
Abstract
Nephrotoxicity is an important problem during methotrexate (MTX) treatment, which has been widely used for the treatment of several cancer types. Females are less susceptible to kidney diseases; however, the reason for this condition has not to be fully clarified. But sex hormones such as estrogen may have a protective effect on the kidney. We aimed to evaluate the possible protective role of estrogen on the MTX-induced renal epithelial cell death. Primary renal proximal tubular epithelial cells (RPTEC) were incubated with MTX (1, 10 and 100 μM), either alone or in combination with the 17β-estradiol, G protein-coupled estrogen receptor 1 (GPER1) agonist G-1, estrogen receptor alpha agonist propyl pyrazole triol (PPT), estrogen receptor beta agonist diarylpropionitrile (DPN). Cell viability was determined by MTT assays. Interleukin (IL)-1β, IL-6, superoxide dismutase (SOD) and malondialdehyde (MDA) levels were determined in RPTEC. Approximately half of the cell death was observed with 10 μM MTX incubation for 48 h. The cell death was prevented by co-incubating with17β-estradiol, PPT and G-1. MTX was significantly induced IL-1β and IL-6.17β-estradiol, PPT and G-1 significantly decreased effects of MTX. SOD activity was significantly decreased treatment with MTX compared to control group. SOD activity was increased with co-incubation with 17β-estradioland G-1 compared to treatment with MTX. MDA levels significantly increased in treatment with MTX compared with the control group. Increased MDA levels by MTX-induced was decreased significantly by the treatment with 17β-estradiol and G-1. These data indicate that especially 17β-estradiol and G-1 may be useful in preventing undesirable effects of MTX in renal failure.
Background
Methotrexate (MTX) is a folic acid antagonist, which is extensively used to treat a variety of disorders such as leukemia, lymphoma, osteosarcoma and autoimmune diseases. In addition, it is the most common antirheumatic drug used for the treatment of rheumatic disorders.Citation1 However, MTX toxicity limits its use. High doses of MTX have been associated with renal, gastrointestinal, hepatic and nervous toxicity.Citation2 Since almost all MTX is removed from the kidneys, nephrotoxicity is an important side effect of treatment with MTX.Citation3,Citation4
MTX-induced toxicity appears to be a consequence of the interaction of many factors: dosing schedule and length of treatment, type of disease and the presence of molecular apoptotic factors.Citation5 The generation of reactive oxygen species (ROS) and the release of proinflammatory cytokines such as interleukin (IL)-1β are considered to be responsible for MTX-induced nephrotoxicity.Citation6–8 Additionally, MTX caused free radicals released by stimulated polymorphonuclear neutrophils, leading to toxicity and cellular damage.Citation9
Estrogen-mediated biological effects are entirely manifested via the known estrogen receptors, ERβ (NR3A2) and ERα (NR3A1). These ERs belong to the steroid hormone receptor superfamily and function as ligand-activated transcription factors. However, these ERs may promote non-genomic signaling events by estrogen. Within this framework, 17β-estradiol induces estrogenic signals by binding to G protein-coupled estrogen receptor 1 (GPER-1).Citation10,Citation11
GPER1 is a recently identified G protein-coupled receptor that binds estrogen with high affinity and has been characterized as a putative membrane ER. Activation of GPER1 can result in both rapid non-genomic signaling events and transcriptional regulation.Citation12 GPER1 is expressed in human brain, liver, pancreatic, placental, blood vessel, bone, lymphoid, endometrial, ovarian, breast and lung cancer tissues.Citation13 The expression of GPER-1 has been determined in another part of the cardiovascular and renal system.Citation14–16 This receptor may be localized to the cell membrane, nucleus, endoplasmic reticulum, mitochondria or Golgi apparatus, and its effects vary depending on this specific intracellular localization.Citation13 It has been suggested that G-1 (chemical name, 1-[4-(6-bromo-1,3-benzodioxol-5-yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinolin-8-yl]-ethanone) acts a specific GPER1 agonist.Citation17
This estrogenic receptor evokes rapid non-genomic cellular effects such as mitogen-activated protein kinase activation and Ca2+ mobilization.Citation18 As genomic effects of G1, Chan et al. found that treatment with 1 μM G-1 for 48 h substantially induced phosphorylation of Erk1/2 and upregulation of p21 expression in PC-3 cell.Citation19
It has been shown that estrogens protect from oxidative damage in tissues including breast, brain and myocardium.Citation20 It is known that men inclined to progression of chronic kidney diseaseCitation21, but this situation, the basic mechanisms are not known. Some studies suggest that the progression of renal disease is slower in women compared to men.Citation22,Citation23 Furthermore, premenopausal diabetic women have a lower risk of developing renal disease compared with age-matched diabetic males, and this protection is lost after menopause.Citation23
In this study, we aimed to examine the mechanism of 17β-estradiol mediated cytoprotection in renal epithelial cells and to describe the ERs that mediating these preservative effects. We administrated MTX-induced oxidative stress in renal epithelial cells as the in vitro pattern. This research presents the actual evidence that activation of ER agonists protects renal epithelial cells from oxidative stress, which is assumed to play a role in the pathogenesis of MTX-induced nephrotoxicity.
Materials and methods
Renal epithelial cell culture
Human renal proximal tubular epithelial cells (RPTEC) were obtained from Lonza (Cologne, Germany). The RPTEC were grown in medium supplemented with renal epithelial cell growth medium Bullet Kit (Lonza) in a standard cell culture condition (humidified atmosphere with 5% CO2 maintained at 37 °C).
MTT assay
Cell viability was assessed by MTT assay (Sigma, St Louis, MO). Cells were seeded into 96-well transparent flat bottom plates (Greiner Bio-One, Frickenhausen, Germany) at a concentration of 1 × 105 cells/well for 24 h. After 1 day, the cells were handled with MTX (1, 10 and 100 μM), either alone or in combination with 100 nm 17β-estradiol (Sigma), diarylpropionitrile (DPN), propyl pyrazole triol (PPT) or G-1 (Tocris Bioscience, Bristol, UK). After 48 h of incubation, 20 μL of a solution including 4,5-dimethylthiazol-2-yl-2,5-diphenyltetrazoliumbromide (MTT, Sigma) was added to each microplate well. After 4 h, the medium was removed, then 100 μL of dimethylsulfoxide added, occurred solvable the formazan crystals was measured with an ELISA microplate reader (Biotek ELx800, Winooski, VT) at a wavelength of 570 nm.
Measurements of SOD activity and MDA levels
The cells were treated with MTX, either alone or in combination with 17β-estradiol, DPN, PPT or G-1. After incubation for 48 h, the medium was removed and then cell pellets were lysed in 50 mm phosphate buffer (PBS, pH 7.0), followed by sonication for 2 min on ice. The mixture was then centrifuged for 10 min at 14,000× g and the supernatant was assayed for enzyme activities and protein concentration. Protein levels were estimated as described by Lowry et al.Citation24 The superoxide dismutase (SOD) activity was determined with Fridovich method.Citation25 The level of malondialdehyde (MDA) was determined by thiobarbituric acid reaction according to Ohkawa et al. method.Citation26 The principle of the method is based on measuring absorbance of the pink color produced by the interaction of TBA with MDA at 532 nm.
Measurements of IL-1 and IL-6 level
The cells were treated with MTX, either alone or in combination with 17β-estradiol, DPN, PPT or G-1. After incubation for 48 h, the medium was removed and then cell pellets were lysed in 50 mm PBS (pH 7.0), followed by sonication for 2 min on ice. The mixture was then centrifuged for 10 min at 14,000× g and the supernatant was assayed forIL-1 and IL-6. IL-1 and IL-6 levels were measured by commercially available ELISA kits (eBioscience Company, Vienna, Austria).
Statistical analysis
Statistical analysis was performed by use of GraphPad Prism 3.0 (GraphPad software, San Diego, CA). For all data, statistical analysis was performed using repeated measurements of ANOVA followed by post hoc analysis with the Bonferroni test to detect differences between the groups. Results are expressed as mean and standard errors of the mean. A p value less than 0.05 was considered significant.
Results
Effects of MTX either alone or in combination with 17β-estradiol and estrogen receptor agonists on cell viability
The dose-dependent effect of MTX (1, 10 and 100 μM) demonstrated that approximately 62% of renal epithelial cells were killed at the highest concentration (100 μM), but only about 26% of renal epithelial cells died at the lowest concentration (1 μM) (. The results showed that 17β-estradiol, PPT and G-1 increased significantly the cell viability compared to treatment with MTX (. We found that DPN was increased the cell viability compared to treatment with MTX, but the difference was not statistically significant.
Figure 1. Renal proximal tubular epithelial cells were treated Methotrexate (1, 10 and 100 μM, n = 6), assessed by MTT. ***p < 0.001 versus vehicles.
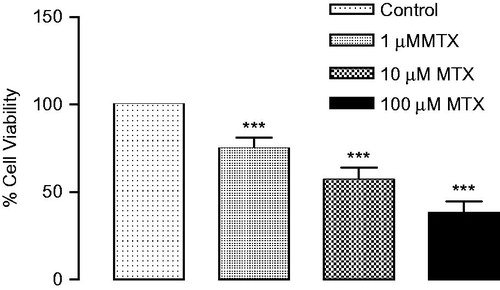
Figure 2. Renal proximal tubular epithelial cells were treated with 17β-estradiol (100 nm, n = 6), GPER-1 agonist G-1 (100 nm, n = 6), ERα agonist PPT (100 nm, n = 6), ERβ agonist DPN (100 nm, n = 6) for 30 min prior to treatment with methotrexate (10 μM, n = 6) for 48 h. Cell viability was determined by MTT assay. ***p < 0.001 versus vehicles, ++p < 0.01, +++p < 0.001 versus MTX.
Changes in cytokine levels after MTX either alone or in combination with 17β-estradiol and estrogen receptor agonists treatments on cells
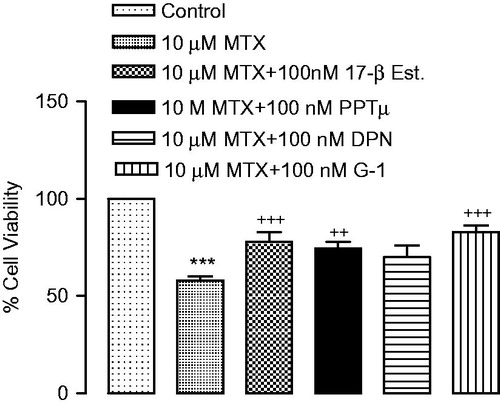
17β-estradiol, PPT and G-1 were significantly decreased the high levels of IL-1β and IL-6 induced by MTX (). IL-1β and IL-6 levels were not statistically significant between the DPN and the MTX group ().
Figure 3. (a) IL-1β and (b) IL-6 levels in renal proximal tubular epithelial cells. ***p < 0.001 versus vehicles, +p < 0.01, ++p < 0.01, +++p < 0.001 versus MTX.
Changes in SOD activity and MDA levels after MTX either alone or in combination with 17β-estradiol and estrogen receptor agonists treatments on cells
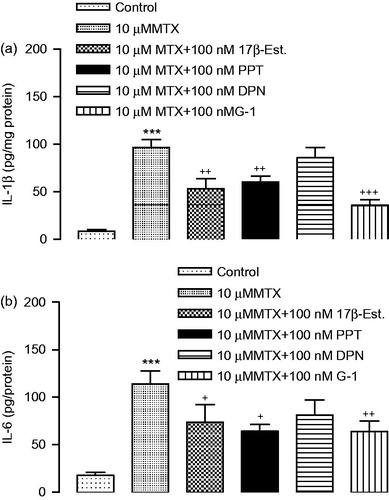
The activities of SOD were found to be dramatically decreased in the MTX-induced group compared to control group. The decreased activity of the SOD in MTX-induced cells was significantly increased by treatment with 17β-estradiol, PPT and G-1. SOD activity was not statistically significant between the DPN and the MTX group (). MDA levels significantly increased in treatment with MTX compared with the control group. Increased MDA levels by MTX-induced were decreased significantly by the treatment with 17β-estradiol and G-1 ().
Figure 4. (a) SOD and (b) MDA activities in renal proximal tubular epithelial cells. ***p < 0.001 versus vehicles, +p < 0.01, ++p < 0.01 versus MTX.
Discussion
The mechanisms of renal injury exist within a large system of signaling pathways driven by the interplay of apoptotic factors and inflammatory cytokines. We evaluated the therapeutic effects of 17β-estradiol, ER1 agonist G-1, ERα agonist PPT, ERβ agonist DPN on MTX-induced nephrotoxicity. In a preliminary study, we observed that the cell proliferation was not statistically significant the treatment of 17β-estradiol and ER agonists (100 nm) compare with control group. But, we found that 17β-estradiol and PPT increased cell proliferation approximately 10% compare to control group, DPN and G1 not effects on cell proliferation (data not shown). According to the results of our study, the cell death was the treatment of MTX combine with 17β-estradiol and ER agonists lower than alone MTX group.
We observed that the levels of IL-1β and IL-6 were increased in MTX group compared to the control. 17β-estradiol, PPT and G-1 were significantly decreased IL-1β and IL-6 induced by MTX. A protective role of estrogen in chronic renal disease such as polycystic kidney disease, hypertensive angio nephrosclerosis, chronic tubulointerstitial nephritis and chronic glomerulonephritis has been established in a meta-analysis by Neugarten et al.Citation23 Particularly, the latter two studies indicate that estradiol at low concentrations might stimulate IL-1β secretion whereas it inhibits its secretion at pregnancy levels.Citation27 It has been showed that estrogen and 17β-estradiol improved the inflammatory status in peritoneal macrophages of mice.Citation28,Citation29 However, production of IL-1β and IL-6 was significantly enhanced by endogenous estrogens.Citation29 Many studies have showed the influence of estrogen on cytokine production.Citation30 With regards to inflammatory cytokine production, the literature is incompatible with 17β-estradiol increasing or reducing secretion of IL-1β. This situation is relative to the experimental design and period of estrogen exposure, the cell models. Furthermore, there is no study on the effect of 17β-estradiol and ER agonists on the production of IL-1β and IL-6 by MTX-induced nephrotoxicity. To our knowledge, this laboratory study is the first to evaluate the protective role of estrogen on the MTX-induced renal epithelial cell death. According to our results, the 17β-estradiol ameliorates progressive nephrotoxicity mediated to ERs of GPER-1 and ERα. ER agonists such as PPT and G-1 are employed at physiological doses for replacement therapy when endogenous production is impaired. Additionally, there is undesirable effect of 17β-estradiol such as heart attacks, strokes and blood clots.Citation31,Citation32
Estrogen alone, estrogen plus progestin and progestins alone all appear to raise the risk of breast cancer.Citation33 The effects of estradiol on several more recently recognized risk markers for cardiovascular disease have been reviewed. Fibrinogen, plasma viscosity, plasminogen activator inhibitor-1, tissue plasminogen activator, insulin sensitivity, homocysteine and markers of platelet aggregation and endothelial cell activation are favorably affected by estrogen therapy. Moreover, estrogen inhibits intimal hyperplasia and smooth muscle migration, promotes angiogenesis and has antioxidant properties. Increases in factor VII, prothrombin fragments 1 and 2 and activated protein C resistance and a decrease in antithrombin III are also seen.Citation34 A recent Cochrane database systematic review, including 19 trials and 41,904 women, showed a significant increase in the risk of venous thromboembolism or a coronary event (after 1 year’s use), stroke (after 3 years’ use) and breast cancer and gallbladder diseases in relatively healthy women after continuous early or delayed endoscopic papillotomy, and an increase in all risks (thromboembolism: after 1–2 years’ use; stroke: after 3 years’ use; gallbladder disease: after 7 years’ use), except breast cancer after estrogen-only treatment.Citation35
Expression of ERα, β and GPER1 are increased in renal tubulesCitation36,Citation37, as well as in renal epithelial cells.Citation38 The ERα and β was mediated by a reduction in levels of proinflammatory cytokines induced by lipopolysaccharide.Citation39 In humans, the GPER1 is associated with low-renin hypertensionCitation40, which leads to kidney failure and vascular dysfunction.Citation14 The GPER1 activation reduces proteinuria and improves creatinine clearance despite hypertension. These findings suggest the renal protective potential for GPER1 agonists in hypertensive nephropathy.Citation12 Our results have shown the increased release of IL-1β and IL-6 to be responsible for nephrotoxicity. Also, PPT and G-1 of ER agonists prevent MTX-induced nephrotoxicity. These ER agonists may suppress the development of nephrotoxicity by decreasing the production of proinflammatory cytokines.
GPER1-dependent antiproliferative and proapoptotic effects of G-1 have been observed in prostate cancer (PC-3) and breast cancer (MCF-7) cells.Citation19,Citation41 However, GPER1-independent antiproliferative and proapoptotic effects of G-1 have been observed in ovarian cancer (KGN) and breast cancer (MDA-MB 231) cells.Citation42 GPER1 selective agents that mimic the beneficial effects of 17β-estradiol without its associated feminizing or other adverse effects could represent an important new family of drugs. Therefore, GPER1 agonist, G-1 can be used as potential therapeutic agents for the treatment of renal injuries.
Recent studies have showed that estrogen can increase mitochondrial biogenesis and function.Citation20,Citation43–45 Estrogen has been shown to substantially restrain mitochondrial production of ROS in endothelial cells.Citation46–48 The high level of reactive oxygen radicals is described as one of the significant causes of MTX-induced renal toxicity. The use of MTX causes increased levels of MDA and decreased SOD in the kidneys and in other tissues.Citation9 Researchers suggested that several antioxidants are protective in MTX-induced renal toxicity. In our study, we observed that the level of SOD was obviously decreased in the MTX group compared to the control group. Also, MDA levels was increased in the MTX. We found that MTX treatment significantly reduced levels of endogen antioxidant enzymes SOD. This decreased SOD could result in aggravated oxidative stress of the cellular structures, probably due to an imbalance of resynthesis machinery. It has been demonstrated that MTX leads to direct oxidative injury. MTX inhibits cytosolic NADP-dependent dehydrogenase and NADP-malic enzyme and causes a decrease in intracellular NADPH levels. NADPH is essential for glutathione (GSH) reductase enzyme that sustains the levels of reduced GSH, which is an important cytosolic antioxidant substance.Citation49 Thus, the reduction in the levels of GSH due to MTX leads to a weakening of the effectivity of the antioxidant defense system protecting the cell against ROS. The decreased activity of the SOD in MTX-induced cells was significantly increased by treatment with 17β-estradiol, PPT and G-1. Increased MDA levels by MTX-induced was decreased significantly by the treatment with 17β-estradiol and G-1. Our research may suggest that the protective effect of ER agonists on MTX-induced renal toxicity from their antioxidant activity.
Taylor et al. observed that the finding of specific ERβ staining to basal surface kidney collecting duct tubules and ERα is found in kidney interstitial cells.Citation50 Because ERβ and ERα are located in different residential areas of kidney, ERβ density may be lower in cells which we use the cells. Therefore, DPN may be ineffective. Consequently, cytokines, oxidant and antioxidant parameters could be not affected.
In conclusion, this study provides the concern of ER agonists in renal protection. ER agonists may be a potent blocker that inhibits cytokine release and prevents cellular death by both cytokine-mediated apoptosis and ROS formation pathways. It may have a protective effect against nephrotoxicity by regulating levels of cytokines, MDA and SOD enzyme. Our study suggests that ER1 agonist G-1 can be used as potential therapeutic agents for the treatment of renal injuries and kidney failure.
Funding information
This study was supported by Kahramanmaras Sutcu Imam University Research Foundation (2013/6-35M).
Ethical approval
All the procedures are performed in studies involving human participants were in accordance with the ethical standards of the institutional ethical committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Disclosure statement
The authors state no conflict of interest.
References
- Jolivet J, Cowan KH, Curt GA, Clendeninn NJ, Chabner BA. The pharmacology and clinical use of methotrexate. N Engl J Med. 1983;309:1094–1104.
- Kolli VK, Abraham P, Isaac B, Selvakumar D. Neutrophil infiltration and oxidative stress may play a critical role in methotrexate-induced renal damage. Chemotherapy 2009;55:83–90.
- Hempel L, Misselwitz J, Fleck C, et al. Influence of high-dose methotrexate therapy (HD-MTX) on glomerular and tubular kidney function. Med Pediatr Oncol. 2003;40:348–354.
- Izzedine H, Launay-Vacher V, Karie S, Caramella C, de Person F, Deray G. Is low-dose methotrexate nephrotoxic? Case report and review of the literature. Clin Nephrol. 2005;64:315–319.
- Neuman MG, Cameron RG, Haber JA, Katz GG, Malkiewicz IM, Shear NH. Inducers of cytochrome P450 2E1 enhance methotrexate-induced hepatocytoxicity. Clin Biochem. 1999;32:519–536.
- Ibrahim MA, El-Sheikh AA, Khalaf HM, Abdelrahman AM. Protective effect of peroxisome proliferator activator receptor (PPAR)-alpha and -gamma ligands against methotrexate-induced nephrotoxicity. Immunopharmacol Immunotoxicol. 2014;36:130–137.
- Morsy MA, Ibrahim SA, Amin EF, Kamel MY, Rifaai RA, Hassan MK. Curcumin ameliorates methotrexate-induced nephrotoxicity in rats. Adv Pharmacol Sci. 2013;2013:387071.
- Abdel-Raheem IT, Khedr NF. Renoprotective effects of montelukast, a cysteinyl leukotriene receptor antagonist, against methotrexate-induced kidney damage in rats. Naunyn Schmiedebergs Arch Pharmacol. 2014;387:341–353.
- Gressier B, Lebegue S, Brunet C, et al. Pro-oxidant properties of methotrexate: Evaluation and prevention by an anti-oxidant drug. Pharmazie 1994;49:679–681.
- Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14:1649–1660.
- Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630.
- Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. 2011;7:715–726.
- Mizukami Y. In vivo functions of GPR30/GPER-1, a membrane receptor for estrogen: From discovery to functions in vivo. Endocr J. 2010;57:101–107.
- Lindsey SH, Cohen JA, Brosnihan KB, Gallagher PE, Chappell MC. Chronic treatment with the G protein-coupled receptor 30 agonist G-1 decreases blood pressure in ovariectomized mRen2.Lewis rats. Endocrinology. 2009;150:3753–3758.
- Kurt AH, Buyukafsar K. Vasoconstriction induced by G1, a G-protein-coupled oestrogen receptor1 (GPER-1) agonist, in the isolated perfused rat kidney. Eur J Pharmacol. 2013;702:71–78.
- Kurt AH, Tiftik RN, Un I, Ulker S, Buyukafsar K. G protein-coupled estrogen receptor1 (GPER1) may mediate Rho-kinase (ROCK-2) up-regulation in coronary endothelial cells. Endocr Regul. 2013;47:75–84.
- Bologa CG, Revankar CM, Young SM, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;2:207–212.
- Prossnitz ER, Sklar LA, Oprea TI, Arterburn JB. GPR30: A novel therapeutic target in estrogen-related disease. Trends Pharmacol Sci. 2008;29:116–123.
- Chan QK, Lam HM, Ng CF, et al. Activation of GPR30 inhibits the growth of prostate cancer cells through sustained activation of Erk1/2, c-jun/c-fos-dependent upregulation of p21, and induction of G(2) cell-cycle arrest. Cell Death Differ. 2010;17:1511–1523.
- Chen JQ, Cammarata PP, Baines CP, Yager JD. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim Biophys Acta. 2009;1793:1540–1570.
- Baylis C. Sexual dimorphism in the aging kidney: Differences in the nitric oxide system. Nat Rev Nephrol. 2009;5:384–396.
- Chin M, Isono M, Isshiki K, et al. Estrogen and raloxifene, a selective estrogen receptor modulator, ameliorate renal damage in db/db mice. Am J Pathol. 2005;166:1629–1636.
- Neugarten J, Acharya A, Silbiger SR. Effect of gender on the progression of nondiabetic renal disease: A meta-analysis. J Am Soc Nephrol. 2000;11:319–329.
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275.
- Fridovich I. Superoxide radical: An endogenous toxicant. Annu Rev Pharmacol Toxicol. 1983;23:239–257.
- Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–358.
- Straub RH. The complex role of estrogens in inflammation. Endocr Rev. 2007;28:521–574.
- Calippe B, Douin-Echinard V, Delpy L, et al. 17Beta-estradiol promotes TLR4-triggered proinflammatory mediator production through direct estrogen receptor alpha signaling in macrophages in vivo. J Immunol. 2010;185:1169–1176.
- Calippe B, Douin-Echinard V, Laffargue M, et al. Chronic estradiol administration in vivo promotes the proinflammatory response of macrophages to TLR4 activation: Involvement of the phosphatidylinositol 3-kinase pathway. J Immunol. 2008;180:7980–7988.
- Pektaş M, Kurt AH, Un I, Tiftik RN, Buyukafsar K. Effects of 17β-estradiol and progesterone on the production of adipokines in differentiating 3T3-L1 adipocytes: Role of Rho-kinase. Cytokine. 2015;72:130–134.
- Cho L. Estrogen raises risk of blood clots and stroke. Replacing this powerful hormone after menopause has more risks than first thought. Heart Advis. 2008;11:7.
- Lekontseva O, Chakrabarti S, Jiang Y, Cheung CC, Davidge ST. Role of neuronal nitric-oxide synthase in estrogen-induced relaxation in rat resistance arteries. J Pharmacol Exp Ther. 2011;339:367–375.
- Colditz GA, Hankinson SE, Hunter DJ, et al. The use of estrogens and progestins and the risk of breast cancer in postmenopausal women. N Engl J Med. 1995;332:1589–1593.
- Mosca L. The role of hormone replacement therapy in the prevention of postmenopausal heart disease. Arch Intern Med. 2000;160:2263–2272.
- Marjoribanks J, Farquhar C, Roberts H, Lethaby A. Long term hormone therapy for perimenopausal and postmenopausal women. Cochrane Database Syst Rev. 2012;7:CD004143.
- Hazell GG, Yao ST, Roper JA, Prossnitz ER, O’Carroll AM, Lolait SJ. Localisation of GPR30, a novel G protein-coupled oestrogen receptor, suggests multiple functions in rodent brain and peripheral tissues. J Endocrinol. 2009;202:223–236.
- Esqueda ME, Craig T, Hinojosa-Laborde C. Effect of ovariectomy on renal estrogen receptor-alpha and estrogen receptor-beta in young salt-sensitive and -resistant rats. Hypertension. 2007;50:768–772.
- Sanden C, Broselid S, Cornmark L, et al. G protein-coupled estrogen receptor 1/G protein-coupled receptor 30 localizes in the plasma membrane and traffics intracellularly on cytokeratin intermediate filaments. Mol Pharmacol. 2011;79:400–410.
- Yang YH, Ngo D, Jones M, Simpson E, Fritzemeier KH, Morand EF. Endogenous estrogen regulation of inflammatory arthritis and cytokine expression in male mice, predominantly via estrogen receptor alpha. Arthritis Rheum. 2010;62:1017–1025.
- Lafferty AR, Torpy DJ, Stowasser M, et al. A novel genetic locus for low renin hypertension: Familial hyperaldosteronism type II maps to chromosome 7 (7p22). J Med Genet. 2000;37:831–835.
- Ariazi EA, Brailoiu E, Yerrum S, et al. The G protein-coupled receptor GPR30 inhibits proliferation of estrogen receptor-positive breast cancer cells. Cancer Res. 2010;70:1184–1194.
- Wang C, Lv X, Jiang C, Davis JS. The putative G-protein coupled estrogen receptor agonist G-1 suppresses proliferation of ovarian and breast cancer cells in a GPER-independent manner. Am J Transl Res. 2012;4:390–402.
- Duckles SP, Krause DN, Stirone C, Procaccio V. Estrogen and mitochondria: A new paradigm for vascular protection? Mol Interv. 2006;6:26–35.
- Klinge CM. Estrogenic control of mitochondrial function and biogenesis. J Cell Biochem. 2008;105:1342–1351.
- Mattingly KA, Ivanova MM, Riggs KA, Wickramasinghe NS, Barch MJ, Klinge CM. Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol Endocrinol. 2008;22:609–622.
- Stirone C, Duckles SP, Krause DN, Procaccio V. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol Pharmacol. 2005;68:959–965.
- Razmara A, Sunday L, Stirone C, et al. Mitochondrial effects of estrogen are mediated by estrogen receptor alpha in brain endothelial cells. J Pharmacol Exp Ther. 2008;325:782–790.
- Guo J, Krause DN, Horne J, Weiss JH, Li X, Duckles SP. Estrogen-receptor-mediated protection of cerebral endothelial cell viability and mitochondrial function after ischemic insult in vitro. J Cereb Blood Flow Metab. 2010;30:545–554.
- Jahovic N, Cevik H, Sehirli AO, Yeğen BC, Sener G. Melatonin prevents methotrexate-induced hepatorenal oxidative injury in rats. J Pineal Res. 2003;34:282–287.
- Taylor AH, Al-Azzawi F. Immunolocalisation of oestrogen receptor beta in human tissues. J Mol Endocrinol. 2000;24:145–155.