
Abstract
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis is a rare autosomal recessive renal disease caused by mutations in genes for the tight junction transmembrane proteins Claudin-16 (CLDN16) and Claudin-19 (CLDN19). We present the first case report of a Mexican family with three affected sisters carrying a p.Gly20Asp mutation in CLDN19 whose heterozygous mother showed evident hypercalciuria and normal low magnesemia without any other clinical, laboratory, and radiological symptoms of renal disease making of her an unsuitable donor. The affected sisters showed variable phenotypic expression including age of first symptoms, renal urinary tract infections, nephrolithiasis, nephrocalcinosis, and eye symptoms consisting in retinochoroiditis, strabismus, macular scars, bilateral anisocoria, and severe myopia and astigmatism. End stage renal disease due to renal failure needed kidney transplantation in the three of them. Interesting findings were a heterozygous mother with asymptomatic hypercalciuria warning on the need of carefully explore clinical, laboratory, kidney ultrasonograpy, and mutation status in first degree asymptomatic relatives to avoid inappropriate kidney donors; an evident variable phenotypic expression among patients; the identification of a mutation almost confined to Spanish cases and a 3.5 Mb block of genomic homozygosis strongly suggesting a common remote parental ancestor for the gene mutation reported.
Introduction
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC; MIM#248250) is a rare autosomal recessive disorder, characterized by renal failure due to excessive Ca2+ and Mg2+ wasting in the urine.Citation1 Two causative genes, CLDN16 (chr 3q28) and CLDN19 (Chr 1p34.2), have been identified.Citation2 In the kidney, both genes are mainly expressed in the thick ascending limb (TAL) and distal convoluted tubules.Citation2 In mammals, claudins generally localize to tight junctions (TJs), interacting each other on adjacent cells to form an ion paracellular selective barrier, playing a critical role in maintaining ionic composition of different tissue compartments. CLDN16 and CLDN19 TJs facilitate efflux of Na+ into the TAL lumen providing an electrochemical gradient driving the resorption of Ca2+ and Mg2+.Citation3 A growing number of different mutations in both genesCitation2,Citation4–10 were identified in patients with FHHNC, with variable phenotypic expression among patients with the same mutation, but with a private characteristic of CLDN19 gene mutations that differentiate them from CLDN16 mutations, the important and variable visual involvement.Citation9 Mutations in CLDN16 and CLDN19 occur throughout the coding region and can affect proper folding, intracellular trafficking of the protein, or its paracellular pore forming function.Citation11–13 Herein, we present the first case report of a Mexican family with three affected sisters carrying a p.Gly20Asp mutation in CLDN19.
Patients and methods
Patient 1 (P1)
The index case is a 33-year-old Mexican female born to the third uneventful pregnancy to non-consanguineous healthy parents. She had retinochoroiditis, eye surgery due to strabismus at age 3 and laser surgery due to myopia and astigmatism at age 26. Started with recurrent urinary tract infections (UTIs) at age 17 experiencing a nephrolithiasis lithotripsy at age 19. She was diagnosed with an end-stage renal disease (ESRD) of unknown etiology with markedly hypomagnesemia, hypercalciuria, and nephrocalcinosis at age 29. Due to her symptomatology and family history of two older sisters having a kidney transplant owed to an ESRD also of unknown etiology, she was sent to our Genetic Clinics. At that time, she was in a transplantation protocol being her mother the selected donor. However, due to an increased urinary calcium excretion she was ruled out, receiving instead a cadaveric kidney transplant.
Patient 2 (P2)
She is a 36-year-old woman product of the second uneventful pregnancy of the same parents. First symptoms were bilateral anisocoria, macular scars, and severe myopia and astigmatism. At the age of 31, she had a tonic-clonic generalized seizure. Routine laboratory analyses revealed an ESRD of unknown etiology, soon after she underwent a kidney transplant from a not related donor.
Patient 3 (P3)
She is a 38-year-old female, product of the first uneventful pregnancy of the same parents. At the age of 12, she had articular pain affecting elbows and knees receiving symptomatic treatment. Three years later started with recurrent UTIs and nephrolithiasis. Diagnosis of idiopathic ESRD was made at 33-years of age, undergoing a kidney transplant from an unrelated donor. Macular scars were found in both eyes without vision impairment.
The transplant outcome in the three sisters has been successful, without complications except from the usual expected ones during the first year following the procedure. They have normal daily life with programmed biannual follow-up after 1, 5, and 6 years post-transplantation.
Clinical and laboratory findings of the sisters and their mother are shown in . Exome sequencing of genomic DNA from the index patient using SeqCap EZ Human Exome Library v3.0 (Nimblegen, Madison, WI) and Illumina HiSeq followed by BWA mapping and GATKv2.0 variant calling revealed that she was homozygous for the CLDN19 p.Gly20Asp mutation. Sanger sequencing using primers chr1_43205676_F 5′-GCGCAGATAGCAGACTGTGA-3′ and chr1_43205676_R 5′-CTCTGCCTCTGACCCTCCTT-3′ confirmed the mutation in a homozygous state in the three sisters and in their heterozygous parents ().
Figure 1. Sanger sequencing chromatograms of chromosome 1 at position 43,205,656–43,205,696 for the index patient (a) and the mother (b) are shown. The nucleotide changes C > T at chr1:43,205,676 which result in CLDN19 p.Gly20Asp are highlighted by boxes. The chromatogram of the index patient shows homozygous T/T at chr1:43,205,676 while the chromatogram of the mother shows heterozygous C/T. Genomic coordinates are based on the February 2009 human reference sequence (hg19).
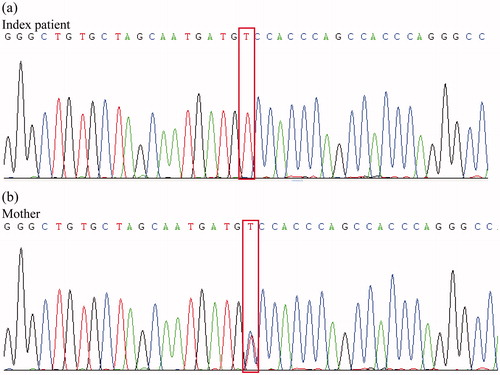
Table 1. Clinical findings and laboratory results.
Discussion
Clinical, laboratory, imagine, and molecular studies confirmed the diagnosis of FHHNC disease in the three sisters and although they share the same CLDN19 gene mutation, an evident variable phenotypic expression was observed. Differences were observed in their age of onset, 12–31 years old, the type of first symptoms presented, such as UTIs and back pain in P1 and P3 and severe myopia, astigmatism, macular scars, and anisocoria in P2 (). Post-transplantation laboratory studies showed similar magnesemia, but dissimilar calciuria and serum creatinine values. ESRD and nephrocalcinosis were common features to all of them but nephrolithiasis only observed in P1 and P3 (). CLDN19 is expressed with highest levels in the kidney and the eyeCitation2,Citation9 being both, renal and ocular manifestations in the affected sisters in agreement with previous reports.Citation2,Citation8,Citation9 The above-mentioned endorse the described phenotypic variability,Citation10 making difficult to define a clear genotype–phenotype correlation.Citation3 Interestingly, the heterozygous mother, although did not exhibited clinical and ultrasound manifestations of renal disease, showed a significant hypercalciuria even higher than their three affected daughters and normal but low reference values of serum magnesium, thus being discarded as a possible donor. Their father studies were normal.
Although FHHNC is considered a rare hereditary cause of ESRD, probably is underdiagnosed due to its variable phenotypic expression. Mutations in CLDN19 should be suspected in the presence of renal failure, nephrocalcinosis, hypomagnesemia, and hypercalciuria of unknown etiology, associated with variable severity of ocular pathology. Although the disease is usually a pediatric diagnosis, in the three sisters herein reported, etiologic diagnosis was delayed until their fourth decade of life. Interestingly the mutation identified in this family, a glycine to asparagine substitution, at position 20 (p.Gly20Asp) of the protein,Citation2,Citation8,Citation9 has been almost exclusively reported in families from Spain and Southwest France origin, where a founder effect was established.Citation9
To our knowledge, this is the first report of a Mexican family with FHHNC and although for the p.Gly20Asp mutation endogamy and a founder effect has been described for the disease parental consanguinity was denied in the family reported herein. The heterozygotes parents that share the same mutation are from different states of the country and although they ignore their Spanish descent, their surnames have a Spanish origin from relatively close regions of Spain, which is the geographical area of origin of the described founder effect in previously reported families. One of them of Basque origin was identified in Latin American people early after colonization and the other of Aragonese origin was also reported in the Latin American population, but less frequently. Moreover, a remarkable finding derived from the exome sequencing data of the index patient is a 3.5 Mb block of homozygosis that strongly suggest that the parents of the affected sisters may have a remote common ancestor.
In conclusion, the described variable clinical expression of the disease among the three sisters and the finding of a heterozygous mother with evident hypercalciuria characteristic of FHHNC suggest that probably other genetic or non-genetic modifier factors may be involved in the mechanism and development of the disease. Although renal features like nephrocalcinosis were reported in heterozygous carriers of a CLDN19 gene mutation,Citation11 we were unable to find reported families in which a heterozygous carrier showed to have an evident hypercalciuria and low normal magnesemia but no other disease symptoms. This findings alert on the need to carefully explore the FHHNC clinical, laboratory, kidney ultrasonography, and mutation status in first degree relatives of patients with CLDN19 gene mutations to avoid unsuitable kidney donors.
Acknowledgments
We thank the family members for their cooperation. W.H. is an adjunct faculty member at the Department of Physiology at the National University of Singapore and the Singapore Eye Research Institute. We thank Yao Fei for technical assistance, Tat Hung Koh for exome sequencing library construction and Foo Jia Nee and Herty Liany for bioinformatics support.
Declaration of interest
The authors declare no conflicts of interest. The results presented in this paper have not been published previously in whole or part. This work was supported by the Department of Genetics of the National Institute of Medical Sciences and Nutrition “Salvador Zubirán, México and the Agency for Science and Technology and Research (A*STAR), Singapore.
References
- Günzel D, Yu ASL. Function and regulation of claudins in the thick ascending limb of henle. Pflugers Arch. 2009;458:77–88
- Konrad M, Schaller A, Seelow D, et al. Mutations in the tight-junction gene claudin 19 (cldn19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet. 2006;79:949–957
- Hou J, Renigunta A, Konrad M, et al. Claudin-16 and claudin-19 interact and form a cation-selective tight junction complex. J Clin Invest. 2008;118:619–628
- Godron A, Harambat J, Boccio V, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: Phenotype–genotype correlation and outcome in 32 patients with cldn16 or cldn19 mutations. Clin J Am Soc Nephrol: CJASN. 2012;7:801–809
- Kang JH, Choi HJ, Cho HY, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis associated with cldn16 mutations. Pediatr Nephrol. 2005;20:1490–1493
- Peru H, Akin F, Elmas S, Elmaci AM, Konrad M. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: Report of three Turkish siblings. Pediatr Nephrol. 2008;23:1009–1012
- Seeley HH, Loomba-Albrecht LA, Nagel M, Butani L, Bremer AA. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis in three siblings having the same genetic lesion but different clinical presentations. World J Pediatrics: WJP. 2012;8:177–180
- Al-Shibli A, Konrad M, Altay W, Al Masri O, Al-Gazali L, Al Attrach I. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (fhhnc): Report of three cases with a novel mutation in cldn19 gene. Saudi J Kidney Dis Transpl: An Official Publication of the Saudi Center for Organ Transplantation, Saudi Arabia. 2013;24:338–344
- Claverie-Martin F, Garcia-Nieto V, Loris C, et al. Claudin-19 mutations and clinical phenotype in Spanish patients with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. PLoS One. 2013;8:e53151
- Naeem M, Hussain S, Akhtar N. Mutation in the tight-junction gene claudin 19 (cldn19) and familial hypomagnesemia, hypercalciuria, nephrocalcinosis (fhhnc) and severe ocular disease. Am J Nephrol. 2011;34:241–248
- Müller D, Kausalya PJ, Claverie-Martin F, et al. A novel Claudin 16 mutation associated with childhood hypercalciuria abolishes binding to ZO-1 and results in lysosomal mistargeting. Am J Hum Genet. 2003;73:1293–1301
- Müller D, Kausalya PJ, Meij IC, Hunziker W. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: Blocking endocytosis restores surface expression of a novel Claudin-16 mutant that lacks the entire C-terminal cytosolic tail. Hum Mol Genet. 2006;15:1049–1058
- Kausalya PJ, Amasheh S, Günzel D, et al. Disease-associated Claudin-16 mutants display intracellular trafficking or paracellular magnesium permeability defects. J Clin Invest. 2006;116:878–891