
Abstract
Background. Ganglioneuromas (GNs) are neural crest cell-derived tumors and rarely occur in the adrenal gland. They are usually asymptomatic and hormonally silent. The majority of cases are detected incidentally during work-up for unrelated conditions. Hormone-secreting pure adrenal GNs in adults are extremely rare. To date, only four cases have been reported in the English literature. Case report. We describe an adult case of endocrinologically active adrenal GN incidentally diagnosed in a 64-year-old male patient with history of uncontrolled hypertension. On physical examination, he had a blood pressure (BP) of 160/100 mmHg. Abdominal computed tomography and magnetic resonance imaging showed a large solid tumor (8.5 × 7.5 × 7 cm) in the right adrenal gland. Urinary levels of norepinephrine, normetanephrine, vanillylmandelic acid and dopamin were elevated, although urinary level of epinephrine was suppressed. Right adrenalectomy was performed for treatment purposes. The histological diagnosis of the resected tumor was adrenal GN. Conclusions. Hormone-secreting pure adrenal GN occurs very rarely in adults and preoperative diagnosis is difficult. Adrenal GN may present with hormonal activity such as increased secretion of catecholamines and their metabolites. There are no specific diagnostic signs and symptoms discriminating GN and pheochromocytoma. Therefore, histopathological examination need for a definitive diagnosis of adrenal GN. The prognosis after completed surgical resection without further therapy seems to be excellent. To our knowledge, the present case is the second report that describes hormone-secreting pure adrenal GN in an adult from Turkey in the English literature. We discuss this case and review the literature on this unusual entity.
Introduction
Ganglioneuromas (GNs) are rare benign, well differentiated, slow-growing neoplasms arising from neural crest tissue and are composed of mature ganglion cells and Schwann's cells in a fibrous stroma (Citation1–7). The incidence of GNs is unknown. They occur most commonly in children and young adults, and rarely occurs in adults (Citation2,Citation8,Citation9). GNs may arise anywhere along the paravertebral sympathetic plexus (Citation2,Citation10). They most commonly occur within the posterior mediastinum (60–80%) and retroperitoneum (32–37.5%), and less commonly in the adrenal medulla (10–15%) (Citation1,Citation2,Citation5,Citation6,Citation11–15). Because imaging procedures such as ultrasonography (US) and computed tomography (CT) have become more widely performed, the number of GNs found incidentally has increased (Citation4,Citation8,Citation16,Citation17).
Characteristically, GNs do not secrete excess catecholamines or steroid hormones. They are generally considered to be non-secretory (hormonally silent or inactive), and can therefore asymptomatic even when the size of tumor is large (Citation4,Citation7,Citation8,Citation12,Citation17). However, some GNs are endocrinologically active, and secrete catecholamines and their metabolites, and rarely adrenocorticotropin (ACTH), cortisol, androgen and vasoactive intestinal polypeptide (VIP) (Citation1,Citation4,Citation18–24). Hormonally active GNs may cause some symptoms such as diarrhea, flushing, diaphoresis, cough, abdominal pain, dyspnea, headache, palpitations, tremor, anxiety, hypertension, or virilization related to secreting hormones (Citation1,Citation3, Citation19–25). In addition, there is no single reliable imaging modality or biochemical test that can accurately differentiate GN from pheochromocytoma (PHEO), neuroblastoma (NB), ganglioneuroblastoma (GNB) or paraganglioma (Citation1,Citation2,Citation4). Therefore, it is generally difficult to diagnose these tumors precisely as GN before surgery. Definitive diagnosis is done by histological examination. Assessment and management of the adrenal GNs are similar to those of other adrenal tumors (Citation2). In this report, we present an adult case of endocrinologically active adrenal GN incidentally diagnosed in a 64-year-old male patient. Histopathological examination of the adrenal mass confirmed the diagnosis.
Case report
A 64-year-old Turkish man was hospitalized at our hospital for further examinations of a heterogeneous, hypoechogenic right adrenal solid mass, measuring 8.5 × 7.5 × 7.0 cm, with well defined borders and fine calcifications that was incidentally discovered by abdominal US during uncontrolled hypertension research in another hospital. Medical history included hypertension for 20 years, hyperglycemia for 6 months and hyperlipidemia for 1 year managed with irbesartan (300 mg/day), hydrochlorothiazide (12.5 mg/day), amlodipine (10 mg/day), metformin (2000 mg/day), gliclazide modified release (60 mg/day) and rosuvastatin (20 mg/day). Hypertension was not controlled with a combination of above antihypertensive drugs. On physical examination (PE), he had a blood pressure (BP) of 160/100 mmHg, a regular pulse of 80 beats/min, and a weight of 70 kg at a height of 164 cm. BP was measured with the patient in the sitting position using a standardized aneroid sphygmomanometer by a physician. The patient had been resting for 30 min before the measurement. He had no features of Cushing's syndrome or virilization. Grade 2 hypertensive retinopathy was found on examination of the fundus. The remainder of the PE was within normal limits. Twenty- four-hour ambulatory BP monitoring (ABPM) measurements were performed with Schiller BR-102 Plus ABP Holter System. The system performs three measurements/hour. Normal 24-h ABP is defined as < 130/80 mmHg. Concurrently, normal daytime and night-time BP levels are defined as < 135/ 85 mmHg and < 120/70 mmHg, respectively. ABPM revealed maximum systolic BP of 155 mmHg and maximum diastolic BP of 87 mmHg with non-dipper pattern. Echocardiography showed left ventricle hypertrophy and diastolic dysfunction. Routine laboratory tests were normal. Laboratory values were as follows: urine norepinephrine (NE): 98 μg/24 h (normal: 20–81), urine epinephrine (E): 1.7 μg/24 h (normal: 2.0–22), urine normetanephrine: 876 μg/24 h (normal: 88–444), urine metanephrine: 126 μg/24 h (normal: 52–341), urine vanillylmandelic acid (VMA): 6.9 mg/24 h (normal: 1.8–6.7), urine dopamine: 444 μg/24 h (normal: 40–400) and homovanillic acid (HVA): 6.1 mg/24 h (normal: 0.5–6.2). The results of other endocrine tests, including the plasma aldosterone concentration (PAC), plasma renin activity (PRA), PAC/PRA ratio, intact parathyroid hormone (iPTH), serum calcitonin, cortisol and ACTH levels, diurnal cortisol rhythms, 24-h urinary free cortisol and androgen hormones, were within normal ranges.
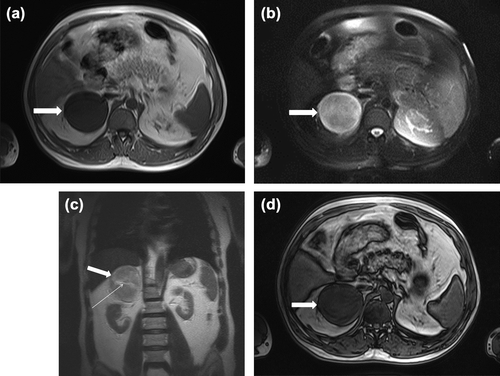
Abdominal CT showed a well demarcated, homogeneous, hypodense right adrenal solid mass (8.5 × 7.5 × 7.0 cm) with faint calcifications (). The non-enhanced attenuation of the tumor was 20 Hounsfield units (HU) on CT, consistent with a non-adenomatous mass. There were no features suggesting the invasion of surrounding structures, or enlarged lymph nodes. On T1-weighted abdominal magnetic resonance imaging (MRI), the tumor was visualized as a homogeneous mass (8.5 × 7.5 × 7.0 cm) with a low signal intensity (hypointense) below that of the liver, located in the right adrenal gland (). T2-weighted MRI revealed a heterogeneous hyperintense mass measuring 8.5 × 7.5 × 7.0 cm, located in the right adrenal gland ( and ). Out-of-phase MRI did not show significant signal loss in the lesion when compared with in-phase MRI (). Characteristics on MRI were not in favor of the diagnosis of adrenocortical adenoma. A 131iodine-meta-iodobenzylguanidine (131I-MIBG) scan could not be performed.
Figure 1. Abdominal computed tomography (CT) shows a regular homogeneous right adrenal mass measuring 8.5 × 7.5 × 7 cm, with attenuation of 20 HU (arrow).
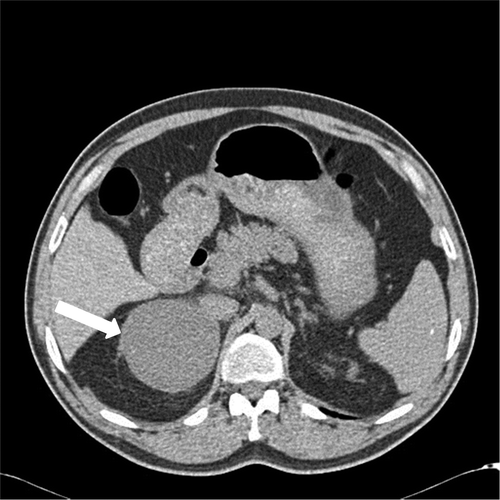
Figure 2. (a) Transverse T1-weighted magnetic resonance imaging (MRI) shows a round, slightly lobulated right adrenal mass that measures 8.5 × 7.5 × 7 cm (arrow). The mass is homogeneous, with signal intensity less than that of liver (hypointense). (b) Transverse T2-weighted MRI scan demonstrating round heterogeneous right adrenal mass (arrow) with markedly high signal intensity greater than that of liver (c) Coronal T2-weighted MRI scan demonstrating round heterogeneous right adrenal mass (thick arrow) with slightly high signal intensity greater than that of liver and with central crescent-shaped calcifications in adrenal mass (slightly hyperintensity) (thin arrow) (d) Out-of-phase MRI no showing significant signal loss in the lesion when compared with in-phase MRI (arrow).
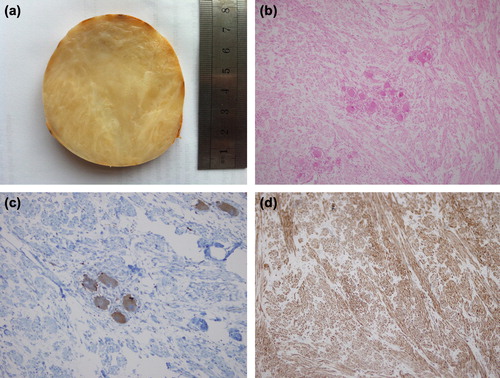
From these findings, we suspected the tumor to be a PHEO as a higher probability or adrenal carcinoma (AC) as a lower probability arising from the right adrenal gland. Following the administration of adequate α-receptor blocking agents with phenoxybenzamine 80 mg/day, a β-adrenoceptor blocker (propranolol, 80 mg/day) was added to the therapy. Normotension was reached on tension Holter monitoring. Right adrenalectomy was performed by the transabdominal route under general anesthesia. The resected tumor was well circumscribed, measured 9 × 8× 7 cm, weighted 240 g and it was encapsulated. The cut surface of the tumor was solid with whitish grey color without evidence of hemorrhage or necrosis (). Microscopically, the sections showed irregular proliferation by spindle-shaped cells and occasionally scattered mature ganglionic cells (). Spindle-shaped cells were strongly stained with monoclonal antibody to S-100 protein and ganglionic cells were stained with monoclonal antibody to neuron-specific enolase (NSE), synaptophysin and chromogranin-A ( and ). The specimen did not show any evidence of malignant degeneration histologically. The tumor was diagnosed as a right adrenal mature GN. Postoperative course was uneventful, and no recurrence was detected at the 12-month follow-up visit. Hyperglycemia disappeared. Hypertension was controlled with only one drug (amlodipin, 10 mg/day).
Figure 3. (a) Gross pathological appearance of postoperative right adrenal tumor. It is encapsulated, 9 × 8× 7 cm in dimensions. The tumor shows a whitish grey and gelatinous appearance in the cut surface without evidence of hemorrhage or necrosis. (b) The tumor showing clusters of mature ganglion cells in Schwannian cell dominant stroma (hematoxylin and eosin staining, original magnification × 100). (c) Neoplastic ganglionic cells were positive for synaptophysin (immunoperoxidase staining, × 200). (d) Spindle-shaped Schwannian cells-rich stroma stained positive for S-100 protein (immunoperoxidase staining, × 100).
Discussion
In this report, we presented a case of hormone- secreting adrenal GN that was differentially diagnosed from PHEO and adrenal carcinoma in a 64-year-old male patient. The adrenal incidentalomas may be adrenocortical adenomas and carcinomas, cysts, myelolipomas, PHEOs, GNs and adrenal metastases from other malignant tumors (Citation26); 1–6% of adrenal incidentalomas are GNs (Citation4,Citation7,Citation12). GNs are rare and well differentiated benign tumors in the NB series of tumors originating from the neural crest tissue of the sympathetic nervous system (Citation1,Citation2,Citation4,Citation27). They should be distinguished from the other groups because they are considered benign and constituted by mature sympathetic ganglion cells (Citation2). GN is generally considered to occur more frequently in older children or young adults, and rarely occurs in adults, which is consistent with the age of the present case (64 years) (Citation2,Citation8,Citation9).
Most of the adrenal GNs are usually hormonally silent, and therefore can be asymptomatic, even when the size of tumor is large (Citation4,Citation7,Citation8,Citation11). Although GNs are generally considered non-secretory, some GNs have been reported to be endocrinologically active (Citation1,Citation3). They occasionally secrete catecholamines and their metabolites, because they can occur as a composite tumor with PHEO, and they rarely secrete ACTH, cortisol, androgen and VIP (Citation1,Citation4,Citation18–24). Hormone-secreting pure adrenal GNs in adults are extremely rare, and only four cases have been reported in the English literature (Citation1,Citation4,Citation28,Citation29). Our case is the second report that describes hormone-secreting pure adrenal GN in an adult from Turkey in the English literature. In the present case, elevated levels of serum cortisol and androgen were not shown, while the urinary NE, normetanephrine, dopamin and VMA levels were elevated. Also, we had first reported a case of dopamine-secreting adrenal GN associated with paroxismal hypertension attacks (Citation1). In this case, we have identified a small increase in the urinary levels of both dopamine and its urinary metabolite, HVA.
Radiological examination has no diagnostic value in most cases of adrenal GN (Citation30). Radiographically, GNs are relatively homogeneous, encapsulated tumors with well margined borders (Citation4,Citation9,Citation11). The tumors generally do not invade adjacent structures (Citation3,Citation9). However, it is difficult to diagnose these tumors precisely as GN preoperatively. On CT imaging, GNs appear as well circumscribed homogeneous masses with lower attenuation (hypodense) than that of muscle (Citation2,Citation4,Citation9,Citation31) (). In some cases, the tumor may be heterogeneous. At CT, calcification has been reported in approximately 20–60% of GN cases and is typically punctate, fine and speckled as opposed to the coarse pattern seen with NBs and GNBs (Citation31–33). In differential diagnosis between adrenal PHEO and adrenal GN, a significant enhancement of CT scan is frequently seen in adrenal PHEO, while the most significant imaging feature adrenal GN is a < 40 HU on CT, as seen in the present case (Citation4,Citation34). The size of adrenal GN varies (range 1–22 cm) (Citation7,Citation8,Citation12), and the median diameter was 6.2 cm in a previous report of 17 cases (Citation7).
On MRI, GNs appear as homogeneous masses with low and, less commonly intermediate, signal intensity on T1-weighted images (Citation2,Citation31,Citation32,Citation35) (). In contrast, on T2-weighted images, the signal intensity is usually heterogeneous and either intermediate to high or markedly high than that of liver (Citation2,Citation4,Citation8,Citation16,Citation35) ( and ). Several reports indicate that relatively high signal intensity on T2-weighted images correlates with GN (); the appearance is presumed to be caused by a combination of abundant myxoid stroma and relatively low amounts of ganglion cells (Citation31,Citation33,Citation35). GNs reveal no absolute change in signal intensity on chemical shift imaging (out-of-phase MRI) (Citation2) (). For the pure GNs, heterogeneous high signal intensity on T2-weighted MR images may be helpful in the differential diagnosis of other masses that have high signal intensity on T2-weighted images (Citation2).
These imaging characteristics on CT and MRI were consistent with the present case. However, in addition to the hormone-secreting activity of adrenal GNs, some of these imaging characteristics on CT and MRI were similar to other adrenal tumors, such as PHEO and/or adrenal carcinoma. For example, a tumor size > 5 cm, heterogeneity and calcification were suggestive of AC, and high signal intensity on T2-weighted MRI images was suggestive of PHEO or AC (Citation4,Citation36). A recent study from China reported that the misdiagnosis rate of adrenal GNs on CT and MRI before an operation was 64.7% (Citation7). In that study, among 11 misdiagnosed cases, five were inconclusive, three were diagnosed as malignant and three were diagnosed as other benign tumors. Therefore, determining a precise diagnosis of adrenal GN on CT and MRI before surgery is difficult (Citation4). However, it is very important to distinguish GNs from other adrenal tumors such as PHEO and/or adrenal carcinoma, before surgery for several reasons (Citation4). First, resection of PHEO is a high-risk surgical procedure. Undiagnosed patients with PHEO who underwent surgical treatment have high mortality rate secondary to hypertensive attack, malignant ventricular arrhythmia and multiple organ failures (Citation4,Citation37). Therefore, preoperative medical preparation is essential to prevent these lethal complications. Second, AC is a rare and deadly disease, and the primary curative treatment for AC is complete surgical resection (Citation4). However, controversy remains regarding whether open or laparoscopic adrenalectomy is the more appropriate treatment modality (Citation4). In addition, AC surgery should be performed by an experienced surgeon. For the above reasons, adrenal GNs should be distinguished from PHEO and/or AC before the operation.
Grossly, GNs are large, well circumscribed, solid, encapsulated masses of firm consistence with a homogeneous whitish grey and gelatinous appearance in the cut surface without evidence of hemorrhage or necrosis, as was seen in the present case (Citation1,Citation2,Citation3,Citation31,Citation33). Microscopically, they are of two subtypes. The mature subtype consists of a spindle cell tumor resembling a NB but has fascicles composed of neuritic processes, Schwann cells and perineural cells and show numerous ganglion cells (Citation2,Citation4,Citation30). Ganglion cells are fully mature cells with abundant cytoplasm, rounded contour and large nuclei with distinct and prominent nucleoli (Citation33). The maturing subtype has a similar stroma but with ganglion cells of differing maturation, from fully mature ones to neuroblasts (Citation2,Citation3). Immature elements (such as neuroblasts), intermediate cells, cellular atypia, mitotic figures and necrosis are not features of GN (Citation3). On immunohistochemical analysis, they are characterized by reactivity with S-100 and neuronal markers such as NSE and synaptophysin (Citation2). The present case was consisted with the mature subtype of adrenal GN. In differential diagnosis between adrenal PHEO and adrenal GN, adrenal PHEO varies in shape and content ranging from solid to cystic and cystosolid and is often accompanied by hemorrhage and necrosis, while adrenal GN is, in most cases, nodular or sublobar in shape with gray or grayish yellow sections and intact capsules, as seen in the present case (Citation34).
The definitive treatment of choice for GN is usually completed surgical resection for localized, non-secretory or non-metastatic tumors when possible (Citation4,Citation12,Citation14,Citation18,Citation30,Citation33,Citation38,Citation39). If the tumor is catecholamine secreting, the acute and chronic effects of increased plasma catecholamines should be reversed prior to the surgical excision of the tumor. Combined α-and β-adrenergic blockades are required preoperatively to control high BP and to prevent intraoperative hypertensive crises. An α-adrenergic blockade (e.g. phenoxybenzamine or doxazosin) should be started at least 7 days preoperatively to allow for expansion of the contracted blood volume. A liberal salt diet is advised during the preoperative period. Once adequate α-adrenergic blockade is achieved, β-adrenergic blockade (e.g. propranolol or labetalol) is initiated (e.g. at least 3 days preoperatively). Laparoscopic surgery for abdominal GNs may be a better substitute for traditional open surgery due to its minimal invasive procedure. However, in the present case, we performed an open tumor excision because the tumor size was above 6 cm, and the possibility of AC could not be completely ruled out (Citation16). Prognosis of completely resected mature adrenal GNs is excellent without further therapies (Citation5,Citation12). The recurrence rate for adrenal GN is near zero, and postoperative complications are rare. However, malignant transformation of GN into malignant peripheral nerve sheath tumors or NBs (Citation40,Citation41) have been reported. Therefore, GNs should be surgically excised completely and should be followed for a long period after the operation (Citation19). In addition, life-long clinical and biochemical follow-up patients with hormone-secreting GNs and adrenal composite PHEO-GN is essential.
In conclusion, hormone-secreting pure adrenal GN occurs very rarely in adults and preoperative diagnosis is very difficult. Adrenal GN may present with hormonal activity such as increased secretion of catecholamines and their metabolites. Its hormonal activity and imaging characteristics are occasionally very similar to those of other adrenal tumors, especially PHEO and AC. Therefore, careful evaluation by endocrine tests and multiple imaging procedures is needed to provide a differential diagnosis. However, histopathological examination is required for a definitive diagnosis of adrenal GN.
Declaration of interest: The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
References
- Erem C, Kocak M, Cinel A, Ersoz HO, Reis A. Dopamine-secreting adrenal ganglioneuroma presenting with paroxysmal hypertension attacks. Saudi Med J. 2008;29:122–125.
- Erem C, Ucuncu O, Nuhoglu I, Cinel A, Cobanoglu U, Demirel A, et al. Adrenal ganglioneuroma: Report of a new case. Endocrine. 2009;35:293–296.
- Geoerger B, Hero B, Harms D, Grebe J, Scheidhauer K, Berthold F. Metabolic activity and clinical features of primary ganglioneuromas. Cancer. 2001;91:1905–1913.
- Sasaki S, Yasuda T, Kaneto H, Otsuki M, Tabuchi Y, Fujita Y, et al. Large adrenal ganglioneuroma. Intern Med. 2012; 51:2365–2370.
- Oderda M, Cattaneo E, Soria F, Barreca A, Chiusa L, Morelli B, et al. Adrenal ganglioneuroma with multifocal retroperitoneal extension: A challenging diagnosis. Sci World J. 2011;11:1548–1553.
- Kamoun M, Mnif MF, Rekik N, Belguith N, Charfi N, Mnif L, et al. Ganglioneuroma of adrenal gland in a patient with Turner syndrome. Ann Diagn Pathol. 2010;14:133–136.
- Qing Y, Bin X, Jian W, Li G, Linhui W, Bing L, et al. Adrenal ganglioneuromas: A 10-year experience in a Chinese population. Surgery. 2010;147:854–860.
- Rondeau G, Nolet S, Latour M, Braschi S, Gaboury L, Lacroix A, et al. Clinical and biochemical features of seven adult adrenal ganglioneuromas. J Clin Endocrinol Metab. 2010;95:3118–3125.
- Lora M.S, Waguespack SG, Moley JF, Walvoord EC. Adrenal ganglioneuromas in children with multiple endocrine neoplasia type 2: A report of two cases. J Clin Endocrinol Metab. 2005;90:4383–4387.
- Chen CL, Huang ST, Chang PL, Ng KF. Adrenal ganglioneuroma: Report of five cases. Chang Gung Med J. 2000;23:550–554.
- Tarantino RM, Lacerda AM, Cunha Neto SH, Violante AH, Vaisman M. Adrenal ganglioneuroma. Arq Bras Endocrinol Metabol. 2012;56:270–274.
- Linos D, Tsirlis T, Kapralou A, Kiriakopoulos A, Tsakayannis D, Papaioannou D. Adrenal ganglioneuromas: Incidentalomas with misleading clinical and imaging features. Surgery. 2011;149:99–105.
- Jain M, Shubha BS, Sethi S, Banga V, Bagga D. Retroperitoneal ganglioneuroma: Report of a case diagnosed by fine-needle aspiration cytology, with review of the literature. Diagn Cytopathol. 1999;21:194–196.
- Zografos GN, Kothonidis K, Ageli C, Kopanakis N, Dimitriou K, Papaliodi E, et al. Laparoscopic resection of large adrenal ganglioneuroma. JSLS. 2007;11:487–492.
- Sucandy I, Akmal YM, Sheldon DG. Ganglioneuroma of the adrenal gland and retroperitoneum: A case report. N Am J Med Sci. 2011;3:336–338.
- Yamaguchi K, Hara I, Takeda M, Tanaka K, Yamada Y, Fujisawa M, et al. Two cases of ganglioneuroma. Urology. 2006;67:622.e1–622.e4.
- Maweja S, Materne R, Detrembleur N, de Leval L, Defechereux T, Meurisse M, et al. Adrenal ganglioneuroma. A neoplasia to exclude in patients with adrenal incidentaloma. Acta Chir Belg. 2007;107:670–674.
- Reddy S. JR., Purushottam G, Pandurangarao K, Ravi Chander PT. Para aortic ganglioneuroma presenting as Cushing's syndrome. Indian J Urol. 2007;23:471–473.
- Tosaka A, Ando M, Arisawa C, Okano T. Endocrinologically active retroperitoneal ganglioneuroma with positive iodine-131-metaiodobenzylguanidine scintigraphy. Int J Urol. 1999; 6:471–474.
- Koch CA, Brouwers FM, Rosenblatt K, Burman KD, Davis MM, Vortmeyer AO, et al. Adrenal ganglioneuroma in a patient presenting with severe hypertension and diarrhea. Endocr Relat Cancer. 2003;10:99–107.
- Corcuff JB, Deminiere C, Trouillas J, Puel O, Perel Y, Barat P. Ectopic Cushing's syndrome due to an adrenal ganglioneuroma. Horm Res Paediatr. 2010;73:405–408.
- Camelo M, Aponte LF, Lugo-Vicente H. Dopamine- secreting adrenal ganglioneuroma in a child: Beware of intraoperative rebound hypertension. J Pediatr Surg. 2012;47: E29–E32.
- Contreras LN, Budd D, Yen TS, Thomas C, Tyrrell JB. Adrenal ganglioneuroma-pheochromocytoma secreting vasoactive intestinal polypeptide. West J Med. 1991;154:334–337.
- Godlewski G, Nguyen Trong AH, Tang J, Semler-Collery R, Joujoux JM, Gaujoux AF. Virilizing adrenal ganglioneuroma containing Leydig cells. Acta Chir Belg. 1993;93:181–184.
- Lucas K, Gula MJ, Knisely AS, Virgi MA, Wollman M, Blatt J. Catecholamine metabolites in ganglioneuroma. Med Pediatr Oncol. 1994;22:240–243.
- Erem C, Ucuncu O, Nuhoglu I, Turkyilmaz S, Yildiz K, Civan N, et al. Large adrenocortical oncocytoma with uncertain malignant potential: Report of a new case and review of the literature. Acta Endocrinol-Bucharest. 2012;8:295–306.
- Celik V, Ünal G, Özgültekin R, Göksel S, Ünal H, Çerçel A. Adrenal ganglioneuroma. Br J Surg. 1996;83:263.
- Taylor AR, Chulajata D, Jones DH, Whitwam JG. Adrenal tumour secreting vasoactive intestinal peptide and noradrenaline. Anaesthesia. 1977;32:1012–1016.
- Aguirre P, Scully RE. Testosterone-secreting adrenal ganglioneuroma containing Leydig cells. Am J Surg Pathol. 1983; 7:699–705.
- Gültekin M, Dursun P, Salman C, Ozyüncü O, Saglam A, Küçükali T, et al. Ganglioneuroma mimicking ovarian tumor: A report of a case and review of the ganglioneuromas. Arch Gynecol Obstet. 2005;271:66–68.
- Duffy S, Jhaveri M, Scudierre J, Cochran E, Huckman M. MR imaging of a posterior mediastinal ganglioneuroma: Fat as a useful diagnostic sign. AJNR Am J Neuroradiol. 2005;26:2658–2662.
- Ichikawa T, Ohtomo K, Araki T, Fujimoto H, Nemoto K, Nanbu A, et al. Ganglioneuroma: Computed tomography and magnetic resonance features. Br J Radiol. 1996;69:114–121.
- Lonergan GJ, Schwab CM, Suarez ES, Carlson CL. Neuroblastoma, ganglioneuroblastoma, and ganglioneuroma: Radiologic-pathologic correlation. Radiographics. 2002;22: 911–934.
- Bing-bing S, Han-Zong L, Cheng C, Shi R, Hua F, Jin W, et al. Differential diagnosis and laparoscopic treatment of adrenal pheochromocytoma and ganglioneuroma. Chin Med J. 2009;122:1790–1793.
- Zhang Y, Nishimura H, Kato S, Fujimoto K, Ohkuma K, Kojima K, et al. MRI of ganglioneuroma: Histologic correlation study. J Comput Asist Tomogr. 2001;25:617–623.
- Fassnacht M, Kenn W, Allolio B. Adrenal tumors: How to establish malignancy?J Endocrinol Invest. 2004;27: 387–399.
- Lo CY, Lam KY, Wat MS, Lam KS. Adrenal pheochromocytoma remains a frequently overlooked diagnosis. Am J Surg. 2000;179:212–215.
- Gilshtein H, Peled Z, Grunner S, Fischer D, Kakiashvili E, Kluger Y. Ganglioneuroma of the adrenal gland: A rare tumor in a rare location. Case Rep Oncol. 2012;5:487–489.
- Carrión López P, Martínez Ruiz J, Martínez Sanchiz C, Perán Teruel M, Atienzar Tobarra M, Donate Moreno MJ, et al. Adrenal ganglioneuroma. Arch Esp Urol. 2012;65: 773–776.
- Ghali VS, Gold JE, Vincent RA, Cosgrove JM. Malignant peripheral nerve sheath tumor arising spontaneously from retroperitoneal ganglioneuroma: A case report, review of the literature, and immunohistochemical study. Hum Pathol. 1992;23:72–75.
- Kulkarni AV, Bilbao JM, Cusimano MD, Muller PJ. Malignant transformation of ganglioneuroma into spinal neuroblastoma in an adult. Case report. J Neurosurg. 1998;88: 324–327.