering from a C1INH-HAE/AAE attack as compared to steady state (Citation13,Citation14); second, C1-INH-HAE patients with unilateral edema of extremities had significantly increased BK concentration in the affected extremity as compared to the non-affected extremity, respectively (Citation14); third, blockage of BK-receptor 2 abrogates the vasoeffective effects of BK and prevented angioedema (Citation15,Citation16) and fourth, mice lacking BK-receptor 2 (BKR2) as well as C1-Inh do not have evidence for increased vascular permeability, whereas C1-Inh deficient mice with normal BKR2 suffer from increased vascular permeability (Citation17). Although LysBK can increase vasopermeability and can be measured in circulation, the contribution of LysBK and its desArg forms in the pathogenesis of C1-INH-dependent angioedema is not clear (Citation18).
BK and Lys-BK are released by proteolytic cleavage of high molecular weight kininogen (HK) or low-molecular weight kininogen (LK), respectively () (Citation19). HK is a 626 amino acid single-chain protein with six domains (D1–D6). The light chain consists of D1–D3 and the heavy chain of D5 and D6. The light- and heavy chain are linked by D4, which serves as a substrate for proteases. Cleavage of D4 at Lys362-Arg363 and Arg371-Ser372 results in the release of the nonapeptide BK (Citation20). In contrast, LK is a 409 amino acid splice site variant of HK lacking domain D6, which has been described as binding site for prekallikrein (PK) and factor XI (FXI), respectively. Proteolytic cleavage of HK occurs by kallikrein (KK), which is produced through the activation of PK via the CP system or directly by prolylcarboxypeptidase (PRCP) on endothelial cells () (Citation20–22). CP system consists of the zymogens FXII, PK, HK and FXI. Upon contact with negatively charged surface FXII is autoactivated by a conformational change resulting in traces of activated FXII (FXIIa), which subsequently induces cleavage of PK to KK () (Citation21). A plethora of activators have been reported to activate FXII, such as polyanionic surfaces (e.g. kaolin, ellagic acid, glycosaminoglycans), mest cell heparin, misfolded proteins, RNA, inorganic polyphosphates, as well as bacteria (Citation23–28). KK in turn reciprocally activates FXII in order to produce more FXIIa resulting in the generation of additional KK (positive feedback), which can subsequently cleave HK to release BK () (Citation21). KK can also generate plasmin by direct cleavage of plasminogen or indirectly by activating single-chain urokinase to two-chain urokinase, which subsequently cleaves plasminogen to plasmin (Citation21). Plasmin at high concentration in turn has been shown to activate FXII and hence may therefore theoretically contribute to CP mediated BK generation (Citation12,Citation29). Factor VII-activating protease (FSAP), which is activated upon contact with dead cells, has been demonstrated induce BK generation from HK; however the contribution to BK formation in vivo is not established yet (Citation30,Citation31). Tissue kallikrein 1 (KLK1) has been shown to induce LK cleavage resulting in the liberation of Lys-BK, which may subsequently processed to BK by aminopeptidases (Citation32,Citation33). Pancreas, salivary glands and kidney have been demonstrated to express KLK1 (Citation32,Citation33). Similarly to the other members of the tissue kallikrein family, KLK1 is synthesized as zymogen and activated by proteolytic cleavage releasing a hydrophobic residue (Citation33). Despite this unusual activation pattern, it is still not known on whether autoactivation and/or activation by a yet unidentified protease result in KLK1 activation in vivo (Citation33). The role of this mainly tissue-restricted kinin generation in the form of Lys-BK in the pathogenesis of C1-inh-mediated angioedema remains to be established.
Figure 1. Bradykinin and Lys-bradykinin formation from high- and low-molecular weight kininogen. High-molecular weight kininogen (HK) has six domains (D1–D6). In contrast, low-molecular weight kininogen (LK), a splicing variant of HK has five domains lacking D6. Proteolytic lavage in D4 of HK by kallikrein (KK) or factor VII activating protease (FSAP) results in the generation of the nonapeptide bradykinin (BK). Cleavage of LK by tissue kallikrein (KLK1) in D4 results in the liberation of the decapeptide lys-bradykinin (lys-BK), which is a potent stimulator of bradykinin-receptor 2 (BKR2). Proteolytic cleavage of BK and lysBK by carboxypeptidases N/M (CPN/CPM) results in the formation of desArg forms (desArg-BK, desArg-lysBK) which are potent stimulators of bradykinin receptor 1 (BKR1). The desArg forms are rapidly degraded into inactive peptide fragments by aminopeptidase P (AAP), neutral endopeptidase (NEP), dipeptidyl peptidase IV (DPPIV) and angiotensin-converting enzyme (ACE). Adapted from Refs. (Citation12,Citation32).
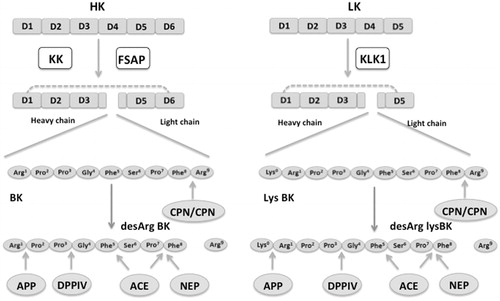
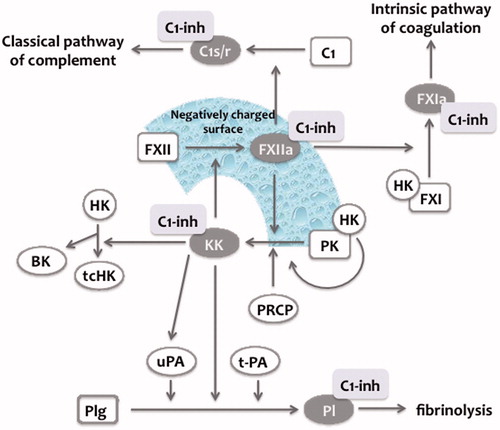
Figure 2. Contact phase system. Factor XII (FXII) is (auto-)activated to activated FXII (FXIIa) upon contact with negatively charged surfaces (see text). In plasma, prekallikrein (PK) circulates in complex with high molecular weight kininogen (HK). FXIIa can activate PK to kallikrein (KK), which in turn can activate FXII to generate more FXIIa (positive feedback). Prolylcarboxypeptidase (PRCP) has also been shown to activate PK to KK. FXIIa can activate the classical pathway of complement as well as FXI from the intrinsic pathway of coagulation. KK cleave single chain HK to two-chain HK tc(HK) thereby releasing bradykinin (BK). In addition, KK can directly activate plasminogen (Plg) to plasmin (Pl) or indirectly by the activation of single chain urokinase (u-PA) to two-chain uPA. C1-inhibitor (C1-Inh) is a crucial regulator of the contact phase system, since it inhibits FXIIa and KK, respectively. In addition, it regulates the classical pathway of complement (via C1s/r), coagulation via FXIa and fibrinolysis via plasmin, respectively. Figure adapted from Refs (Citation21,Citation41).
BK and Lys-BK exert their biological effects via ligation of the constitutively expressed BKR2 resulting in relaxation of smooth muscle cells and increased vasopermeability, respectively ( and ) (Citation12). BK and Lys-BK may further be processed by plasmatic carboxypeptidase N (CPN) and/or membrane-bound carboxypeptidase M (CPM) to desArg-BK9 and desArg-lysBK9, respectively (). The des-Arg forms of BK and Lys-BK are potent simulators of the inducible bradykinin receptor 1 (BKR1) (Citation12). However, the clinical significance of inducible BKR1 in the pathogenesis of increased vasopermeability is still not fully understood. Although BKR1 has been reported to play a minor role, recent publication challenges its dispensable role in the pathogenesis of increased vasopermeability (Citation34).
Figure 3. Effects of estrogens and androgens on the bradykinin system. Bradykinin (BK) and lys-bradykinin (lys-BK) generation via kallikrein (KK) and tissue kallikrein (KLK1) from high molecular weight kininogen (HK) and low molecular weight kininogen (LK) increases vasopermeability via ligation of bradykinin-receptor 2 (BKR2). Estrogens decrease C1-inhibitor (C1-Inh) concentrations and levels of angiotensin-converting enzyme (ACE) ①. In contrast, estrogens increase Factor XII (FXII) levels ①. (Attenuated) androgens increase C1-Inh and aminopeptidase P levels ②.
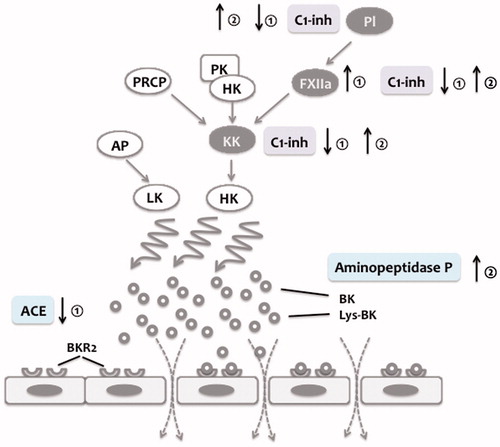
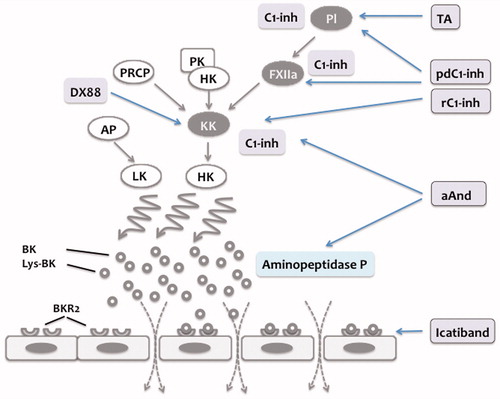
Figure 4. Summary of treatment options in hereditary and acquired C1-inhibitor-mediated angioedema. C1-inhibitor (C1-Inh) regulates contact phase activation by inhibition of activated factor XII (FXIIa) and kallikrein (KK). Treatment of plasma-derived C1-Inh (pd-C1-Inh) and recombinant C1-Inh (rC1-Inh) restores the regulation of contact phase activation and hence reduces BK formation. Tranexamic acid (TA) inhibits plasmin generation thereby attenuating the positive feedback loop resulting in generation of additional FXIIa. Ecallantide (DX88) is a direct inhibitor of KK. Attenuated androgens (aAnd) increase C1-Inh and aminopeptidase P levels. Icatiband selectively blocks BK effects on bradykinin-receptor 2 (BKR2), thereby preventing BK-mediated increase in permeability.
In circulation both, BK and Lys-BK and their des-Arg forms are short-lived being immediately degraded into inactive products by aminopeptidase P (AAP), neutral endopeptidase (NEP), dipeptidyl peptidase IV (DPPIV) and angiotensin-converting enzyme (ACE), respectively () (Citation12,Citation35–40). The short half-life also explains why the measurement of BK in plasma is troublesome and critically depends on preanalytical handling of patient plasma samples (Citation13).
C1-inhibitor regulates BK generation
C1-Inh is a regulator of many plasmatic cascade systems, such as CP system, the classical pathway of complement, fibrinolysis and the intrinsic coagulation system. C1-Inh is a crucial regulator of the CP system, since it is the most important inhibitor of FXIIa and besides α2-macroglobulin the most important inhibitor of KK in fluid phase (). Through the inhibition of KK and FXIIa, C1-Inh efficiently controls BK generation. C1-Inh inhibits the classical pathway of complement and has been described as an efficient inhibitor of FXIa, representing the initial protease of the intrinsic pathway of coagulation. In addition, C1-Inh has been reported as an efficient inhibitor of FSAP and plasmin in the fluid phase, respectively (). C1-Inh belongs to the SERine Protease Inhibitor (serpin) family, which also includes α1-antitrypsin, antithrombin, α2-antiplasmin, and plasminogen-activator inhibitor type-1 (PAI-1). C1-Inh is as heavily glycosylated single chain polypeptide of 478 amino acid residues with a reported molecular weight of 76 kDa (105 kDa on sodiumdodecyl sulfate–polyacrylamide gel electrophoresis due to its abundant glycosylation). Its physiological plasma concentration is 240 mg/L with an estimated half-life of 67–72 h. Serpins offer a “fake-substrate” located on the carboxy-terminal part of the serpin domain (“reactive loop”) consisting in a peptidyl-bond (P1–P1′), which matches with the substrate specificity of the target protease (Citation41). Proteolytic cleaveage of this P1–P1′ bond (Arg444-Thr445) by the target protease results in an SDS resistant covalent complex of C1-Inh with the target protease, which is instantly followed by a significant structural change of the serpin characterized by an insertion of the cleaved reactive center loop into the central β-sheet of C1-Inh. The serpin-protease complexes have an estimated half-life of 20–47 min and are efficiently removed from the circulation in the reticoluendotheilal system, among others by via the low-density lipoprotein receptor (Citation41). In contrast to other serpins, C1-Inh has an extraordinary long and heavily glycosylated N-terminal part, which does not directly inhibit serine protease activity. The function of the N-terminal part is not clear yet. Publications form one single group claim that the heavy glycosylated N-terminal part of C1-Inh has an important role in the attenuation of the inflammatory response by preventing via its expressing Sialyl-lewis X moieties the selectin-mediated interaction of leukocytes with endothelial cells (Citation42,Citation43). However, others have not confirmed these results yet.
Hereditary and acquired C1-inhibitor-dependent angioedema: inadequate regulation of BK formation by an absolute or relative deficiency of C1-inhibitor
An absolute or relative decrease in functional levels of active C1-Inh results in C1-INH- HAE/AAE. C1-INH-HAE type 1 is characterized by a decreased level C1-Inh antigen and accounts for approx. 85% of the cases. C1-INH-HAE type II is caused by dysfunctional C1-Inh and characteristically presents with a discrepant low C1-Inh activity in relation to the normal C1-Inh antigen concentration. C1-INH-HAE type 2 accounts for approx. 15% of the cases (Citation41,Citation44). The inheritance is autosomal dominant and patients are heterozygous for the deficiency (Citation41,Citation44). The molecular pathology underlying C1-INH-HAE types I and II is heterogeneous and complex. Failure of C1-Inh production and/or the production of dysfunctional C1-Inh due to large gene deletions, mis-sense or point mutations affecting the serpin domain have been reported (Citation45,Citation46). Whatever the underlying molecular mechanism, the final result is a decreased activity of C1-Inh in the circulation. However, there is no clear relationship between specific molecular mutations in the C1-Inh gene (genotype) and the severity of the clinical picture (phenotype). Being a heterozygous disorder, C1-Inh activity in affected subject is considered to be 50% of normal. Interestingly, C1-INH-HAE patients normally present with markedly lower C1-Inh activity levels around 10–30% (Citation41,Citation47,Citation48). It is still not entirely clear why the observed activity lies beyond the expected C1-Inh activity. Increased turnover of C1-Inh due to complex formation with C1s and/or a decreased cellular synthesis of C1-Inh in HAE affected subjects are among the discussed mechanisms (Citation41,Citation47–49).
Recently, HAE characterized by normal levels of both C1-Inh activity and antigen has been described (Citation50). It affects – with some exceptions – women (Citation50). Hypothetically, estrogens are thought to play a crucial role in the pathogenesis of – at least – some of these angioedema forms. Indeed, estrogens have been reported to increase FXII levels and to decrease both C1-Inh and ACE concentrations () (Citation51–55). In a significant percentage of these patients FXII mutations have been identified, which may result in an increased FXIIa activity and/or an increased susceptibility of FXII for (auto)activation (FXII-HAE) (Citation56–59). A gain-of-function mutation in FXII gene (Thr328Lys) results in an increased activity of FXIIa leading to an in uncontrolled activation of the CP system and hence BK generation (Citation57,Citation59). Another mutation in the FXII gene (e.g. Thr309Lys and Thr309Arg) has been described in patients presenting with the clinical symptoms of C1-INH-HAE with normal levels of C1-Inh (Citation56,Citation58). Conceptually, given the fact that these mutations affect the prolin-rich domain of FXII, which is considered to play a role in the interaction with negatively charged surfaces, one might speculate that these forms of FXII are more prone to activation upon contact with negatively charged surfaces (Citation58). Spontaneous amidase activity in plasma as a surrogate marker for serine protease activity of the CP and fibrinolytic system has been shown to be increased in patients with FXII-HAE regardless the clinical severity (Citation60). Interestingly, ACE and CPN activity levels inversely correlated with disease severity pointing to the important role of BK-degrading enzymes in the pathogenesis of HAE type III, but most probably in other forms as well (Citation61).
In contrast to C1-INH- or FXII-HAE, C1-INH-AAE is caused by acquired autoantibodies directed to C1-Inh, resulting in a decreased inhibitory capacity toward its target proteases (Citation62,Citation63). In most cases of C1-INH-AAE, an underlying disorder, such as a lymphoproliferative disease (B-NHL) or autoimmune disease, can be found (Citation64,Citation65). However, as in other autoimmune disorders, AAE might also be a herald of future autoimmune disease or lymphoma, respectively. In some cases, the diagnosis of AAE precedes the manifestation of a lymphoproliferative disease by many years.
Diagnosis
Diagnosis of C1-inh-dependent angioedema may be troublesome. This is illustrated by the fact that there is a significant delay (up to 16 years) between the first symptoms and the final diagnosis (Citation2,Citation9,Citation66). The most important factor for a quick diagnosis is a sufficient grade of suspicion by the attending physician. Relapsing subcutaneous swellings with or without prodromal symptoms, which generally evolve slower than allergic edemas, the lack of urticaria, the presence of relapsing unexplained abdominal symptoms and/or a family history of HAE is conspicuous and should prime the attending physician to consider further diagnostic testing for hereditary of AAE forms (Citation2,Citation9,Citation44). Although the mean age of onset of symptoms in C1-INH-HAE is between 8 and 12 years, the first attacks can occur in adult life, which holds for both hereditary and acquired forms (Citation67). Conversely, in families known with HAE, the first symptoms of angioedema may manifest at much younger age, as the family is keener at identifying HAE. A negative family history does not rule out C1-INH-HAE, since in 20–25% neo-mutations in the C1-Inh gene cause C1-Inh deficiency.
In case of a suspected C1-inh-dependent angioedema laboratory diagnosis focuses in a first approach on the measurement of C4 antigen levels and the measurement C1-Inh activity, preferably by a chromogenic assay (Citation9,Citation68). C4 levels are mostly decreased in C1-INH-HAE/AAE and in case of normal C4 levels one should repeat the test or reconsider the diagnosis of C1-INH-HAE/AAE (Citation9). In case of a C1-Inh activity below 50% the reduced activity should be confirmed in a second independent sample. When C1-Inh activity is low, simultaneous testing of C1-Inh antigen should be performed – if possible – in order to make a distinction between C1-INH-HAE types I and II, respectively. Although of academic value, genetic testing is not needed for the diagnosis of C1-INH-HAE types I and II. It is important to realize that the state-of-the-art measurement of C1-Inh activity is based on the inhibitory capacity toward C1s measured with a chromogenic substrate, but not toward the enzymes critically involved in the generation of BK. Establishment of the genetic mutation, however, may be helpful to ascertain the diagnosis in smaller children (in whom interpretation of C1-Inh plasma levels may be difficult) and can be used for prenatal testing and (when desired) immediate assessment of the genetic status of a newborn through cord blood. Besides the decreased C1-Inh activity and low C4 levels C1-INH-AAE is characterized by low levels of C1q, most probably due to consumption by immune complexes (Citation9,Citation45). Specialized laboratories can also identify the autoantibodies toward C1-Inh. Diagnosis of FXII-HAE is troublesome and there is no specific diagnostic test to identify these patients. FXII-HAE is characterized by normal C1-Inh activity and antigen levels. Clotting factor-based assays may help to identify increased activity of FXIIa in plasma (Citation57). In a collective of 100 symptomatic patients suffering from FXII-HAE spontaneous amidase activity (see above) has been shown to correlate with disease severity (Citation60). However, specific genetic testing for the known FXII mutations or sequencing of the FXII gene is the most appropriate diagnostic tools to confirm or exclude HAE type III (Citation9). In some of these patients with a hereditary form no defect can be identified (HAE of unknown origin, U-HAE) (Citation1).
Treatment
The acute treatment of C1-inh-dependent HAE aims to reduce the severity and duration of an angioedema attack. In contrast, prophylactic therapy targets to prevent the occurrence or – at least – reduce the frequency and severity of angioedema attacks (long-term prophylaxis) and to prevent attacks during anticipated stress situations, for example surgical procedures (short-term prophylaxis) (Citation9). The goal of all established treatment is to restore the regulation of BK formation, to improve the efficacy to degrade BK or to antagonize BK-effects on the level of its receptor. In severe C1-INH-AAE, besides symptomatic therapy treatment of the underlying disease is a sine-qua-non in order to attenuate antibody-production to C1-Inh. Elimination of autoreactive B-cells with anti-CD20 (Rituximab) turned out to be effective (Citation69,Citation70).
Attenuated androgens (17-alpha alkylated androgens), such as danazol and stanozol, are used in both, short- and long-term prophylaxis. In randomized, double-blind placebo-controlled crossover studies, attenuated androgens have been shown to reduce both, frequency and severity of attacks (Citation71,Citation72). This reduction in frequency and severity is supported by prospective open-label studies (Citation73–77). In addition, in a significant percentage attenuated androgens also induced a complete remission (Citation73,Citation75,Citation77). The most common side effects of androgen treatments are virilization, weight gain, voice deepening, altered lipid metabolism, hypertension, elevated liver enzymes and, rarely, liver neoplasms (Citation78). Androgens have been demonstrated to increase mRNA expression of C1-Inh (Citation79). Therefore, the beneficial effects of androgens are mainly thought to be due to an increase of C1-Inh levels although the dosages of androgens used in patients are lower than the concentrations needed to stimulate C-Inh gene transcription in vitro. In addition, androgens have been demonstrated to increase the concentration of AAP, thereby contributing to an accelerated degradation of BK ( and ) (Citation80). Tranexamic acid, an antifibrinolytic drug, has been shown to be effective in the treatment of C1-INH-HAE, although not in all cases, and is mainly used for long-term prophylaxis in children, where androgens are contraindicated (Citation9,Citation67,Citation81,Citation82). Being a competitive inhibitor of plasminogen activation tranexamic acid limits additional FXIIa activation and C1-Inh consumption by plasmin in HAE and AAE patients () (Citation83).
Administration of plasma-derived C1-Inh is an established treatment for acute attacks as well as for short- and long-term prophylaxis (Citation9). The rational of this treatment is to increase C1-Inh concentrations in order to restore the regulation of CP-mediated BK generation () (Citation41). C1-Inh purified from pooled human plasma has been successfully used since the early 1970s in Europe as a life-saving agent for the treatment of C1-INH- HAE and -AAE in Europe (Citation84–88). Based on retrospective studies, the efficacy of C1-inh-dependent angioedema has been unquestioned. However, until recently C1-Inh therapy to treat acute attacks in C1-INH-HAE as well as for prophylactic use has not been approved by the FDA, and hence C1-Inh treatment was not available in the USA. Therefore, two randomized placebo-controlled trials have been performed to demonstrate the efficacy in the treatment of HAE attacks. In the first study, nanofiltered plasma-derived C1-Inh (Cinryze) has been shown to decrease the median time to the onset of unequivocal relief from an attack from more than 4 h in the placebo group to 2 h in the C1-Inh group (Citation89). Similarly, the International Multicenter Prospective Angioedema C1-Inhibitor Trial 1 (IMPACT1) demonstrated C1-Inh (Berinert) to significantly decrease the median time to the onset of symptom relief dose-dependently from 1.5 to 0.5 h in the group treated with 20 U/kg C1-Inh and from 1.5 to 1.2 h with 10 U/kg C1-Inh, respectively (Citation90). In an open-label extension study of IMPACT1, 57 patients have been treated with 20 U/kg C1-Inh for a total of 1085 attacks (Citation91). The median time to the onset of symptom relief was 0.46 h and to complete resolution of symptoms 15.5 h, respectively. The shortest time to resolution has been observed for laryngeal edema (5.8 h), for abdominal, peripheral and facial edema time to resolution ranged from 12.8 to 26.6 h (Citation91). The efficacy of prophylactic C1-Inh administration has been demonstrated in a 22-subject crossover study: the frequency of attacks has significantly been reduced from 12.7 in the placebo group to 6.3 over 12 weeks in the group treated with 1000 U nanofiltered C1-Inh twice a week (Citation89). In an open label extension follow-up study prophylactic administration of nanofiltered C1-Inh reduced the frequency of attacks by 93.7% (0.19 attacks/month), 87.7% of the patients had less than one attack per month and 34.9% did not suffer from any attack during prophylactic treatment (Citation92). The studies conducted so far as well as the Berinert patient registry demonstrated plasma-derived C1-Inh to be a safe therapy (Citation84–89,Citation91,Citation92). A potential limitation of C1-inh replacement treatment is the requirement for intravenous administration. However, self-administration programs for patients allowing home treatment by themselves are successful and have proven to reduce time to initiation of treatment markedly (Citation11). Recombinant C1-Inh (Rhucin®) purified from the milk of transgenic rabbits turned out to be effective in the treatment of acute C1-INH-HAE attacks as well (Citation93–95). Despite the very-short half-life recombinant C1-Inh significantly shortened the time to pain relief from 152 to 90 min and the time to minimal symptoms from 483 to 303 min (Citation93,Citation94). No drug-related adverse events or immunogenic reactions to the C1-Inh or rabbit proteins have been observed so far (Citation93–95). Recombinant C1-Inh has also been shown to be effective in long-term prophylaxis (Citation96).
Ecallantide (Dx88) is a recombinant protein expressed in Pichia pastoris specifically inhibiting KK () (Citation97). Ecallantide non-significantly improved the time to improvement of symptoms from 240 min in the placebo group to 165 min in the treatment arm (Citation98). An open-label follow-up study indicates that Ecallantide may indeed shorten the duration of an attack, however, 5% of the patients experienced hypersensitivity reactions (Citation99). As a selective BKR2 antagonist, Icatibant inhibits BK induced vasodilatation in vivo () (Citation17,Citation100). Icatibant does not interact with BKR1. In two randomized, double-blind controlled trials, Icatibant has been shown to be effective in the treatment of acute attacks. In the For Angioedema Subcutaneous Treatment (FAST) 1 study, Icatibant shortened non-significantly the median time to relief of symptoms from 4.6 h in the placebo group to 2.5 h in the treatment group (Citation101). In the FAST-2 trial, Icatibant significantly decreased the median time from 12 h in the tranexemic acid group to 2 h in the treatment group (Citation101). In an open-label extension study, the median time to symptom relief in patients with frequent attacks has been 1–2 h. The subcutaneous administration facilitates administration of the drug in comparison to the intravenous administration of the C1-Inh concentrates, however, mild to moderate injection-site reactions may be common with the use of Icatibant. Besides these dermal adverse events, treatment with Icatibant is considered to be safe (Citation102).
Conclusion
Increasing insight into the pathogenesis of C1-inh-dependent angioedema has resulted in better diagnostic and therapeutic management strategies for patients with congenital and acquired deficiency of C1-inh. It may be assumed that a further increase in the knowledge on the pathogenesis of angioedema formation and the link to precipitating events may further improve the treatment of these patients as well patients with other forms of severe angioedema.
Disclosure statement
Sacha Zeerleder received an unrestricted grant from Viropharma. Marcel Levi reports no conflict of interest.
References
- Cicardi M, Aberer W, Banerji A, Bas M, Bernstein JA, Bork K, et al. Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy. 2014;69:602–16.
- Bygum A. Hereditary angio-oedema in Denmark: a nationwide survey. Br J Dermatol. 2009;161:1153–8.
- Prematta M, Gibbs JG, Pratt EL, Stoughton TR, Craig TJ. Fresh frozen plasma for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol. 2007;98:383–8.
- Hofman ZL, Relan A, Hack CE. Hereditary angioedema attacks: local swelling at multiple sites. Clin Rev Allergy Immunol. 2016;50:34--40.
- Zanichelli A, Vacchini R, Badini M, Penna V, Cicardi M. Standard care impact on angioedema because of hereditary C1 inhibitor deficiency: a 21-month prospective study in a cohort of 103 patients. Allergy. 2011;66:192–6.
- Bouillet-Claveyrolas L, Ponard D, Drouet C, Massot C. Clinical and biological distinctions between type I and type II acquired angioedema. Am J Med. 2003;115:420–1.
- Bork K, Hardt J, Schicketanz KH, Ressel N. Clinical studies of sudden upper airway obstruction in patients with hereditary angioedema due to C1 esterase inhibitor deficiency. Arch Intern Med. 2003;163:1229–35.
- Bork K, Hardt J, Witzke G. Fatal laryngeal attacks and mortality in hereditary angioedema due to C1-INH deficiency. J Allergy Clin Immunol. 2012;130:692–7.
- Bowen T, Cicardi M, Farkas H, Bork K, Longhurst HJ, Zuraw B, et al. 2010 International consensus algorithm for the diagnosis, therapy and management of hereditary angioedema. Allergy Asthma Clin Immunol. 2010;6:24. doi: 10.1186/1710-1492-6-24.
- Aygoren-Pursun E, Bygum A, Beusterien K, Hautamaki E, Sisic Z, Wait S, et al. Socioeconomic burden of hereditary angioedema: results from the hereditary angioedema burden of illness study in Europe. Orphanet J Rare Dis. 2014;9:99. doi: 10.1186/1750-1172-9-99.
- Levi M, Choi G, Picavet C, Hack CE. Self-administration of C1-inhibitor concentrate in patients with hereditary or acquired angioedema caused by C1-inhibitor deficiency. J Allergy Clin Immunol. 2006;117:904–8.
- Campbell DJ. The kallikrein-kinin system in humans. Clin Exp Pharmacol Physiol. 2001;28:1060–5.
- Nussberger J, Cugno M, Amstutz C, Cicardi M, Pellacani A, Agostoni A. Plasma bradykinin in angio-oedema. Lancet. 1998;351:1693–7.
- Nussberger J, Cugno M, Cicardi M, Agostoni A. Local bradykinin generation in hereditary angioedema. J Allergy Clin Immunol. 1999;104:1321–2.
- Griesbacher T, Legat FJ. Effects of FR173657, a non-peptide B2 antagonist, on kinin-induced hypotension, visceral and peripheral oedema formation and bronchoconstriction. Br J Pharmacol. 1997;120:933–9.
- Griesbacher T, Sametz W, Legat FJ, Diethart S, Hammer S, Juan H. Effects of the non-peptide B2 antagonist FR173657 on kinin-induced smooth muscle contraction and relaxation, vasoconstriction and prostaglandin release. Br J Pharmacol. 1997;121:469–76.
- Han ED, MacFarlane RC, Mulligan AN, Scafidi J, Davis AE 3rd. Increased vascular permeability in C1 inhibitor-deficient mice mediated by the bradykinin type 2 receptor. J Clin Invest. 2002;109:1057–63.
- Campbell DJ, Krum H, Esler MD. Losartan increases bradykinin levels in hypertensive humans. Circulation. 2005;111:315–20.
- Jacobsen S. Separation of 2 different substrates for plasma kinin-forming enzymes. Nature. 1966;210:98–9.
- Sainz IM, Pixley RA, Colman RW. Fifty years of research on the plasma kallikrein-kinin system: from protein structure and function to cell biology and in-vivo pathophysiology. Thromb Haemost. 2007;98:77–83.
- Caliezi C, Wuillemin WA, Zeerleder S, Redondo M, Eisele B, Hack CE. C1-Esterase inhibitor: an anti-inflammatory agent and its potential use in the treatment of diseases other than hereditary angioedema. Pharmacol Rev. 2000;52:91–112.
- Shariat-Madar Z, Mahdi F, Schmaier AH. Identification and characterization of prolylcarboxypeptidase as an endothelial cell prekallikrein activator. J Biol Chem. 2002;277:17962–9.
- Cochrane CG, Griffin JH. The biochemistry and pathophysiology of the contact system of plasma. Adv Immunol. 1982;33:241–306.
- Herwald H, Moergelin M, Olsen A, Rhen M, Dahlbäck B, Müller-Esterl W, et al. Activation of the contact-phase system on bacterial surfaces – a clue to serious complications in infectious diseases. Nat Med. 1998;4:298–302.
- Hojima Y, Cochrane CG, Wiggins RC, Austen KF, Stevens RL. In vitro activation of the contact (Hageman factor) system of plasma by heparin and chondroitin sulfate E. Blood. 1984;63:1453–9.
- Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci USA. 2007;104:6388–93.
- Muller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139:1143–56.
- Maas C, Govers-Riemslag JW, Bouma B, Schiks B, Hazenberg BP, Lokhorst HM, et al. Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J Clin Invest. 2008;118:3208–18.
- Ewald GA, Eisenberg PR. Plasmin-mediated activation of contact system in response to pharmacological thrombolysis. Circulation. 1995;91:28–36.
- Etscheid M, Beer N, Fink E, Seitz R, Johannes D. The hyaluronan-binding serine protease from human plasma cleaves HMW and LMW kininogen and releases bradykinin. Biol Chem. 2002;383:1633–43.
- Stephan F, Hazelzet JA, Bulder I, Boermeester MA, van Till JO, van der Poll T, et al. Activation of factor VII-activating protease in human inflammation: a sensor for cell death. Crit Care. 2011;15:R110.
- Kaplan AP, Joseph K. Pathogenic mechanisms of bradykinin mediated diseases: dysregulation of an innate inflammatory pathway. Adv Immunol. 2014;121:41–89.
- Pathak M, Wong SS, Dreveny I, Emsley J. Structure of plasma and tissue kallikreins. Thromb Haemost. 2013;110:423–33.
- Bossi F, Fischetti F, Regoli D, Durigutto P, Frossi B, Gobeil F Jr, et al. Novel pathogenic mechanism and therapeutic approaches to angioedema associated with C1 inhibitor deficiency. J Allergy Clin Immunol. 2009;124:1303–10e4.
- Connelly JC, Skidgel RA, Schulz WW, Johnson AR, Erdos EG. Neutral endopeptidase 24.11 in human neutrophils: cleavage of chemotactic peptide. Proc Natl Acad Sci USA. 1985;82:8737–41.
- Dorer F, Ryan JW, Stewart JM. Hydrolysis of bradykinin and its higher homologues by angiotensin-converting enzyme. Biochem J. 1974;141:915–7.
- Heins J, Neubert K, Barth A, Canizaro PC, Behal FJ. Kinetic investigation of the hydrolysis of aminoacyl p-nitroanilides by dipeptidyl peptidase IV from human and pig kidney. Biochim Biophys Acta. 1984;785:30–5.
- Orawski AT, Susz JP, Simmons WH. Aminopeptidase P from bovine lung: solubilization, properties, and potential role in bradykinin degradation. Mol Cell Biochem. 1987;75:123–32.
- Skidgel RA, Davis RM, Tan F. Human carboxypeptidase M. Purification and characterization of a membrane-bound carboxypeptidase that cleaves peptide hormones. J Biol Chem. 1989;264:2236–41.
- Tan F, Jackman H, Skidgel RA, Zsigmond EK, Erdos EG. Protamine inhibits plasma carboxypeptidase N, the inactivator of anaphylatoxins and kinins. Anesthesiology. 1989;70:267–75.
- Zeerleder S. C1-inhibitor: more than a serine protease inhibitor. Semin Thromb Hemost. 2011;37:362–74.
- Cai S, Davis AE 3rd. Complement regulatory protein C1 inhibitor binds to selectins and interferes with endothelial-leukocyte adhesion. J Immunol. 2003;171:4786–91.
- Cai S, Dole VS, Bergmeier W, Scafidi J, Feng H, Wagner DD, et al. A direct role for C1 inhibitor in regulation of leukocyte adhesion. J Immunol. 2005;174:6462–6.
- Longhurst H, Cicardi M. Hereditary angio-oedema. Lancet. 2012;379:474–81.
- Cugno M, Zanichelli A, Foieni F, Caccia S, Cicardi M. C1-inhibitor deficiency and angioedema: molecular mechanisms and clinical progress. Trends Mol Med. 2009;15:69–78.
- Wagenaar-Bos IG, Hack CE. Structure and function of C1-inhibitor. Immunol Allergy Clin North Am. 2006;26:615–32.
- Cugno M, Nuijens J, Hack CE, Eerenberg A, Frangi D, Agostoni A, et al. Plasma levels of C1 inhibitor complexes and cleaved C1 inhibitor in patients with hereditary angioneurotic edema. J Clin Invest. 1990;85:1215–20.
- Quastel M, Harrison R, Cicardi M, Alper CA, Rosen FS. Behavior in vivo of normal and dysfunctional C1 inhibitor in normal subjects and patients with hereditary angioneurotic edema. J Clin Invest. 1983;71:1041–6.
- Katz Y, Strunk RC. Synthesis and regulation of C1 inhibitor in human skin fibroblasts. J Immunol. 1989;142:2041–5.
- Binkley KE. Factor XII mutations, estrogen-dependent inherited angioedema, and related conditions. Allergy Asthma Clin Immunol. 2010;6:16.
- Cohen AJ, Laskin C, Tarlo S. C1 esterase inhibitor in pregnancy. J Allergy Clin Immunol. 1992;90:412–3.
- Farsetti A, Misiti S, Citarella F, Felici A, Andreoli M, Fantoni A, et al. Molecular basis of estrogen regulation of Hageman factor XII gene expression. Endocrinology. 1995;136:5076–83.
- Gordon EM, Ratnoff OD, Saito H, Donaldson VH, Pensky J, Jones PK. Rapid fibrinolysis, augmented Hageman factor (factor XII) titers, and decreased C1 esterase inhibitor titers in women taking oral contraceptives. J Lab Clin Med. 1980;96:762–9.
- Ogston D, Walker J, Campbell DM. C1 inactivator level in pregnancy. Thromb Res. 1981;23:453–5.
- Stevenson JC, Oladipo A, Manassiev N, Whitehead MI, Guilford S, Proudler AJ. Randomized trial of effect of transdermal continuous combined hormone replacement therapy on cardiovascular risk markers. Br J Haematol. 2004;124:802–8.
- Bork K, Wulff K, Hardt J, Witzke G, Staubach P. Hereditary angioedema caused by missense mutations in the factor XII gene: clinical features, trigger factors, and therapy. J Allergy Clin Immunol. 2009;124:129–34.
- Cichon S, Martin L, Hennies HC, Muller F, Van Driessche K, Karpushova A, et al. Increased activity of coagulation factor XII (Hageman factor) causes hereditary angioedema type III. Am J Hum Genet. 2006;79:1098–104.
- Dewald G, Bork K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun. 2006;343:1286–9.
- Martin L, Raison-Peyron N, Nothen MM, Cichon S, Drouet C. Hereditary angioedema with normal C1 inhibitor gene in a family with affected women and men is associated with the p.Thr328Lys mutation in the F12 gene. J Allergy Clin Immunol. 2007;120:975–7.
- Defendi F, Charignon D, Ghannam A, Baroso R, Csopaki F, Allegret-Cadet M, et al. Enzymatic assays for the diagnosis of bradykinin-dependent angioedema. PLoS One. 2013;8:e70140.
- Charignon D, Ghannam A, Defendi F, Ponard D, Monnier N, Lopez Trascasa M, et al. Hereditary angioedema with F12 mutation: factors modifying the clinical phenotype. Allergy. 2014;69:1659–65.
- Geha RS, Quinti I, Austen KF, Cicardi M, Sheffer A, Rosen FS. Acquired C1-inhibitor deficiency associated with antiidiotypic antibody to monoclonal immunoglobulins. N Engl J Med. 1985;312:534–40.
- Jackson J, Sim RB, Whelan A, Feighery C. An IgG autoantibody which inactivates C1-inhibitor. Nature. 1986;323:722–4.
- Zingale LC, Castelli R, Zanichelli A, Cicardi M. Acquired deficiency of the inhibitor of the first complement component: presentation, diagnosis, course, and conventional management. Immunol Allergy Clin North Am. 2006;26:669–90.
- Castelli R, Deliliers DL, Zingale LC, Pogliani EM, Cicardi M. Lymphoproliferative disease and acquired C1 inhibitor deficiency. Haematologica. 2007;92:716–8.
- Roche O, Blanch A, Caballero T, Sastre N, Callejo D, Lopez-Trascasa M. Hereditary angioedema due to C1 inhibitor deficiency: patient registry and approach to the prevalence in Spain. Ann Allergy Asthma Immunol. 2005;94:498–503.
- Farkas H, Harmat G, Fust G, Varga L, Visy B. Clinical management of hereditary angio-oedema in children. Pediatr Allergy Immunol. 2002;13:153–61.
- Wagenaar-Bos IG, Drouet C, Aygoren-Pursun E, Bork K, Bucher C, Bygum A, et al. Functional C1-inhibitor diagnostics in hereditary angioedema: assay evaluation and recommendations. J Immunol Methods. 2008;338:14–20.
- Branellec A, Bouillet L, Javaud N, Mekinian A, Boccon-Gibod I, Blanchard-Delaunay C, et al. Acquired C1-inhibitor deficiency: 7 patients treated with rituximab. J Clin Immunol. 2012;32:936–41.
- Bygum A, Vestergaard H. Acquired angioedema – occurrence, clinical features and associated disorders in a Danish nationwide patient cohort. Int Arch Allergy Immunol. 2013;162:149–55.
- Sheffer AL, Fearon DT, Austen KF. Methyltestosterone therapy in hereditary angioedema. Ann Intern Med. 1977;86:306–8.
- Gelfand JA, Sherins RJ, Alling DW, Frank MM. Treatment of hereditary angioedema with danazol. Reversal of clinical and biochemical abnormalities. N Engl J Med. 1976;295:1444–8.
- Sheffer AL, Fearon DT, Austen KF. Hereditary angioedema: a decade of management with stanozolol. J Allergy Clin Immunol. 1987;80:855–60.
- Sheffer AL, Fearon DT, Austen KF. Clinical and biochemical effects of stanozolol therapy for hereditary angioedema. J Allergy Clin Immunol. 1981;68:181–7.
- Hosea SW, Santaella ML, Brown EJ, Berger M, Katusha K, Frank MM. Long-term therapy of hereditary angioedema with danazol. Ann Intern Med. 1980;93:809–12.
- Fust G, Farkas H, Csuka D, Varga L, Bork K. Long-term efficacy of danazol treatment in hereditary angioedema. Eur J Clin Invest. 2011;41:256–62.
- Rosen FS, Beyler A. Hereditary angioneurotic edema and its correction with androgen therapy. Birth Defects Orig Artic Ser. 1980;16:499–507.
- Banerji A, Sloane DE, Sheffer AL. Hereditary angioedema: a current state-of-the-art review, V: attenuated androgens for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol. 2008;100:S19–22.
- Pappalardo E, Zingale LC, Cicardi M. Increased expression of C1-inhibitor mRNA in patients with hereditary angioedema treated with Danazol. Immunol Lett. 2003;86:271–6.
- Drouet C, Desormeaux A, Robillard J, Ponard D, Bouillet L, Martin L, et al. Metallopeptidase activities in hereditary angioedema: effect of androgen prophylaxis on plasma aminopeptidase P. J Allergy Clin Immunol. 2008;121:429–33.
- Sheffer AL, Austen KF, Rosen FS. Tranexamic acid therapy in hereditary angioneurotic edema. N Engl J Med. 1972;287:452–4.
- Blohme G. Treatment of hereditary angioneurotic oedema with tranexamic acid. A random double-blind cross-over study. Acta Med Scand. 1972;192:293–8.
- Tengborn L, Blomback M, Berntorp E. Tranexamic acid – an old drug still going strong and making a revival. Thromb Res. 2015;135:231–42.
- Agostoni A, Bergamaschini L, Martignoni G, Cicardi M, Marasini B. Treatment of acute attacks of hereditary angioedema with C1-inhibitor concentrate. Ann Allergy. 1980;44:299–301.
- Cicardi M, Bergamaschini L, Marasini B, Boccassini G, Tucci A, Agostoni A. Hereditary angioedema: an appraisal of 104 cases. Am J Med Sci. 1982;284:2–9.
- Bork K, Barnstedt SE. Treatment of 193 episodes of laryngeal edema with C1 inhibitor concentrate in patients with hereditary angioedema. Arch Intern Med. 2001;161:714–8.
- Bork K, Meng G, Staubach P, Hardt J. Treatment with C1 inhibitor concentrate in abdominal pain attacks of patients with hereditary angioedema. Transfusion. 2005;45:1774–84.
- Bork K, Witzke G. Long-term prophylaxis with C1-inhibitor (C1 INH) concentrate in patients with recurrent angioedema caused by hereditary and acquired C1-inhibitor deficiency. J Allergy Clin Immunol. 1989;83:677–82.
- Zuraw BL, Busse PJ, White M, Jacobs J, Lumry W, Baker J, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med. 2010;363:513–22.
- Craig TJ, Levy RJ, Wasserman RL, Bewtra AK, Hurewitz D, Obtulowicz K, et al. Efficacy of human C1 esterase inhibitor concentrate compared with placebo in acute hereditary angioedema attacks. J Allergy Clin Immunol. 2009;124:801–8.
- Craig TJ, Bewtra AK, Bahna SL, Hurewitz D, Schneider LC, Levy RJ, et al. C1 esterase inhibitor concentrate in 1085 Hereditary Angioedema attacks – final results of the I.M.P.A.C.T.2 study. Allergy. 2011;66:1604–11.
- Zuraw BL, Kalfus I. Safety and efficacy of prophylactic nanofiltered C1-inhibitor in hereditary angioedema. Am J Med. 2012;125:938e1–7.
- van Doorn MB, Burggraaf J, van Dam T, Eerenberg A, Levi M, Hack CE, et al. A phase I study of recombinant human C1 inhibitor in asymptomatic patients with hereditary angioedema. J Allergy Clin Immunol. 2005;116:876–83.
- Riedl MA, Bernstein JA, Li H, Reshef A, Lumry W, Moldovan D, et al. Recombinant human C1-esterase inhibitor relieves symptoms of hereditary angioedema attacks: phase 3, randomized, placebo-controlled trial. Ann Allergy Asthma Immunol. 2014;112:163–9e1.
- Choi G, Soeters MR, Farkas H, Varga L, Obtulowicz K, Bilo B, et al. Recombinant human C1-inhibitor in the treatment of acute angioedema attacks. Transfusion. 2007;47:1028–32.
- Reshef A, Moldovan D, Obtulowicz K, Leibovich I, Mihaly E, Visscher S, et al. Recombinant human C1 inhibitor for the prophylaxis of hereditary angioedema attacks: a pilot study. Allergy. 2013;68:118–24.
- Schneider L, Lumry W, Vegh A, Williams AH, Schmalbach T. Critical role of kallikrein in hereditary angioedema pathogenesis: a clinical trial of ecallantide, a novel kallikrein inhibitor. J Allergy Clin Immunol. 2007;120:416–22.
- Cicardi M, Levy RJ, McNeil DL, Li HH, Sheffer AL, Campion M, et al. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med. 2010;363:523–31.
- Lumry WR, Bernstein JA, Li HH, MacGinnitie AJ, Riedl M, Soteres DF, et al. Efficacy and safety of ecallantide in treatment of recurrent attacks of hereditary angioedema: open-label continuation study. Allergy Asthma Proc. 2013;34:155–61.
- Cockcroft JR, Chowienczyk PJ, Brett SE, Bender N, Ritter JM. Inhibition of bradykinin-induced vasodilation in human forearm vasculature by icatibant, a potent B2-receptor antagonist. Br J Clin Pharmacol. 1994;38:317–21.
- Cicardi M, Banerji A, Bracho F, Malbran A, Rosenkranz B, Riedl M, et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med. 2010;363:532–41.
- Aberer W, Maurer M, Reshef A, Longhurst H, Kivity S, Bygum A, et al. Open-label, multicenter study of self-administered icatibant for attacks of hereditary angioedema. Allergy. 2014;69:305–14.