Abstract
A 70-year-old man with uremia was referred because of hemoptysis. A chest X-ray showed diffuse infiltration in the right lung field. Laboratory data were remarkable for renal failure, anemia, and thrombocytopenia. Furthermore, laboratory evidence of microangiopathic hemolytic anemia was present. A kidney biopsy revealed diffuse crescentic glomerulonephritis with linear staining of IgA along the glomerular basement membrane (GBM). No thrombotic microangiopathy was noted on renal biopsy. Circulating IgG anti-GBM antibody was not detected, and IgA anti-GBM antibody was not tested. The patient was treated with plasmapheresis and pulse steroid therapy, which resulted in an immediate improvement in the pulmonary hemorrhage and hematological abnormalities. However, the patient did not regain renal function and remained on hemodialysis.
INTRODUCTION
Anti-glomerular basement membrane (GBM) disease is a rare but aggressive form of glomerulonephritis (GN) mediated by circulating autoantibodies reactive with the GBM. These antibodies bind in a linear pattern to the non-collagenous domain of the α3 chain of type IV collagen of GBM.Citation1 The immunoglobulins are almost exclusively of the IgG subtype. The occurrence of GN characterized by linear staining of non-IgG class immunoglobulin along the GBM is quite rare, with only 13 previously reported cases in the worldwide literature, including 11 IgA, 2 IgM, and 1 lambda light chain.Citation2–12 Herein, we report an unusual case of IgA anti-GBM disease presenting with rapidly progressive GN, pulmonary hemorrhage (PH), and microangiopathic hemolytic anemia (MAHA).
CASE REPORT
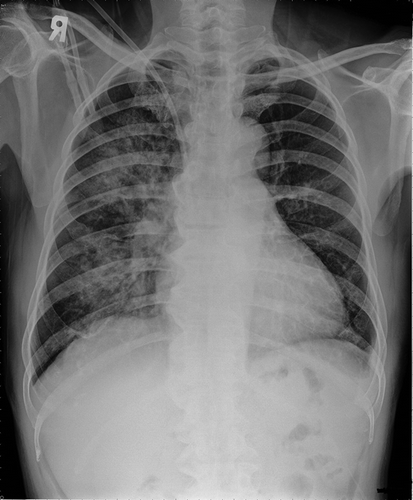
A 70-year-old man who had been diagnosed with uremia with undetermined cause and had begun hemodialysis for 3 months was referred to our hospital because of hemoptysis. He had a previous history of hypertension. Renal dysfunction was first detected at a local hospital in August 2010, with a serum creatinine level of 2.8 mg/dL. Urinalysis at that time showed hematuria and proteinuria. The second report of renal function test was recorded at the same hospital in March 2011, with a serum creatinine level of 4.0 mg/dL and urine protein-to-creatinine ratio of 2283 mg/g. The patient was lost to follow-up until he presented to another hospital in August 2011 because of uremia requiring renal replacement therapy (unavailable data of renal function test). Then, he was put into hemodialysis program. The patient was referred to our hospital in November 2011 with a 1-week history of hemoptysis. A chest X-ray showed diffuse infiltration in the right lung field (). There was no fever, arthralgia, or skin lesion. The patient had clear consciousness. Crackles were heard in the right basal lung field. Laboratory studies revealed the following: white blood cell count of 5600/μL, hemoglobin of 6.4 g/dL, hematocrit of 18.1%, platelet count of 31,000/μL, corrected reticulocyte count of 4.1%, lactate dehydrogenase of 850 U/L, haptoglobin of <7.2 mg/dL, creatinine of 9.99 mg/dL, total protein of 5.1 g/dL, and albumin of 2.7 g/dL. Schistocytes were detected on peripheral smear. Prothrombin time and partial thromboplastin time were normal. Coombs’ tests were negative. The patient had been nearly anuric, and his urinalysis showed protein of 300 mg/dL and red blood cell of >100/high-power field. Serum complements C3 and C4 were 86.1 mg/dL (normal range 90–180 mg/dL) and 26.2 mg/dL (normal range 10–40 mg/dL), respectively. Serology was negative for antinuclear antibody, antineutrophil cytoplasmic antibody (ANCA), and IgG anti-GBM antibody (using indirect immunofluorescence). Tests for IgA anti-GBM were unavailable. Cryoglobulin was not detected. Microbiological studies of sputum specimens collected by spontaneous expectoration were negative for bacteria, fungi, and mycobacteria.
Figure 1. A chest X-ray on admission showing diffuse infiltration in the right lung field.
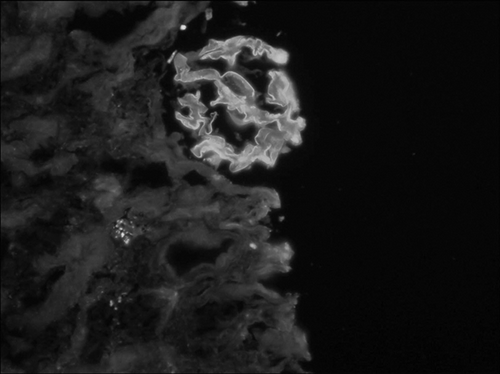
With a presumed diagnosis of pulmonary-renal syndrome, a kidney biopsy was performed on the 7th hospital day. The specimen contained 23 glomeruli, of which 18 were globally sclerotic. Of the remaining five viable glomeruli, three had fibrocellular crescents and two had fibrous crescents. No thrombotic microangiopathy was noted. Immunofluorescence studies revealed staining of IgA (2+) and IgG (1+) in a linear pattern along the peripheral capillary loops (). Immunofluorescence staining was negative for IgM, C3, and C1q. No electron-dense deposits were identified on electron microscopy. The findings of renal biopsy suggested anti-GBM disease. The patient was treated with two sessions of plasmapheresis and pulse methylprednisolone (500 mg/day × 3 days) followed by oral prednisone (1 mg/kg/day) and oral cyclophosphamide (1 mg/kg/day), which resulted in an immediate improvement in the PH and hematological abnormalities. However, the patient did not regain renal function and remained on hemodialysis.
Figure 2. Immunofluorescence studies of renal biopsy showing 2+ staining of IgA in a linear pattern along the peripheral capillary loops.
DISCUSSION
Pulmonary-renal syndrome is defined as a combination of PH and GN. A variety of autoimmune disorders can cause this syndrome. Similar to the differential diagnosis of rapidly progressive GN, the immunopathology of pulmonary-renal syndrome comprises three major categories: anti-GBM antibody-mediated disease, which has a linear immunoglobulin deposits; immune complex-mediated diseases, which have granular immune deposits; and ANCA-associated diseases, which have a paucity of immune deposits. In our patient, the presence of significant IgA deposition in a linear pattern along the GBM suggested an anti-GBM antibody-mediated mechanism.
A rare form of anti-GBM GN mediated by IgA autoantibodies has been described in 11 reports that are reviewed and summarized in . Anti-GBM antibodies may be found in the circulation by indirect immunofluorescence or radioimmunoassay. However, the circulating anti-GBM antibodies may be transitory such that the antibodies are not always detected. Furthermore, standard assays for circulating anti-GBM antibodies are designed to detect only IgG. Therefore, serum containing a non-IgG class antibody would be reported as negative by standard assays, and IgA anti-GBM antibodies need to be tested separately. In addition, the specificity of IgA anti-GBM antibodies has seldom been characterized, and it is uncertain whether the epitope of α3(IV) collagen specific to IgG anti-GBM antibodies is the target. In previous reports, one patient with recurrent anti-GBM disease had a monoclonal IgA1-kappa antibody targeting collagenase-sensitive epitopes within α1/α2(IV) collagen,Citation13 and another patient with crescentic GN and subepidermal blisters developed IgA autoantibodies against the α5 and α6(IV) collagen.Citation11
Table 1. Clinical and histological features of patients with IgA form of anti-GBM GN.
The spectrum of renal manifestations in IgA anti-GBM GN ranges from isolated hematuria with normal renal function to rapidly progressive renal failure requiring renal replacement therapy. Hypocomplementemia was present in two of nine patients with available data. There is also a broad spectrum of glomerular histological findings in IgA anti-GBM GN, including normal glomeruli,Citation5 focal mesangial GN,Citation5 thin basement membrane glomerulopathy,Citation7 focal segmental glomerulosclerosis,Citation9 focal necrotizing and crescentic GN,Citation2,3,10,12 and diffuse crescentic GN.Citation8,11
Organ damage in anti-GBM disease can be confined to the kidneys, the lungs, or both. In the classic IgG form of anti-GBM disease, the spectrum of clinical presentations ranges from isolated renal disease without overt pulmonary involvement in approximately 35% of patients to PH without renal involvement in <5% of patients, and PH accompanies GN in approximately 60% of patients. There have been reports of four cases, including ours, of IgA anti-GBM disease with pulmonary involvement, of which all concomitantly had clinical signs of renal involvement.Citation2,3,9
MAHA has been reported also in association with anti-GBM disease.Citation14,15 Stave et al.Citation14 reported a series of 12 patients with anti-GBM disease, of which 6 had clinical laboratory evidence of MAHA and 6 had histological evidence of thrombotic microangiopathy on renal biopsy. Notably, there were two patients who had MAHA alone without thrombotic microangiopathy on renal biopsy, as was the case with our patient. In some instances, anti-GBM disease coincided with thrombotic thrombocytopenic purpura.Citation16–18 It has been speculated that interleukin-12 might be involved in the common pathogenesis of both anti-GBM disease and thrombotic thrombocytopenic purpura.Citation19
In conclusion, similar to the classic IgG form of anti-GBM disease, IgA variant of anti-GBM GN could be associated with PH. Early diagnosis of the disease responsible for pulmonary-renal syndrome is crucial because of the danger of irreversible loss of renal function or mortality from fulminant PH, which could be prevented in many cases by appropriate treatment. Furthermore, we describe the first case of IgA anti-GBM disease concomitant with MAHA.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Borza DB, Neilson EG, Hudson BG. Pathogenesis of Goodpasture syndrome: A molecular perspective. Semin Nephrol. 2003;23:522–531.
- Border WA, Baehler RW, Bhathena D, Glassock RJ. IgA antibasement membrane nephritis with pulmonary hemorrhage. Ann Intern Med. 1979;91:21–25.
- Espinosa-Meléndez E, Forbes RD, Hollomby DJ, Ahuja J, Katz MG. Goodpasture’s syndrome treated with plasmapheresis. Report of a case. Arch Intern Med. 1980;140:542–543.
- Savage CO, Pusey CD, Bowman C, Rees AJ, Lockwood CM. Antiglomerular basement membrane antibody mediated disease in the British Isles 1980–4. Br Med J. 1986;292:301–304.
- Fivush B, Melvin T, Solez K, McLean RH. Idiopathic linear glomerular IgA deposition. Arch Pathol Lab Med. 1986;110:1189–1191.
- Savige JA, Yeung SP, Bierre AR, Kincaid-Smith P. Lambda-light-chain-mediated anti-GBM disease. Nephron. 1989;52: 144–148.
- de Caestecker MP, Hall CL, MacIver AG. Atypical antiglomerular basement membrane disease associated with thin membrane nephropathy. Nephrol Dial Transplant. 1990;15:909–913.
- Gris P, Pirson Y, Hamels J, Vaerman JP, Quoidbach A, Demol H. Antiglomerular basement membrane nephritis induced by IgA1 antibodies. Nephron. 1991;58:418–424.
- Fervenza FC, Terreros D, Boutaud A, . Recurrent Goodpasture’s disease due to a monoclonal IgA-kappa circulating antibody. Am J Kidney Dis. 1999;34:549–555.
- Shaer AJ, Stewart LR, Cheek DE, Hurray D, Self SE. IgA antiglomerular basement membrane nephritis associated with Crohn’s disease: A case report and review of glomerulonephritis in inflammatory bowel disease. Am J Kidney Dis. 2003;41: 1097–1109.
- Ghohestani RF, Rotunda SL, Hudson B, . Crescentic glomerulonephritis and subepidermal blisters with autoantibodies to alpha5 and alpha6 chains of type IV collagen. Lab Invest. 2003;83:605–611.
- Ho J, Gibson IW, Zacharias J, Fervenza F, Colon S, Borza DB. Antigenic heterogeneity of IgA anti-GBM disease: New renal targets of IgA autoantibodies. Am J Kidney Dis. 2008;52: 761–765.
- Borza DB, Chedid MF, Colon S, Lager DJ, Leung N, Fervenza FC. Recurrent Goodpasture’s disease secondary to a monoclonal IgA1-kappa antibody autoreactive with the alpha1/alpha2 chains of type IV collagen. Am J Kidney Dis. 2005;45:397–406.
- Stave GM, Croker BP. Thrombotic microangiopathy in anti-glomerular basement membrane glomerulonephritis. Arch Pathol Lab Med. 1984;108:747–751.
- Stallworthy E, Yehia M. Thrombotic microangiopathy in a patient with anti-glomerular basement membrane antibody disease. Nephrology (Carlton). 2006;11:375–376.
- Terryn W, Benoit D, Van Loo A, . Goodpasture’s syndrome associated with autoimmune thrombotic thrombocytopenic purpura an unusual case. Nephrol Dial Transplant. 2007;22: 3672–3673.
- Torok N, Niazi M, Al Ahwel Y, Taleb M, Taji J, Assaly R. Thrombotic thrombocytopenic purpura associated with anti-glomerular basement membrane disease. Nephrol Dial Transplant. 2010;25:3446–3449.
- Watanabe H, Kitagawa W, Suzuki K, . Thrombotic thrombocytopenic purpura in a patient with rapidly progressive glomerulonephritis with both anti-glomerular basement membrane antibodies and myeloperoxidase anti-neutrophil cytoplasmic antibodies. Clin Exp Nephrol. 2010;14:598–601.
- Shin JI, Park SJ, Kim JH. Is there a link between thrombotic thrombocytopenic purpura and anti-glomerular basement membrane disease? Nephrol Dial Transplant. 2011;26:2058–2059.