Abstract
Fabry disease—a genetic disorder characterized by the accumulation of globotriaosylceramide in cell lysosomes resulting from an X-linked deficiency of α-galactosidase A activity—presents with multiorgan manifestations, including progressive renal disease. Recently, its prevalence has been reported to be higher in hemodialysis (HD) patients than in the general population. We, therefore, examined patients on maintenance dialysis living in the Nagasaki Prefecture, Japan, to clarify the prevalence of Fabry disease. We screened 933 patients on maintenance dialysis, who were residents of Nagasaki Prefecture in Japan, for α-galactosidase A activity using a dried blood spot on filter paper. Patients with low α-galactosidase A activity were clinically assessed; subsequently, genetic analysis of the α-Galactosidase A gene (MIM:30064) was performed in these patients. Of the 933 patients, 55 had low α-galactosidase A activity; of these, one male and two females had α-Galactosidase A mutations. The prevalence of Fabry disease was thus 0.32%, which was similar to that reported previously. However, one mutation was newly identified, while the E66Q mutation observed in two patients was as previously identified. These two patients with the E66Q mutation were excluded because of the possibility of polymorphism; the prevalence of Fabry disease in the HD population was finally calculated to be 0.11%. The prevalence of Fabry disease in patients on maintenance dialysis living in Nagasaki Prefecture was 0.32%. Dried blood spot screening was considered as a simple and effective method for screening patients on maintenance dialysis for Fabry disease.
INTRODUCTION
Fabry disease is an X-linked recessive lysosomal storage disorder caused by mutations in α-Galactosidase A (α-Gal A) gene (MIM:30064) that lead to deficient activity of this enzyme.Citation1 Absence or reduced activity of α-galactosidase A (α-Gal A) leads to the intracellular accumulation of glycosphingolipids, mainly globotriaosylceramide, in various tissues.Citation2 Distinctive cytoplasmic inclusions can be observed in renal epithelial cells, endothelial cells, pericytes, vascular smooth muscle cells, cardiomyocytes, and neurons of the autonomic nervous system.Citation3 The clinical manifestations of Fabry disease differ between the classic and variant forms in hemizygotes. Male patients with the classic form present with acroparesthesia, hypohidrosis, corneal opacities, stroke, cardiac abnormalities, and renal disorders. The mortality rate in patients with the classic form is extremely high. The variant form of Fabry disease presents with manifestations limited to the heartCitation4–6 or kidneysCitation7–9 with few or none of the clinical symptoms observed in the classic form. These patients have relatively higher plasma α-Gal A activity and a milder phenotype than those with the classic form. However, most of such patients are at risk for end-stage renal disease (ESRD), eventually requiring renal replacement therapy. It has been reported that the prevalence of Fabry disease is 1 in 40,000–117,000 males.Citation3,10 Because not all patients undergo renal biopsy before renal replacement therapy, the above-mentioned prevalence may be lower than the actual prevalence. To date, several reports have indicated that in dialysis patients, the prevalence of Fabry disease may be as high as 0.5–1.2%.Citation11–17 Furthermore, it appears that the prevalence of Fabry disease in females is also much higher among hemodialysis (HD) patients. Because specific enzyme replacement therapy (ERT) is now available for patients with Fabry disease,Citation18 the identification of such patients is critical.
This study aimed to define the prevalence of Fabry disease in patients from the Nagasaki Prefecture, Japan, undergoing chronic HD or continuous ambulatory peritoneal dialysis (CAPD) by a screening test using a dried blood spot on filter paper. In addition, we also examined the clinical characteristics of these patients and investigated the mutations in the α-Gal A gene in patients with confirmed low enzyme activity.
MATERIALS AND METHODS
Study Population and Protocol
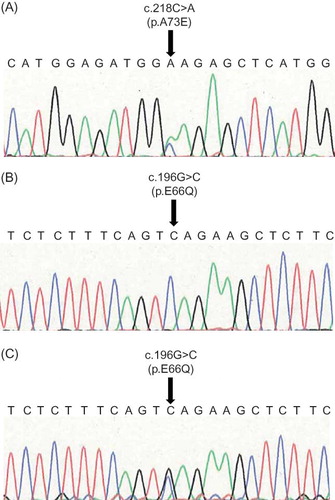
Our selection criterion was patients on maintenance dialysis (HD and CAPD) at Nagasaki University Hospital and its affiliated hospitals in Nagasaki Prefecture, Japan. After obtaining informed consent, we screened 933 patients (557 men and 376 women) on maintenance dialysis from December 2006 to March 2009. The patients we screened in this study account for approximately a quarter of all maintenance dialysis patients in Nagasaki. This study was designed with the following objectives: (1) to screen patients on maintenance dialysis for Fabry disease by an enzyme activity assay using a dried blood spot, (2) to confirm positive blood spot test results by repeated assays, and (3) to confirm the diagnosis of Fabry disease by molecular analysis of the α-Gal A gene. The study protocol was approved by the Human Ethics Review Committee of Nagasaki University School of Medicine, and written informed consent was obtained from each subject. The results of the enzyme activity assay were directly communicated to the patient by the doctor. We provided genetic information regarding Fabry disease to the patients in whom the deficiency of α-Gal A activity was confirmed; genotyping was performed for those patients who provided written informed consent. Once diagnosed with Fabry disease, the patient was referred to the genetic counselor of the Nagasaki University Hospital for genetic counseling if the patient desired the same.
Measurement of α-Galactosidase A Activity
Venous blood was collected from patients before the initiation of renal replacement therapy. Four drops of blood were spotted on filter paper, allowed to dry at room temperature, and stored at 2–4°C until they were sent to Kumamoto University (Kumamoto, Japan) for analysis. Blood spot α-Gal A activity was determined using a fluorescent substrate, as previously described by Chamoles et al.Citation19 Briefly, 40 μL of McIlvaine buffer (0.1 M citrate:0.2 M NaH2PO4, 36.8:63.2; pH 6.0) was added to 96-microwell plates. Then, 3 mm punched dried blood spots were added to the buffer and processed for extraction at room temperature for 2 h. Subsequently, 30 μL of this blood extract was transferred into another 96-microwell plate, and 100 μL of the reaction mixture (3.5 mM 4-MU galactosylpyranoside, 100 mM citrate, 200 mM phosphate, 100 mM N-acetylgalactosamine) was added to each well and incubated at 37°C for 24 h. The reaction was terminated with 150 μL of termination solution (300 mM glycine, NaOH, pH 10.6) immediately after the reaction. The fluorescence intensity from the 4-methylumbelliferones in the wells was measured at 450 nm using a fluorescent plate reader (BIO-TEK, Winooski, VT, USA). One unit (Agal U) of enzymatic activity was equal to 0.34 pmol of 4-methylumbelliferyl-d-galactopyranoside cleaved per hour per disc. Samples with activities <20 Agal U were retested by the same method. Samples with activities less than the same cut-off were considered as “screening positive.” Patients showing low α-Gal A activity in both assays were clinically assessed, and a genetic study of the α-Gal A gene was performed, provided that the patient provided consent.
Genetic Study of α-Galactosidase A Gene
Genomic DNA was extracted from peripheral blood leucocytes for α-Gal A mutation analyses.Citation20 DNA regions of the α-Gal A gene were analyzed by polymerase chain reaction after amplifying each of the even α-Gal A exons and sequencing the opposite strand.
Statistical Analysis
Values are presented as the mean ± standard deviation (SD).
RESULTS
α-Galactosidase A Activity and Genetic Analysis
The average α-Gal A activity of the patients was 23.4 ± 15.4 Agal U. The first assay revealed 178 of the 933 dialysis patients to have low plasma α-Gal A activity (14.6 ± 3.3 Agal U), while the mean enzyme activity of 755 patients was within the normal range at 26.0 ± 16.3 Agal U. Among the 178 patients with low activity, 55 (24 men and 31 women) were screened for low enzyme activity by a repeated assay using the dried blood spot (14.0 ± 3.0 Agal U). We performed genetic analysis in 36 patients who consented to the same. The results revealed missense mutations in one male and two females ( and ). The average α-Gal A activity of these three patients was 11.9 ± 3.3 Agal U, while that of the remaining 33 patients was 14.9 ± 2.6 Agal U. Patients 2 and 3 had the same mutation (c.196G>C, p.E66Q) that has been described previously.11,21 Patient 1 had a missense mutation (c.218C>A, p.A73E) that, to the best of our knowledge, has not been previously reported.
Figure 1. Flow chart for screening dialysis patients for Fabry disease.
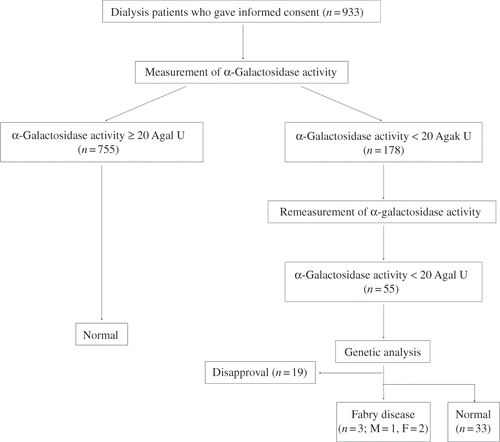
Figure 2. Direct nucleotide sequencing of PCR-amplified DNA from the α-Gal A gene. (A) Sense strand of the DNA from patient 1. Arrow indicates nucleotide 218, where a C>A transversion resulted in the amino acid substitution p.Ala73Glu. (B, C) Sense strands of the DNA from patient 2 (B) and patient 3 (C). Arrow indicates nucleotide 196, where a G>C transversion resulted in the amino acid substitution p.Glu66Gln.
Clinical Evaluation of the Patients
shows the clinical characteristics of the three patients newly diagnosed with Fabry disease. Patient 1 was diagnosed with ESRD in 2006 and with initiation of CAPD and diagnosis of left ventricular hypertrophy (LVH) in 2007. The patient’s mother had a history of renal disease and the elder brother suffered from hearing impairment. In 2008, the patient suffered a stroke, and the dialysis modality was changed from CAPD to HD. Unfortunately, the patient refused to undergo ERT; the consent for having her family screened for Fabry disease was also refused.
Table 1. Patient characteristics.
Patient 2 was diagnosed with focal segmental glomerulosclerosis by renal biopsy in 1976; CAPD was started in 1995. At 2 months after CAPD initiation, the dialysis modality was changed to HD because of fungal peritonitis. Sequence analysis of the α-Gal A gene in this patient revealed that his two daughters also had the same missense mutation (E66Q). The patient accepted ERT and has suffered no side effects thus far.
Patient 3 had a history of acute myocardial infarction in 1998 with renal insufficiency diagnosed. She was started on maintenance HD in 2001. Subsequent to genetic analysis, the patient is undergoing ERT with no side effects reported thus far. Family screening and genetic analysis also showed the same heterozygous mutation in her two daughters.
DISCUSSION
Fabry disease is considered as a rare disorder, with the estimated prevalence of classic hemizygous disease being 1 in 40,000–117,000 males (0.0025–0.00085%).Citation3,10 However, Fabry disease is reported as the cause of ESRD in 0.0167% and 0.0188% of dialysis patients in the United States.Citation16 To date, a few variant types of Fabry disease have been identified; their manifestations are primarily limited to the heart, kidneys, or brain.Citation8,20–24 It is, however, very difficult to identify Fabry disease in dialysis patients because patients with variants of Fabry disease often lack symptoms that are observed in the classic form. Recently, several studies have investigated the prevalence of Fabry disease in dialysis patients. Kotanko et al.Citation14 found Fabry disease in 4 out of 2480 (0.16%) Austrian patients on maintenance dialysis. More recently, Merta et al.Citation15 found Fabry disease in 5 out of 3370 (0.15%) HD patients in the Czech Republic. Based on the findings of these studies, the prevalence of Fabry disease in dialysis patients appears to be 10–50 times higher than that in the general population. Further, the prevalence of Fabry disease among Japanese patients on maintenance dialysis has been reported to be 0.16–1.2%.Citation8,11,12,17
In our study, we examined the prevalence of Fabry disease in patients from the Nagasaki Prefecture undergoing HD or CAPD, assessing a dried blood spot on filter paper in the screening test. Among 933 patients, 3 patients were diagnosed with Fabry disease (0.32%); this prevalence was similar to that reported previously for dialysis patients.Citation8,11,12,17 We believe that there are no regional idiosyncrasies of Fabry disease in terms of prevalence rate or inherited mutations, which can be explained by the fact that Fabry disease is panethnic. In our study, one patient had the novel A73E mutation, while two patients had the previously reported E66Q mutation. All three patients had low α-Gal A activity, and Fabry disease had not been previously diagnosed in any of them. In addition, the E66Q patients had no symptoms of Fabry disease apart from renal failure and LVH. The family members (children) of these two patients, who were asymptomatic, were also identified as heterozygotes for Fabry disease. Previous reports included patients with E66Q mutations, indicating the substitution of glutamine for glutamic acid at residue 66;Citation11,17,21 this mutation is currently under debate as to whether it is a disease-caused mutation or not. Lee et al. Citation25 reported that the allele frequency of E66Q was approximately 1% among 833 newborns in Korea, suggesting that E66Q is a polymorphism because no globotriaosylceramide is found in patients with E66Q.The current definition of Fabry disease is a measured decrease or deficit of α-Gal A enzyme activity or the detection of any mutation of the α-Gal A gene. In view of this definition, patients with E66Q should be included among those with Fabry disease. Thus, the frequency of Fabry disease in this study would be 0.32% if all the three patients were included. However, if E66Q were indeed a polymorphism, the previously reported prevalence of Fabry disease would need to be reviewed. After excluding the E66Q mutation, the prevalence of Fabry disease in our HD patients would be 0.11% (1/933 patients).
α-Gal A can be measured in a variety of materials such as dried whole blood spots, blood leukocytes, serum, or plasma. Since approximately 300,000 patients are under maintenance dialysis in Japan, large-scale screening of all of these patients would be difficult. However, diagnosis of this condition is important because an efficacious treatment—ERT—is now available. Therefore, it is important to have an easy, safe, economical, and simple method for the screening of Fabry disease. Most of the previous screening studies used a plasma α-Gal A assay, and others measured α-Gal A activity in leukocytes. The dried blood spot test, which we used in the present study, is easy to carry out because it can be performed directly using whole blood, which only needs to be dried, and it is inexpensive, whereas serum or plasma must be centrifuged and frozen to be sent for testing, the resulting cost of which would be higher than that of the dried blood spot test. Therefore, we believe that the dried blood spot test is a useful and simple screening tool for the detection of Fabry disease; hence, we used dried blood spots for the α-Gal A test in our study. Recently, however, it has been reported that the average α-Gal A activity in dried blood spot samples prepared using EDTA tubes was higher when compared with those spotted directly irrespective of disease status.Citation26 Therefore, further studies would be necessary for validating this assay to determine the best method for screening Fabry disease.
ERT is now available for patients with Fabry disease in many countries. ERT can delay disease progression in the heart and kidneys; however, early diagnosis prior to the onset of irreversible pathologic changes is essential for successful treatment. In our study, two out of the three patients diagnosed with Fabry disease were females. Fabry disease management guidelines in 2006 recommended that female patients should be offered ERT if they manifest significant symptoms or show evidence of progressive organ involvement.Citation27 Even for patients on maintenance dialysis, it is crucial to anticipate and treat the manifestations of cardiac and cerebrovascular involvements.
In conclusion, we consider the dried blood spot test to be a useful and simple screening tool for the detection of Fabry disease. It is important to identify patients with Fabry disease and the complications of the disease as early as possible for an early intervention, such as ERT, which may delay the disease progression.
ACKNOWLEDGMENTS
The authors are grateful to the following facilities that participated in this study: Kouseikai Hospital, Maeda Clinic, Sakuramachi Clinic, Kawatomi Clinic, Isahaya Health Insurance General Hospital, Hokusho Central Hospital, Tanaka Clinic, Shinzato Clinic, Sakuramachi Hospital, and Nagasaki Municipal Medical Center.
Declaration of interest:The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry’s disease. Ceramide trihexosidase deficiency. N Engl J Med. 1967;276:1163–1167.
- Sweeley CC, Klionsky B. Fabry’s disease: Classification as a sphingolipidosis and partial characterization of a novel glycolipid. J Biol Chem. 1963;238:3148–3150.
- Desnick RJ, Ioannou YA, Eng CM. α-Galactosidase A deficiency: Fabry disease. In: Scriber CR, Beauder AL, Sly WS, ., eds. The Metabolic Bases of Inherited Disease. 8th ed. New York: Mc Graw-Hill; 2001:3733–3774.
- Nakao S, Takenaka T, Maeda M, . An atypical variant of Fabry’s disease in men with left ventricular hypertrophy. N Engl J Med. 1995;333:288–293.
- Sakuraba H, Oshima A, Fukuhara Y, . Identification of point mutations in the alpha-galactosidase A gene in classical and atypical hemizygotes with Fabry disease. Am J Hum Genet. 1990;47:784–789.
- von Scheidt W, Eng CM, Fitzmaurice TF, . An atypical variant of Fabry’s disease with manifestations confined to the myocardium. N Engl J Med. 1991;324:395–399.
- Meroni M, Spisni C, Tazzari S, . Isolated glomerular proteinuria as the only clinical manifestation of Fabry’s disease in an adult male. Nephrol Dial Transplant. 1997;12:221–223.
- Nakao S, Kodama C, Takenaka T, . Fabry disease: Detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int. 2003;64:801–807.
- Sawada K, Mizoguchi K, Hishida A, . Point mutation in the alpha-galactosidase A gene of atypical Fabry disease with only nephropathy. Clin Nephrol. 1996;45:289–294.
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254.
- Tanaka M, Ohashi T, Kobayashi M, . Identification of Fabry’s disease by the screening of alpha-galactosidase A activity in male and female hemodialysis patients. Clin Nephrol. 2005;64:281–287.
- Ichinose M, Nakayama M, Ohashi T, Utsunomiya Y, Kobayashi M, Eto Y. Significance of screening for Fabry disease among male dialysis patients. Clin Exp Nephrol. 2005;9:228–232.
- Linthorst GE, Hollak CE, Korevaar JC, Van Manen JG, Aerts JM, Boeschoten EW. Alpha-galactosidase A deficiency in Dutch patients on dialysis: A critical appraisal of screening for Fabry disease. Nephrol Dial Transplant. 2003;18:1581–1584.
- Kotanko P, Kramar R, Devrnja D, . Results of a nationwide screening for Anderson-Fabry disease among dialysis patients. J Am Soc Nephrol. 2004;15:1323–1329.
- Merta M, Reiterova J, Ledvinova J, . A nationwide blood spot screening study for Fabry disease in the Czech Republic hemodialysis patient population. Nephrol Dial Transplant. 2007;22:179–186.
- Thadhani R, Wolf M, West ML, . Patients with Fabry disease on dialysis in the United States. Kidney Int. 2002;61:249–255.
- Fujii H, Kono K, Goto S, . Prevalence and cardiovascular features of Japanese hemodialysis patients with Fabry disease. Am J Nephrol. 2009;30:527–535.
- Eng CM, Guffon N, Wilcox WR, . Safety and efficacy of recombinant human alpha-galactosidase A—replacement therapy in Fabry’s disease. N Engl J Med. 2001;345:9–16.
- Chamoles NA, Blanco M, Gaggioli D. Fabry disease: Enzymatic diagnosis in dried blood spots on filter paper. Clin Chim Acta. 2001;308:195–196.
- Nakamura K, Robertson M, Liu G, . Complete heart block and sudden death in mice overexpressing calreticulin. J Clin Invest. 2001;107:1245–1253.
- Shimotori M, Maruyama H, Nakamura G, . Novel mutations of the GLA gene in Japanese patients with Fabry disease and their functional characterization by active site specific chaperone. Hum Mutat. 2008;29:331.
- Rolfs A, Böttcher T, Zschiesche M, . Prevalence of Fabry disease in patients with cryptogenic stroke: A prospective study. Lancet. 2005;366:1794–1796.
- Chimenti C, Pieroni M, Morgante E, . Prevalence of Fabry disease in female patients with late-onset hypertrophic cardiomyopathy. Circulation. 2004;110:1047–1053.
- Monserrat L, Gimeno-Blanes JR, Marín F, . Prevalence of Fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2007;50:2399–2403.
- Lee BH, Heo SH, Kim GH, . Mutations of the GLA gene in Korean patients with Fabry disease and frequency of the E66Q allele as a functional variant in Korean newborns. J Hum Genet. 2010;55:512–517.
- Olivova P, van der Veen K, Cullen E, . Effect of sample collection on α-galactosidase A enzyme activity measurements in dried blood spots on filter paper. Clin Chim Acta. 2009;403:159–162.
- Eng CM, Germain DP, Banikazemi M, . Fabry disease: Guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006;8:539–548.