Abstract
The goal of contemporary research in pemphigus vulgaris and pemphigus foliaceus is to achieve and maintain clinical remission without corticosteroids. Recent advances of knowledge on pemphigus autoimmunity scrutinize old dogmas, resolve controversies, and open novel perspectives for treatment. Elucidation of intimate mechanisms of keratinocyte detachment and death in pemphigus has challenged the monopathogenic explanation of disease immunopathology. Over 50 organ-specific and non-organ-specific antigens can be targeted by pemphigus autoimmunity, including desmosomal cadherins and other adhesion molecules, PERP cholinergic and other cell membrane (CM) receptors, and mitochondrial proteins. The initial insult is sustained by the autoantibodies to the cell membrane receptor antigens triggering the intracellular signaling by Src, epidermal growth factor receptor kinase, protein kinases A and C, phospholipase C, mTOR, p38 MAPK, JNK, other tyrosine kinases, and calmodulin that cause basal cell shrinkage and ripping desmosomes off the CM. Autoantibodies synergize with effectors of apoptotic and oncotic pathways, serine proteases, and inflammatory cytokines to overcome the natural resistance and activate the cell death program in keratinocytes. The process of keratinocyte shrinkage/detachment and death via apoptosis/oncosis has been termed apoptolysis to emphasize that it is triggered by the same signal effectors and mediated by the same cell death enzymes. The natural course of pemphigus has improved due to a substantial progress in developing of the steroid-sparing therapies combining the immunosuppressive and direct anti-acantholytic effects. Further elucidation of the molecular mechanisms mediating immune dysregulation and apoptolysis in pemphigus should improve our understanding of disease pathogenesis and facilitate development of steroid-free treatment of patients.
Introduction
Autoimmune pemphigus is a life-threatening mucocutaneous blistering disease associated with IgG antibodies targeting several types of keratinocyte antigens and eliciting epidermal clefting (acantholysis) via intracellular signaling activating apoptotic enzymes (apoptolysis) Citation[1]. Systemic administration of glucocorticosteroid hormones is essential to establish control of disease during the acute stage Citation[2]. Although glucocorticosteroid treatment is life-saving, it may cause severe side effects, including death Citation[3,4]. Therefore, pemphigus patients need drugs that can replace glucocorticosteroids. The development of non-steroidal treatment has been hampered by a lack of clear understanding of the mechanisms leading to keratinocyte detachment and death in pemphigus. This overview of recent advances in the knowledge of pemphigus autoimmunity challenges the existing dogmas, helps resolve old controversies, and identifies new perspectives for treatment. It encompasses knowledge on pemphigus vulgaris (PV) and pemphigus foliaceus (PF), but specifically excludes reports on paraneoplastic pemphigus, or PNP, originally described by Anhalt et al. Citation[5], because this disease is not related to PV and PF. The notion that PNP represents a variant of classical pemphigus stems from the facts that patients with PV or PF sometime have concomitant neoplasms Citation[6–8] and that some patients with PNP develop anti-desmoglein (Dsg) 1 and/or 3 antibodies—the hallmark of classical pemphigus Citation[9]. In fact, PNP represents only one manifestation of the heterogeneous autoimmune syndrome—termed paraneoplastic autoimmune multiorgan syndrome (PAMS)—targeting both tegumental epithelium and internal organs Citation[10]. In marked contrast to classical pemphigus, PAMS has an overall mortality more than 90% despite therapy, with progressive respiratory failure with clinical features of bronchiolitis obliterans being the most frequent cause of death Citation[11]. Sloughing of bronchial epithelial cells contributes to the occlusion of the small airways that provides a potential mechanism for the respiratory failure Citation[10]. Patients with PAMS develop mucocutaneous lesions that resemble pemphigoid, erythema multiforme, lichen planus, and graft vs. host disease, as well as the pemphigus-like variant that was termed PNP in the index patient with PAMS. Oral mucosal lesions of painful, progressive stomatitis are the hallmark of the disease and usually are the initial manifestation of PAMS Citation[12]. The proposed pathogenesis of PAMS continues to evolve. It is clear that the immunopathologic mechanisms differ appreciably from those responsible for the lesions of classical pemphigus. The spectrum of PAMS includes patients with disease predominantly or exclusively mediated by the cell-mediated autoimmunity effectors and those with both autoantibodies and cellular cytotoxicity Citation[13].
Pemphigus autoantigens
Following the discovery of IgG autoantibodies in patients with PV Citation[14] and PF Citation[15], numerous attempts have been made to identify targeted antigens. The patient's serum and isolated IgG fraction were utilized in the immunoprecipitation and immunoblotting experiments using the epidermal or keratinocyte culture proteins as well as saliva and urine as substrates. Although the low-sensitivity approaches, such as fluorography with metabolically labeled keratinocyte proteins preabsorbed with human serum Citation[16], demonstrated single protein bands, a more sensitive but less specific immunoblotting technique revealed more than a dozen of targeted keratinocyte proteins Citation[17]. An enhanced sensitivity immunoprecipitation assay demonstrated that different PV or PF patients produce antibodies recognizing both common and unique antigens Citation[18].
Representative images of protein bands recognized by PV and PF sera are shown in . The full list of “pemphigus antigens” reported to date contains over 40 protein bands with apparent molecular weights (MWs) of 12, 18, 25, 30, 33, 35, 38, 40, 45, 47, 50, 52, 55, 57, 59, 60, 62, 66, 67, 68, 70, 75, 78, 80, 85, 95, 100, 102, 105, 110, 112, 120, 130, 140, 160, 170,180, 185/190, 210, and 260 kD Citation[16,18–37]. Indeed, some of these bands may include the same polypeptides migrated with slightly different MWs, and vice versa, some may include two or more similarly sized but distinct antigens. For instance, in addition to the 130 kD Dsg 3, PV patients develop antibodies recognizing yet unidentified protein(s) of the same 130 kD MW in Dsg3− / − keratinocytes Citation[18] and peripheral blood mononuclear cells Citation[38].
Figure 1. Characterization of anti-keratinocyte antibody profiles of PV and PF sera by immunoprecipitation with proteins from cultures of human epidermal keratinocytes resolved by 7.5% SDS-PAGE. Modified from Ref. Citation[18].
![Figure 1. Characterization of anti-keratinocyte antibody profiles of PV and PF sera by immunoprecipitation with proteins from cultures of human epidermal keratinocytes resolved by 7.5% SDS-PAGE. Modified from Ref. Citation[18].](/cms/asset/e4573c7d-bbb8-4a1a-884c-b051a209623f/iaut_a_606444_f0001_b.gif)
Identification of the nature of proteins targeted by pemphigus autoimmunity is a subject of intense research. Originally, it was assumed that the proteins with the MW of approximately 60 kD or less are “contaminating” keratins that do not represent meaningful targets. However, recent studies demonstrated that only 2% of pemphigus and normal sera contain anti-keratin antibodies Citation[39]. Furthermore, a 66 kD antigen recognized by PV IgG—a membrane glycoprotein composed of two apparently identical subunits of 33 kD—was used to raise rabbit antibody that induced PV-like phenotype in neonatal mouse Citation[27]. Nevertheless, the candidates for the pathophysiologically relevant PV and PF antigens were selected among a few bands migrating with a higher MW, wherein the 130 and 160 polypeptides were most commonly seen Citation[16,29]. The antigens with these MWs were identified as Dsg 3 Citation[17] and Dsg 1 Citation[40], respectively. Thereafter, exploration of the nature of pemphigus antigens has been hampered by a simplistic (or “monopathogenic” Citation[41]) explanation of pemphigus pathophysiology through the “Dsg compensation” hypothesis placing Dsg 1/3 in the center of the pathophysiologic loop Citation[42].
The Dsg compensation hypothesis maintains that anti-Dsg 1 and 3 antibody profiles in pemphigus sera and the normal epidermal distributions of Dsg 1 and 3 determine the sites of blister formation and that either Dsg 1 or Dsg 3 alone is sufficient to maintain keratinocyte adhesion Citation[42]. The three postulates of this hypothesis are as follows: (1) in the superficial epidermis of PF patients, where Dsg 1 without Dsg 3 is expressed, anti-Dsg 1 antibody alone can cause blisters; (2) Dsg 3 antibody alone is sufficient to cause suprabasal split in the oral mucosa of PV patients that lacks Dsg 1; and (3) skin lesions in PV patients develop when both Dsg 1 and Dsg 3 antibodies are present. The major flaw of this hypothesis is an assumption that the integrity of the stratified squamous epithelium enveloping skin and oral mucosa relies entirely on Dsg 1 and 3 molecules. If that would be the case, the epidermis would have disintegrated to a single cell suspension in the PV patients who develop both anti-Dsg 1 and 3 antibodies ().
Figure 2. The imaginary appearances of epidermis in the skin of PV patients that produce both Dsg 1 and 3 antibodies based on the postulates of Dsg compensation hypothesis vs. real appearance of lesional epidermis in PV patients.
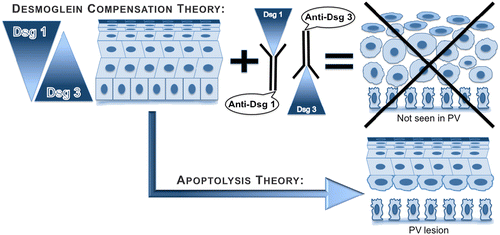
The monopathogenic explanation of localization of intraepidermal clefts in PV and PF through Dsg compensation hypothesis ignores the complexity of homo- and heterophilic interactions of seven known desmosomal cadherins, i.e. Dsg 1–4 and desmocollin (Dsc) 1–3. In reality, Dsg 3 alone cannot sustain epidermal cohesion. This is evident from the facts that Dsg 3 cannot compensate for a loss of Dsc 3 in the conditional Dsc3null mutant mouse that exhibits suprabasal acantholysis and overt skin blistering Citation[43]. Furthermore, the in vitro experiments demonstrated that extracellular domain of Dsg 3 mediates only a weak homophilic adhesion Citation[44]. Lack of skin blisters in patients with striate palmoplantar keratoderma featuring a deletion mutation in the extracellular domain of Dsg 1 and in mice with engineered or spontaneous mutations of Dsg 3 (reviewed in Citation[45,46]) clearly indicate that the integrity of epidermis does not depend solely on Dsg 1 and 3. The electron microscopic studies demonstrated that keratinocytes deprived of endogenous production of Dsg 1 or 3 due to gene silencing via RNA interference continue to form desmosomes Citation[47]. Apparently, the redundancy of desmosomal cadherins renders the external integument sufficient integrity and durability.
The first evidence that keratinocyte antigens other than Dsg 1 and 3 are pathophysiologically relevant in pemphigus was provided by experiments showing the ability to induce suprabasal acantholysis and gross skin blisters in Dsg3− / − neonates by passive transfer of PV patients' antibodies Citation[48]. In this model, murine epidermis lacked Dsg 3 and the passively transferred PV IgGs lacked anti-Dsg 1 antibody. Hence, the injected PV antibodies that caused blisters could target only the non-Dsg 1 and 3 antigens that mediated and/or regulated keratinocyte adhesion. This observation prompted further investigations into the nature of pemphigus antigens (reviewed in Citation[41]). By now, over 50 human proteins have been reported to specifically react with pemphigus IgG (). In addition to the known desmosomal cadherins and several other types of adhesion molecules, the hitherto identified pemphigus antigens include cell membrane (CM) receptors, immunologic/hematologic antigens, neuronal/oncologic antigens, and thyrogastric cluster antigens.
Table I. Self-antigens recognized by pemphigus IgGs.
Of particular interest is a recently discovered autoimmunity against a novel member of the peripheral myelin protein (PMP)-22/gas3 family termed PERP as well as the structurally related PMP-22 Citation[49,50]. Knockout mice lacking PERP display a phenocopy of PV Citation[51], which gave rise to a notion that the biologic function of PERP is limited to desmosomal stabilization Citation[52]. However, PERP is expressed in various types of cells that do not form desmosomes Citation[53], which argues against its exclusive biologic function in desmosomal adhesion. Recent findings and its putative tetraspan transmembrane topology implicate a role for PERP in the extrinsic apoptotic pathway that involves direct interaction between adaptor proteins and the receptor complexes activating caspase 8, i.e. PERP is a novel cell death receptor Citation[54]. Hence, dissolution of desmosomes and PV-like intraepidermal split in Perp− / − mice apparently result from aberrant inside-out signaling along the altered cell death pathways.
In a radioimmunoprecipitation assay of 34 PV and 6 PF serum, 85% of patients precipitated keratinocyte acetylcholine (ACh) receptors (AChRs) Citation[48]. Noteworthy, human keratinocytes express AChRs of both muscarinic (mAChR) and nicotinic (nAChR) classes, and these receptors regulate cell adhesion in a synergistic fashion (reviewed in Citation[55,56]). Blocking of either class of keratinocyte AChRs leads to disassembly of desmosomal and adherence junctions due to phosphorylation of desmosomal and classical cadherins, respectively, whereas cholinergic agonists prevent cell detachment by activating protein phosphatases and upregulating expression of the cadherin genes. Targeting of α3 nAChR by pemphigus antibody was discovered in a patient with coexistent PF, myasthenia gravis, and thymoma Citation[57]. Indeed, pemphigus patients occasionally develop myasthenia gravis, an anti-nAChR autoimmune disease (reviewed in Citation[6]). Based on the epitope-spreading model Citation[58], patients with myasthenia gravis might produce antibodies binding other members of an overall homologous nAChR protein gene family (reviewed in Citation[59]).
The types of other AChRs targeted by pemphigus autoimmunity have been investigated using various experimental approaches. Reactivity of PV IgG with the mixed muscarinic and nicotinic α9 AChR was observed in the immunofluorescence blocking experiments, wherein staining of monkey esophagus by rabbit anti-α9 antibody was prevented due to preincubation of the substrate with PV antibodies Citation[36]. Recently, the proteomics approach has demonstrated that PV antibodies react with α10 subunit that can be a part of the pentameric α9α10 nAChR (unpublished). Therefore, the binding site for α9 antibody in the heteromeric α9α10 nAChR also can be hindered by anti-α10 antibody present in PV sera. When added to keratinocyte monolayer, anti-α9 antibody produced acantholysis Citation[36], indicating that the alteration of either α9 or α10 subunit inactivates functioning of the α9α10 channel coupled to the regulation of keratinocyte adhesion.
Using the proteomics technology, two research groups have independently demonstrated that pemphigus autoimmunity targets the M1 subtype of keratinocyte mAChRs Citation[60,61]. The protein array technology also identified M2, M4, and M5 mAChR subtypes as targets of pemphigus autoimmunity Citation[61]. Probing of keratinocyte λgt11 cDNA library with the PV IgG eluted from a 75 kD band that stained epidermis in pemphigus-like intercellular pattern and caused acantholysis in the keratinocyte monolayers revealed a novel type of AChRs, termed pemphaxin (a.k.a. annexin 9) Citation[62]. Apparently, the AChR-binding pemphaxin is one of the annexin protein family members targeted by pemphigus autoimmunity Citation[63]. Most recently, it has been reported that patients with endemic form of PF have autoantibodies to pilosebaceous units and to their surrounding neurovascular packages Citation[64].
A large spectrum of organ-non-specific antigens that can be targeted by pemphigus autoimmunity () has been recently reviewed Citation[39,65,66]. Although contribution of organ-non-specific antigens to keratinocyte detachment and death in pemphigus remains to be elucidated, it is plausible to speculate that some of them, e.g. tumor necrosis factor (TNF) receptor superfamily member 5, are involved in the activation of the extrinsic apoptotic pathway and others, e.g. NADH dehydrogenase-like protein, in the activation of the intrinsic pathway Citation[37]. Of particular interest is a very high intensity of reactivity of PV IgG with Fc-IgG2 Citation[49]. The fact that it has >95% homology with Fc-IgG1 may explain a hitherto mysterious ability of the chimeric baculoproteins containing the constant region of human IgG1 and Dsg 1 or Dsg 3, in contrast to the extracellular portion of Dsg 3 alone, to absorb out all disease-causing antibodies Citation[67–69]. Thus, it can be concluded that pemphigus autoimmunity is directed against multiple organ-specific and non-organ-specific proteins, some of which are also targeted in other types of autoimmune diseases.
Pemphigus autoantibodies
Exploration of novel self-antigens reacting with pemphigus antibodies remains one of the top priorities in pemphigus research, because binding of patients' IgGs to these antigens triggers keratinocyte detachment and death in PV and PF. Although the mechanism of blistering in patients' skin and mucosa involves various factors, including cell-mediated cytotoxicity, proteolytic enzymes, and pro-inflammatory and pro-apoptotic cytokines, the principal role of anti-keratinocyte antibodies in the pathophysiology of autoimmune pemphigus has been well documented. The major lines of evidence are as follows: (1) occurrence of transient pemphigus-like skin lesions in neonates born by mothers with active pemphigus; (2) induction of pemphigus-like phenotype upon passive transfer of patients' IgGs to neonatal mice; and (3) elimination of disease causing activity of patients' IgG fraction due to absorption with antigenic constructs. Despite enormous efforts to single out an autoantibody responsible for either PV or PF, none of hitherto reported results provides compelling evidence in favor of the monopathogenic hypothesis of pemphigus immunopathology.
A first successful attempt to reproduce disease phenotype in animal model was reported by Peterson and Wuepper Citation[27] who observed small vesicles and limited areas of suprabasal acantholysis in the skin of neonatal mouse 36 h after intraperitoneal injection of a very high (40 mg) dose of rabbit IgG antibody raised against the 66 kD pemphigus antigen. Unfortunately, that antibody was not affinity purified on the antigenic peptide nor was its reactivity with keratinocyte proteins characterized. Some microscopic blisters also could be induced by pemphigus IgG eluted from a recombinant amino-terminus of Dsg 3 Citation[67]. That IgG fraction uniquely recognized a 130 kD protein of SDS-PAGE-resolved keratinocyte proteins. Although the anti-Dsg 3 antibody did not cause gross skin blisters, the obtained phenotype was deemed significant and the antibody termed “pathogenic”. Paradoxically, gross blisters in that study were induced, albeit by the pemphigus IgG fraction depleted of Dsg 3 antibody, indicating that non-Dsg 3 antibodies were actually pathogenic. This finding, however, was interpreted as evidence that disease-causing antibodies are directed to other “conformational” epitopes of Dsg 3 Citation[67].
Next, it was hypothesized that the extracellular domain of Dsg 3 should be combined with the Fc-portion of human IgG1 to create a proper conformation of the disease-specific Dsg 3 antigen Citation[68]. Indeed, the obtained chimeric protein absorbed out all disease-causing antibodies and injection of preabsorbed PV IgG fraction to neonatal mice did not produce extensive skin blisters. Furthermore, the modified chimera that contained His residues attached to the Dsg3-Ig construct eliminated disease-causing antibodies from sera of patients with PAMS, and the eluted antibodies caused gross skin blisters in neonatal mice Citation[70]. That same strategy was applied to create a “proper” conformational epitope of the Dsg 1 antigen capable of eliminating the disease-causing antibodies from PF sera Citation[69]. Surprisingly, the antibodies eluted from neither chimeric construct were examined for their reactivities against keratinocyte proteins to confirm their unique specificity for the 130 kD Dsg 3 and 160 kD Dsg 1 antigens.
Successive studies demonstrated that the results obtained in experiments with recombinant (r)Dsg1-Ig-His and rDsg3-Ig-His chimeras that led to a conclusion that anti-Dsg 1 and anti-Dsg 3 antibodies are pathogenic in PF and PV, respectively, were complicated by the presence of non-Dsg antibodies Citation[18]. shows that antibodies eluted from the chimeric constructs react with a mixture of keratinocyte proteins, including a non-Dsg 3 130 kD protein produced in the Dsg3− / − keratinocytes used as a source of antigens. Noteworthy, the fact that the rDsg3-His construct was recognized by PV antibodies indicated that it actually had a proper confirmation. In contrast, the Fc-containing Dsg constructs evidently did not have proper confirmation.
Figure 3. Profiles of PV IgGs absorbed by recombinant Dsg 1 and Dsg 3 baculoproteins in the Western blots of keratinocyte proteins resolved by 7.5% SDS-PAGE. Lane 1, reaction of PV IgG purified on the rDsg3-His construct with human keratinocytes. Lane 2, no primary antibody control for lane 1. Lane 3, no primary antibody control for lanes 4 and 5. Lane 4, reaction of PV IgG purified on the rDsg1-Ig-His construct with Dsg3null keratinocytes. Lane 5, reaction of PV IgG purified on the rDsg3-Ig-His construct with Dsg3null keratinocytes. The positions of relative molecular mass (Mr) markers run in parallel lanes of each blot are shown to the left of the respective blot. The apparent relative Mr of keratinocyte protein bands visualized due to PV antibody binding is shown to the right of lanes 2 and 5 in the columns designated Mr. Modified from Ref. Citation[18].
![Figure 3. Profiles of PV IgGs absorbed by recombinant Dsg 1 and Dsg 3 baculoproteins in the Western blots of keratinocyte proteins resolved by 7.5% SDS-PAGE. Lane 1, reaction of PV IgG purified on the rDsg3-His construct with human keratinocytes. Lane 2, no primary antibody control for lane 1. Lane 3, no primary antibody control for lanes 4 and 5. Lane 4, reaction of PV IgG purified on the rDsg1-Ig-His construct with Dsg3null keratinocytes. Lane 5, reaction of PV IgG purified on the rDsg3-Ig-His construct with Dsg3null keratinocytes. The positions of relative molecular mass (Mr) markers run in parallel lanes of each blot are shown to the left of the respective blot. The apparent relative Mr of keratinocyte protein bands visualized due to PV antibody binding is shown to the right of lanes 2 and 5 in the columns designated Mr. Modified from Ref. Citation[18].](/cms/asset/3d611e7e-2d5f-4829-9406-ee9d390c78e5/iaut_a_606444_f0003_b.gif)
These constructs were ill designed, because the ability of Fc fragment to mediate the Fc–Fc interactions known as Fc-mediated immune precipitation Citation[71,72] was ignored. It has been recently demonstrated that the CH2 and CH3 domain regions of the Fc fragment used by Amagai et al. Citation[68–70] in their pathogenic antibody elimination experiments provide an interface for the antigen-unspecific binding with another IgG molecule in immune complexes Citation[73]. Thus, an antigen-unspecific, Fc-mediated immune precipitation can explain adsorption of multiple antibodies on the chimeric Dsg baculoproteins. However, one of the antibodies evidently was absorbed specifically. A protein array has revealed that PV patients produce anti-Fc-IgG antibody Citation[49].
Several attempts have been made to produce a PV phenotype in mice by eliciting an innate anti-Dsg 3 antibody production. Immunization with recombinant Dsg 3, however, failed to induce acantholytic lesions despite explicit antibody production Citation[74–76]. On the other hand, the results of experiments with Rag2− / − mice transplanted with the Dsg 3 antibody producing splenocytes from either rDsg3-immunized Citation[77] or naive Dsg3− / − mice Citation[78] suggested the pathogenicity of Dsg 3 antibody in PV. Since the recipient mice produced limited oral acantholysis and erosions, this mouse model was termed “active disease” Citation[79]. A recent report, however, indicate that in the Dsg3− / − splenocyte adoptive transfer model, the efficacy of anti-Dsg 3 antibody in causing acantholysis is < 20% Citation[80]. Such low frequency of functional anti-Dsg 3 antibodies observed in the recipient Rag2− / − mice is consistent with inability of the majority of monoclonal murine Citation[81] and human Citation[82,83] anti-Dsg 3 antibodies to alter keratinocyte adhesion. Some anti-Dsg 3 antibodies, however, do cause acantholysis, albeit morphologically different from that found in PV patient's skin.
Anti-Dsg 3 antibody produces a desmosomal split without keratin retraction, apparently due to steric hindrance of Dsg 3 from opposing cells Citation[84,85]. In marked contrast, numerous classical Citation[86–89] and contemporary Citation[55,90] electron microscopic studies of PV patients' skin vividly demonstrated that desmosomes remain intact till the late stages of acantholysis when they are cleaved behind the desmosomal plaque, due to shearing forces produced by collapsing cells, and float free in the intercellular space. Thus, although the alterations of keratinocyte adhesion in recipient Rag2− / − mice are different from those taking place in patient's skin, these mice are still useful for studying the regulation of adaptive immune response to Dsg 3.
Although anti-Dsg 1 and 3 antibodies, alone or in combination, are not exclusively responsible for triggering intraepidermal blistering in patient's skin, they have a diagnostic utility. The Dsg 1 and Dsg 3 ELISAs provide a simple and highly sensitive approach to confirm the initial diagnosis of autoimmune pemphigus and differentiate it from other blistering diseases. The true value of ELISA results for patient management and prognosis, however, remains uncertain. The Dsg 1 and Dsg 3 IgG antibody titers do not always correlate with pemphigus disease activity Citation[91,92] nor do they predict exacerbation and relapse of the disease Citation[93]. Dsg 3 antibody can be absent in PV patients with active disease and present during remission Citation[94–96]. Furthermore, anti-Dsg 1 or 3 antibodies have been detected in healthy subjects, relatives of pemphigus patients, and patients with irrelevant medical conditions Citation[97–109].
Early reports suggested that levels of the IgG4 subclass of Dsg 3 antibody are associated with active disease and the IgG1 levels with remission Citation[110–112]. The enthusiasm about using the IgG4 autoantibody titer for management of the disease, however, was dampened by results of a recent study. Although in patients with active pemphigus, IgG4 and IgG1 were the dominant subclasses (96% and 76%, respectively), in clinical remission the autoantibodies predominantly belonged to the IgG2 (75%) and IgG4 (37.5%) subclass. Circulating IgG2 and IgG4 subclass autoantibodies were also observed in 60% and 23.3%, respectively, of healthy relatives Citation[101]. Additionally, some PV patients develop IgA and IgE classes of Dsg 3 antibodies Citation[113,114].
It was originally thought that the clinical phenotype of pemphigus is defined by the anti-Dsg autoantibody profile as follows: anti-Dsg 1 antibody alone is associated with PF, anti-Dsg 3 antibody alone—with mucosal variant of PV, and both antibodies—with mucocutaneous variant of PV Citation[115]. Although Dsg 1 antibody indeed appears to be a reliable serologic marker of PF and Dsg 3 antibody—that of PV, up to 58% of PF patients and 12% of patients with endemic PF (Fogo Selvagem) were reported to develop antibodies against both Dsg 1 and Dsg 3 Citation[108,116,117]. Furthermore, it has been conclusively demonstrated that Dsg 1 and Dsg 3 testing cannot differentiate between various morphologic subtypes of PV Citation[96,118,119]. In one study, for instance, 46% of PV patients did not have the PV phenotype (mucosal or mucocutaneous) predicted by their Dsg antibody profile Citation[118]. In another study, the Dsg 1+/Dsg 3+ pattern was observed in 15% of PV patients with exclusive mucous membrane involvement Citation[96].
A recent observation that an increase in Dsg 1 antibody titer in a PV patient has occurred already after the patient had started treatment and went into clinical remission Citation[120] supports the notion that anti-Dsg antibodies “witness” rather than trigger PV, i.e. that production of these autoantibodies is the result rather than the cause of epidermal blistering in pemphigus Citation[121]. Reactivity of pemphigus autoantibodies with both extracellular and intracellular domains of Dsg 1 and Dsg 3 Citation[122] suggests that these antibodies are produced already after the whole Dsg molecules have been released from the CM of damaged keratinocytes into the intercellular space and became available to antigen-presenting cells. The presence of the N-terminal portion of Dsg 3 in human sera Citation[123] lends additional support for the Dsg sloughing hypothesis.
Perhaps other desmosomal cadherins are also shed from the CM of damaged keratinocytes. The presence of anti-Dsc 1–3 antibodies in PV patients has been discovered relatively long ago Citation[124,125], but the interest to these antibodies was diminished by the reports that none of 45 Citation[126] or 74 Citation[127] PV patients tested by ELISA had any anti-Dsc antibodies. However, a recent study showing that the Dsc 3 loss of function model exhibits a phenocopy of PV Citation[43] suggested that anti-Dsc 3 antibody contributes to PV. As expected, the monoclonal antibody raised against the extracellular domain of Dsc 3 caused intraepidermal blistering in an in vitro model of human skin and a loss of cell–cell adhesion in the keratinocyte culture Citation[128].
In the most recent study, 6 out of 38 PV and 1 out of 85 normal serum samples immunoprecipitated Dsc 3 Citation[129]. Furthermore, while incubation of patient's IgG with human keratinocytes caused the loss of intercellular adhesion, adsorption with rDsc 3 prevented this effect Citation[129]. Thus, while antibodies to the desmosomal cadherins may be playing a scavenging role by eliminating CM debris from the intercellular spaces of damaged epidermis they may obstruct homophilic and heterophilic binding between the neighboring keratinocytes, thus contributing to acantholysis.
The initial insult that triggers keratinocyte damage in pemphigus is apparently sustained by autoantibodies to the cell membrane receptors whose ligation causes cell shrinkage—the earliest sign of keratinocyte detachment in pemphigus lesions Citation[55,86–90] that leads to ripping desmosomes off the CM. Indeed, it has been demonstrated that rabbit anti-α9 AChR antibody causes keratinocyte shrinkage and rounding up Citation[36]. Noteworthy, pemphigus IgG produces similar morphologic changes in keratinocyte monolayers Citation[130].
Likewise, pharmacologic inhibition of α3 nAChR causes keratinocytes to retract their cytoplasmic aprons, shrink, and round up Citation[131]. Pemphigus-like acantholysis has also been observed due to inhibition of keratinocyte mAChRs Citation[130], in keeping with the synergistic control of keratinocyte adhesion by auto/paracrine ACh that maintains the polygonal shape of keratinocytes and their adhesion by simultaneously activating both classes of keratinocyte cholinergic receptors Citation[132,133]. Hence, blockade of any type of keratinocyte AChRs by pemphigus IgGs can trigger acantholysis.
Although the hypothesis of primary involvement of autoantibodies against canonical AChR subtypes in pemphigus pathophysiology is awaiting its in vivo confirmation, the already completed studies have demonstrated essential role of antibody against the non-canonical ligand of AChRs termed pemphaxin Citation[62]. Preabsorption of PV sera with recombinant pemphaxin eliminated acantholytic activity and eluted antibody immunoprecipitated native pemphaxin. Although anti-pemphaxin antibody alone did not cause skin blisters in vivo, its addition to the preabsorbed PV IgG fraction restored the acantholytic activity of passively transferred antibodies Citation[62].
These observations indicate that pemphaxin is an essential part of the pool of keratinocyte cell surface antigens that should be simultaneously targeted by autoantibodies to induce acantholysis. The anti-mitochondrial antibodies from different PV patients that recognized distinct combinations of antigens with apparent MWs of 25, 30, 35, 57, 60, and 100 kD evidently are also pathogenic, because their absorption abolished the ability of PV IgG to cause keratinocyte detachment both in vitro and in vivo Citation[37].
Involvement of multiple autoantibody specificities in pemphigus pathogenesis is explained through the “multiple hit” hypothesis Citation[134] as follows: anti-AChR antibodies trigger acantholysis by weakening cohesion of neighboring keratinocytes due to inhibition of the physiologic control of their polygonal shape and intercellular attachment. The affected keratinocytes shrink, causing desmosomes to be sloughed in the intercellular space. The adhesion molecules floating free in the intercellular space bring about a reciprocal production of scavenger antibodies that, in turn, saturate epidermis, thus preventing nascent desmosome formation by steric hindrance. Thus, according to the multiple hit hypothesis, pemphigus results from a synergistic and cumulative effects of autoantibodies targeting keratinocyte CM antigens of different kinds including (i) molecules that regulate cell shape and adhesion (e.g. AChRs); and (ii) molecules that mediate cell–cell adhesion (e.g. desmosomal cadherins). Severity of the disease and exact clinical picture depend on the ratio of different kinds of autoantibodies in each particular patient.
In conclusion, different patients develop distinct constellations of autoantibodies which, together with the individual's re-epithelialization abilities, determine clinical severity of disease, its natural course, and response to treatment. The Dsg 1 and 3 antibodies are the sensitive markers of pemphigus, but their primary role in the pathogenesis of PF and PV, respectively, is overestimated. Therefore, not surprisingly, clinical trial of the Dsg 3 peptides (PI-0824 vaccine) has not shown the anticipated clinical or immunologic activity (reviewed in Citation[135]). Apparently, an attack by a constellation of autoantibodies simultaneously targeting several keratinocyte proteins is required to disrupt the integrity of epidermis. The multiple hit hypothesis reconciles findings of anti-AChR autoimmunity with the fact that pemphigus patients also develop autoantibodies to adhesion molecules as well as various other proteins. The hypothetical sequence of immunopathologic and biochemical events leading to acantholysis in pemphigus is shown in . Future studies should define the autoantibodies that sustain an initial insult triggering keratinocyte detachment and those produced as a result of primary cell damage to clean up the proteins released in the intercellular spaces by damaged cells.
Figure 4. Hypothetical scheme of the time course of pathobiologic events leading to acantholysis in pemphigus. In stage I, antibodies to PERP and/or cellular AChR block the physiologic control of polygonal cell shape and intercellular adhesion. This increases phosphorylation of adhesion molecules with their subsequent dissociation from the adhesion units on CM, and also initiates programed cell death. In stage II, the tonofilament (TF). cytoskeleton collapses and keratinocytes shrink with associated sloughing of desmosomes which elicits autoimmune response to the desmosomal antigens. In stage III, anti-Dsg antibodies bind to their targets on the CM of keratinocytes thus precluding formation of new intercellular junctions. Modified from Ref. Citation[55].
![Figure 4. Hypothetical scheme of the time course of pathobiologic events leading to acantholysis in pemphigus. In stage I, antibodies to PERP and/or cellular AChR block the physiologic control of polygonal cell shape and intercellular adhesion. This increases phosphorylation of adhesion molecules with their subsequent dissociation from the adhesion units on CM, and also initiates programed cell death. In stage II, the tonofilament (TF). cytoskeleton collapses and keratinocytes shrink with associated sloughing of desmosomes which elicits autoimmune response to the desmosomal antigens. In stage III, anti-Dsg antibodies bind to their targets on the CM of keratinocytes thus precluding formation of new intercellular junctions. Modified from Ref. Citation[55].](/cms/asset/e3b51a37-ed87-4b0e-9ba7-30b7064f7286/iaut_a_606444_f0004_b.gif)
Regulation of pemphigus autoimmunity
Although autoimmunity is a normal event, autoimmune diseases result from an aberration of the normal phenomenon Citation[136]. The etiology of this switch in pemphigus is apparently multifactorial, with the final common pathway being a loss of normal self-tolerance in the stratified squamous epithelium. Analysis of genetic factors contributing to PV and PF has shown that the same genetic regions can contribute to both forms of the disease (reviewed in Citation[137]). The HLA genes are probably the most significant genetic predisposition factors, because they play an important role in the antigen presentation process, whereas other loci may participate in an additive or epistastic manner.
Population studies have consistently shown a link between certain class II HLA alleles and distinct ethnic groups of pemphigus patients. A recent study involving a large cohort of White European and Indo-Asian patients with PV confirmed associations with the alleles HLA DRβ1*0402 and 1404, and DQβ1*0302 and 0503 Citation[138]. The DRβ1*1404 was the strongest risk factor in the Indo-Asian group and DRβ1*0402—in the White European group. In White Europeans, a significant association was also shown for the novel allele DRβ1*1454 Citation[138]. Also, it has been documented that HLA-DRβ1*0402 is associated with PV in Jewish and HLA-DQβ1* 0503 in non-Jewish populations Citation[139–141].
Although HLA studies have shown that susceptibility to PF also correlates with the presence of DR4, DR14, and DR1 alleles, in contrast to PV, no single DR4 or DR14 allele was found to be overrepresented in PF patients Citation[137]. The HLA-DR/DQ distributions does not differ among PV patients according to the presence or absence of anti-Dsg 1 coexisting with anti-Dsg 3 Citation[142].
Although the basis for autoimmunity in pemphigus remains unrecognized, the regulation of anti-keratinocyte autoimmunity in PV and PF has been studied based on the assumption that Dsg 1/3 antibodies solely represent pemphigus autoimmunity. It was reported that the Th1 and Th2 cell recognition of Dsg 3 peptides is restricted by HLA-DRβ1*0402 and/or HLA-DQβ1*0503, and that the proliferative response of autoreactive Th cells can be blocked by anti-DR and anti-DQ antibodies, respectively Citation[143–146].
A loss of self-tolerance against Dsg 3 in both T and B lymphocytes was found to be required for efficient production of anti-Dsg 3 IgG antibodies Citation[147–149]. The anti-Dsg 3 antibody production in mice was inhibited by the anti-CD154 monoclonal antibody that blocks CD40L–CD40 interaction Citation[150]. The CD8+ T cells specific for Dsg 3 were also detected in PV patients Citation[151], which is in keeping with earlier observation of the autoreactive cytotoxic T lymphocytes are sensitized to putative keratinocyte antigens in PV patients Citation[152].
Autoimmunity to certain epitopes of Dsg 3 may be a normal event, because Dsg 3-reactive B cells as well as Th1 and Th2 cells are present in normal individuals Citation[97,100,101,146,151,153,154]. The presence of autoreactive B cells is evidenced by production of anti-Dsg 3 antibodies by healthy relatives of PV patients Citation[97,100,101,154]. The presence of Dsg 3-reactive Th1 cells has been demonstrated in healthy carriers of PV-associated HLA class II alleles, and the Dsg 3-reactive Th1 clones derived from these individuals were restricted by HLA-DRβ1*0402 and DQβ1*0503 Citation[146,151,153].
There is a predominance of autoreactive Dsg 3-reactive Th1 cells in healthy individuals and Dsg 3-reactive Th2 cells in PV patients Citation[146]. However, Dsg 3-reactive Th2 cells are detected at similar frequencies in acute onset, chronic active, and remittent PV, while the number of autoreactive Th1 cells exceeds that of Th2 cells in chronic active PV Citation[146]. Likewise, Dsg 1-responsive Th1 and Th2 cells were also found both in patients with PF and in healthy individuals Citation[155].
Defects in Tregs have been reported in a wide variety of human organ-specific autoimmune diseases Citation[156]. While the induction of Treg suppressive activity is specific and requires antigenic stimulation through T-cell receptor, the suppression exerted by Tregs is antigen nonspecific Citation[157]. Various Tregs can employ distinct mechanisms to collaboratively regulate the duration and magnitude of an immune response Citation[158]. It has been postulated that active immune suppression operates in healthy individuals possessing Dsg 1- and Dsg 3-reactive T cells, and that an imbalance of the putative relationship between autoreactive Th and Tregs (Tr1) cells is critical to the development of pemphigus Citation[159]. In support, the CD4+CD25 + hi Tregs are decreased in peripheral blood of PV patients Citation[160]. Furthermore, a subset of Dsg 3-reactive, interleukin (IL)-10-secreting Tr1 cells was found in the majority of healthy carriers of PV-associated HLA class II alleles and only in < 20% of PV patients Citation[161]. However, a recent study demonstrated that PV skin lesions contain both Foxp3-expressing cells and IL-17 producing CD4+ cells Citation[162].
Thus, deficiency of Tregs in PV patient's blood is not accompanied by a decrease in Tregs in PV lesions, and a decrease in Tregs in peripheral blood may result from accumulation of Tregs in skin lesions and draining lymph nodes Citation[162]. Alternatively, or additionally, pemphigus autoimmunity may be triggered via the pathway involving activation of toll-like receptors (TLRs). This mechanism has been suggested by a recent demonstration of reversible relapse of PF triggered by the TLR7 agonist imiquimod Citation[163]. In this scenario, B-cell tolerance is broken due to ligation of B-cell receptor and TLR by self-antigen/TLR ligand, leading to breakage of T-cell tolerance and activation of autoreactive B and T cells Citation[164].
In conclusion, regulation of Dsg 1 and 3 antibody production agrees with the basic postulates of fundamental immunology on T cell–B cell cooperation. Th1 and Th2 cells found in patients with PV and healthy carriers of PV-associated HLA class II alleles recognize identical epitopes of the Dsg 3 ectodomain presented by antigen-presenting cells. A decrease in Tregs in peripheral blood of PV patients does not validate the postulated deficiency of the immunosuppressive activity, because Tregs are present in PV lesions. Future studies of the immunoregulatory mechanisms of PV should characterize the reputed interplay between Tregs and Th17 cells (), and identify the role for TLRs that can regulate function of Th1, Th2, Th17 cells, and Tregs.
Figure 5. Hypothetical scheme of immune dysregulation in PV. Abbreviations: APC, antigen-presenting cells; IL, interleukin. Modified from Ref. Citation[339].
![Figure 5. Hypothetical scheme of immune dysregulation in PV. Abbreviations: APC, antigen-presenting cells; IL, interleukin. Modified from Ref. Citation[339].](/cms/asset/03a4e5e1-43fc-4f67-adae-1dcc112a8480/iaut_a_606444_f0005_b.gif)
Molecular mechanisms of keratinocyte detachment in pemphigus
Several hypotheses have been put forward to explain the mechanism of pemphigus acantholysis. The classical explanation through the hypothesis of steric hindrance of Dsg 1- and Dsg 3-mediated adhesion by respective antibodies Citation[165,166] has been challenged by numerous reports showing activation of specific signaling pathways in keratinocytes exposed to pemphigus IgGs. The steric hindrance hypothesis is based on the erroneous assumptions that binding of PV IgG in the epidermis is limited to desmosomes Citation[167] and that the phenocopies of PF and PV in mouse skin can be induced by passive transfer of PV IgGs recognizing uniquely the desmosomal cadherins Dsg 1 and 3, respectively Citation[68,69].
In fact, although Dsg molecules are indeed predominantly localized to desmosomes Citation[168], binding of PV IgG extends well beyond the desmosomes decorating the entire surface of keratinocytes Citation[169]. As already mentioned, the Dsg-Fc-IgG constructs were found to be not specific for Dsg 1 or 3 antibodies Citation[18,36]. Furthermore, PF IgG has recently been shown to cause dissociation of keratinocytes without blocking Dsg 1 homophilic transinteraction Citation[170]. Surprisingly, this cumulative evidence that non-Dsg molecules are targeted by pathogenic PV IgG in the interdesmosomal areas is being interpreted as targeting of Dsg 3 located outside of the desmosomes Citation[171].
The alternative Dsg 3 alteration hypotheses proposed by different authors Citation[171–173] have one common theme. They are based on the assumption that all outside-in signals elicited due to binding of PV IgG to keratinocytes emanate exclusively from Dsg 3. To account for a very broad spectrum of signals, it was inferred that Dsg 3 can act as both adhesion receptor and signal transmitter Citation[171–173]. The following hypothetical chain of events was envisioned: (1) PV IgG signaling is initiated by ligation of nonfunctional pool of Dsg 3 outside of the desmosome; (2) when ligated by an autoantibody, these putative nonfunctional Dsg 3 molecules, either membrane associated or internalized or both, send signals that impair Dsg 3 trafficking into and out of desmosome; and (3) the supposedly impaired Dsg trafficking specifically depletes Dsg 3 from desmosomes without changes in other functional proteins.
The notion about principal role of Dsg 3 in PV IgG signaling stems from experiments in which the whole PV IgG fraction, rather than affinity purified patient's anti-Dsg 3 antibody, was used to elicit biologic responses Citation[174–183]. When interpreting results, it was assumed that the whole plethora of PV IgG effects, including Dsg 3 endocytosis and activation of various signaling cascades, resulted exclusively from Dsg 3 ligation by an autoantibody. However, Jennings et al. Citation[184] have recently demonstrated an explicit biologic effect of PV IgG on the keratinocytes expressing the Dsg 3 molecules whose “pathogenic” epitopes were hidden due to cell preincubation at 4°C with the anti-Dsg 3 monoclonal antibody AK23 that reportedly reproduces PV phenotype Citation[81,185] and induces PV IgG-like signaling Citation[186].
Therefore, depletion of Dsg 3 is apparently a secondary event resulting from an inside-out signaling caused by keratinocyte response to the pathogenic autoantibodies that deliver an initial insult. The primary role of anti-PEPR antibody in the depletion of Dsg 3 was suggested by an observation that binding of PV antibodies triggers internalization of PERP, which enhances depletion of desmosomal Dsg 3 and intercellular adhesion defects Citation[187]. Thus, endocytosis of the immune complexes containing non-junctional Dsg 3 and depletion of Dsg 3 from desmosomes apparently represent two independent events, with the former being a natural outcome of formation of antigen–antibody complex on the CM and the latter resulting from the inside-out signaling altering function and trafficking of desmosomal cadherins.
The outside-in signaling elicited due to binding of PV IgGs to keratinocytes proceeds via different pathways, consistent with simultaneous ligation of several types of cell surface receptors by distinct antibodies produced by pemphigus patients. Different research groups reported engagement of Src, epidermal growth factor receptor (EGFR) kinase (EGFRK), cAMP, protein kinases A and C (PKC), phospholipase C, mTOR, p38 MAPK, JNK, other tyrosine kinases, and calmodulin Citation[37,47,175,188–191]. The preferential signaling pathway downstream of targeted self-antigens is apparently determined by a unique composition of the pool of anti-keratinocyte antibodies produced by each PV patient, because IgG fractions from different PV patients exhibit distinctive time patterns of kinase activation Citation[37].
Timing of kinase activation is critical for understanding the hierarchy of signaling events leading to acantholysis. The time course studies demonstrated that the activities of Src and EGFRK peak at 30–60 min after exposure to PV IgG Citation[37,47], suggesting that engagement of Src/EGFRK is a key step that relays signals emanating from ligated antigens to the intracellular effectors affecting keratinocyte adhesion and viability.
Activation of EGFRK due to binding of PV IgG to keratinocytes is followed by phosphorylation of its downstream substrates, the MAP kinase ERK and the transcription factor c-Jun Citation[189]. Activation of PKC is also one of the earliest events in PV IgG-induced acantholysis Citation[192]. The elevation of p38 MAPK activity caused by antibodies from some PV patients can be observed already at 15 min, while the majority of PV IgGs activate p38 MAPK after a prolonged incubation Citation[37]. Thus, it is becoming evident that an array of interconnected signaling cascades emanating from different cell surface antigens simultaneously targeted by a constellation of patients' anti-keratinocyte antibodies trigger acantholysis and keratinocyte death in pemphigus.
Two different approaches have been used to elucidate involvement of Dsg 3 in the PV IgG-mediated signaling and define relevant pathways. Through one approach, the Dsg1 or Dsg3 genes in cultured human keratinocytes were silenced using the RNA interference technology Citation[47]. Transfection with small interfering RNAs that inhibited expression of either Dsg 1 or Dsg 3 or both in all cases blocked approximately 50% of p38 MAPK activity, but only slightly altered the PV IgG-dependent raise in Src and EGFRK activities. To avoid any possible contribution to PV IgG signaling by residual Dsg 3 protein, a separate series of experiments employed keratinocytes grown from the epidermis of neonatal Dsg 3 knockout mice Citation[37]. It was documented that lack of Dsg 3 did not affect the ability of PV IgG to activate Src and EGFRK.
However, in the absence of Dsg 3, the PV IgG-dependent activation of both p38 MAPK and JNK was significantly reduced. Because both p38 MAPK and JNK can be activated secondary to keratinocyte shrinkage and detachment Citation[193–195], and since keratinocyte damage in PV is associated with activation of the cell death program (reviewed in Citation[196]), it was important to determine whether p38 MAPK and JNK activation precedes or follows launching of the apoptotic cascade. Inhibitors of the executioner caspases abolished activation of JNK and the late p38 MAPK peak Citation[37], indicating that these activities were indeed triggered by the cell injury rather than by the PV IgG binding to keratinocyte antigens. This supposition has been recently corroborated by a report that p38 MAPK activation occurs downstream at the loss of intercellular adhesion in PV Citation[197].
Thus, although the pool of anti-keratinocyte antibodies produced by PV patients contains anti-Dsg 1 and/or 3 antibodies, published studies indicate that non-Dsg antibodies are the major contributors to early signaling events. Early activation of the Src/EGFRK and PKC-dependent pathways is apparently pathogenic because it leads to acantholysis Citation[47,189], late activation of p38 MAPK is secondary to cell detachment Citation[197], whereas activation of the cAMP/protein kinase A step appears to have a protective function Citation[191].
The principal signaling event leading to acantholysis is triggered due to antibody interference with the physiologic control of keratinocyte survival, shape, and adhesion. For instance, blocking of keratinocyte AChRs interferes with auto/paracrine control of assembly/disassembly of intercellular junctions, cell shape, motility, proliferation, apoptosis, and differentiation (reviewed in Citation[198]). Both muscarinic and nicotinic antagonists have been shown to widen the intercellular spaces in epidermis and cause overt acantholysis in vitro through the mechanism that may involve alterations of both production and phosphorylation of keratinocyte adhesion molecules (reviewed in Citation[55]). It is well known that phosphorylation of adhesion molecules plays an important role in assembly/disassembly of intercellular junctions Citation[199–203], and that phosphorylation of cadherin Citation[204–206], γ-catenin Citation[207], desmoplakin Citation[208,209], and Dsg Citation[210–212] is associated with a loss of adhesion.
Some intercellular junction proteins are phosphorylated on serine, some on tyrosine, and some on both residues. The seminal works by Aoyama et al. Citation[212,213] demonstrated that binding of PV IgG to cell surface antigens induces phosphorylation of Dsg 3, its dissociation from plakoglobin, and formation of Dsg 3-depleted desmosomes. These findings were corroborated by the results showing that in addition to Dsg 3, PV IgG also increases the level of phosphorylation of keratinocyte E-cadherin as well as β-, γ-, and p120 catenins Citation[214,215]. These experiments also demonstrated that keratinocyte dyshesion correlates closely with the degree of tyrosine phosphorylation of p120-catenin by Src and serine phosphorylation of β-catenin by classic PKC isoforms Citation[215].
The Src-dependent cascade is also responsible for keratinocyte shrinkage (cell volume reduction) and keratin aggregation Citation[47]. The cytoskeletal collapse has been reported as an early event in pemphigus acantholysis that precedes visible separation of keratinocytes Citation[47,179,216–218]. Thus, it appears that PV IgG-induced phosphorylation of adhesion molecules and structural proteins leads to weakening of intercellular junctions and collapse of the cytoskeleton, respectively. The chronological scheme of early signaling and pathobiologic events in keratinocytes exposed to PV IgG are shown in .
Figure 6. Hypothetical scheme of early signaling steps during first 6 h after PV IgG binding to keratinocytes and their correlation with the major intracellular pathobiologic events. Abbreviations: EGFR, epidermal growth factor receptor; Dsg, desmoglein; NDAgs, non-Dsg antigens. From Ref. Citation[47].
![Figure 6. Hypothetical scheme of early signaling steps during first 6 h after PV IgG binding to keratinocytes and their correlation with the major intracellular pathobiologic events. Abbreviations: EGFR, epidermal growth factor receptor; Dsg, desmoglein; NDAgs, non-Dsg antigens. From Ref. Citation[47].](/cms/asset/89a972e0-ba9a-4b6f-acd0-9f555fe6afe3/iaut_a_606444_f0006_b.gif)
Numerous classical and modern clinical and experimental studies in pemphigus demonstrated that desmosomes separate when the intercellular spaces are already widened Citation[87,90,219–222]. Desmosomes do not split and disappear until late in acantholysis when keratinocytes are almost completely separated from each other. At early stages of acantholysis, the half-desmosomes remain invisible because they are firmly adhering to each other. In late acantholysis, some half-desmosomes adhere to each other so strongly that they can be ripped off one cell while remaining adherent to their counterpart on the opposite cells.
Hence, disruption of intercellular bridges results from ripping intact desmosomes off the plasma membrane of collapsing keratinocytes by shearing forces. The intact desmosomes ripped off from neighboring cells can be seen floating free in the intercellular space, which is in keeping with the sloughing of desmosomal cadherins that gives rise to scavenging autoantibodies. The “basal cell shrinkage” hypothesis reconciles the time course of acantholysis in PV with the characteristic appearance of acantholytic epidermis, known as “tombstoning” Citation[223]. According to this hypothesis: (1) keratinocytes separate because they shrink more than can be held together by desmosomes; (2) the suprabasal clefting occurs because basal cells shrink more than suprabasal keratinocytes; and (3) pharmacologic inhibition of the principal signaling pathways leading to cytoskeletal disorganization should prevent pemphigus acantholysis.
One of the most important recent advances in pemphigus research was elucidation of the molecular mechanisms that selectively target basal cells in PV, as predicted by the basal cell shrinkage hypothesis. Pretel et al. Citation[190] demonstrated that pretreatment with the mTOR inhibitor sirolimus prevented suprabasal acantholysis in the epidermis of neonatal mice injected with PV IgG. In this model, PV antibodies caused unopposed upregulation of mTOR selectively in basal keratinocytes, which was associated with the appearance of signs of apoptosis that were also abolished by sirolimus Citation[190].
Downstream of mTOR, the induction of keratinocyte apoptosis by PV antibodies apparently proceeds through the c-Myc-dependent pathway Citation[218]. c-Myc-induced apoptosis involves caspase 9 Citation[224], which is activated in keratinocytes treated with PV IgG Citation[225]. In agreement with the fact that cyclin-dependent kinase 2 (Cdk2) is required by c-Myc to induce apoptosis Citation[226], PV sera induces accumulation of Cdk2 that contributes to acantholysis in the mouse model of PV Citation[227]. Indeed, sirolimus has been shown to inhibit expression of both c-Myc Citation[228] and Cdk2 Citation[229]. Taken together, these observations help explain why epidermal clefting in PV always occurs just above basal cells (tombstoning), despite deposition of IgG antibodies throughout the entire epidermis.
Both extrinsic and intrinsic pathways of cell death triggered in keratinocytes by PV IgGs can lead to the structural damage manifested by basal cell shrinkage. It has been documented by different research groups that in the skin of PV patients, keratinocytes exhibit signs of apoptosis that precede their detachment and blister formation, and that PV IgG and sera induce biomolecular markers of apoptosis and oncosis in keratinocyte monolayers and skin organ cultures Citation[225,230–235]. Two groups of PV patients, each producing autoantibodies activating predominantly either apoptotic or oncotic cell death pathway, have been identified Citation[225].
The anti-PERP PV antibody may launch the cell death pathways in keratinocytes, because PERP expression leads to activation of an extrinsic receptor-mediated apoptotic pathway with a possible subsequent engagement of the intrinsic apoptotic pathway Citation[54]. Anti-mitochondrial PV antibodies also can trigger intrinsic apoptotic cascade in keratinocytes Citation[37]. Other types of autoantibodies and soluble mediators of inflammation can activate cell death pathways in keratinocytes. Dr Pincelli's group demonstrated that Fas ligand (FasL) in pemphigus sera induces keratinocyte apoptosis through activation of caspase 8 Citation[231], and that FasL neutralizing antibody prevents PV IgG-induced apoptosis both in vitro and in vivo Citation[236].
Furthermore, it has been shown that TNFα mRNA is abundantly expressed in PV skin lesions Citation[237,238]; serum TNFα levels correlate closely with disease activity and autoantibody titers Citation[239,240]; and anti-TNFα antibody inhibits acantholysis induced by PV autoantibodies in vitro Citation[237]. The synergistic acantholytic effects of PV IgG, FasL, and TNFα were documented in experiments with keratinocyte monolayers and full thickness equivalents of human epidermis Citation[241]. These mediators of apoptosis as well as the increased amounts of activated kallikreins Citation[242] and several types of inflammatory cytokines Citation[237,239,243,244] found to be elevated in pemphigus sera may account for a minor acantholytic activity of the PV serum depleted of IgG, as reported by Cirillo et al. Citation[245].
Besides PV IgGs, cell death pathways in pemphigus can be triggered by autocrine and paracrine factors released from damaged keratinocytes in situ. PV IgG binding to keratinocytes not only elicits secretion of soluble FasL but also increases expression of Bax, Fas receptor (FasR), coaggregation of FasL and FasR with caspase 8 in a membranal death-inducing signaling complex, and downregulation of the anti-apoptotic Bcl-2, FLIP-l, and the oncosis inhibitor calpastatin Citation[189,225,231,232,234,246,247]. In turn, TNFα induces urokinase plasminogen activator mRNA Citation[237].
It is well known that blister fluid and/or perilesional skin of PV patients contains high levels of proteases and various inflammatory cytokines that may contribute to acantholysis Citation[242,248–250]. Thus, a simultaneous autoimmune attack by the three classes of autoantibodies against desmosomal, mitochondrial, and other keratinocyte autoantigens, such as AChRs and PERP, may be required to induce pathologic changes in patient's skin. The anti-keratinocyte antibodies synergize with the effectors of apoptotic pathway FasL and TNFα as well as proinflammatory/cytotoxic serum and tissue factors, such as serine proteases and cytokines, that altogether overcome the natural resistance and activate cell death pathways in keratinocytes ().
Figure 7. Hypothetical scheme of signaling events mediating keratinocyte damage in PV. Abbreviations: AMPVAb, anti-mitochondrial PV antibody; Cs, caspase; FasL, Fas ligand; FasR, Fas receptor; MRPVAg, mitochondria-related PV antigen; NO, nitric oxide; OPVAb, PV antibodies of other specificities, including anti-AChR and anti-PERP antibodies; OPVAg, other types of putative PV antigens; PVAb, PV antibody; TNFα, tumor necrosis factor α. Modified from Ref. Citation[37].
![Figure 7. Hypothetical scheme of signaling events mediating keratinocyte damage in PV. Abbreviations: AMPVAb, anti-mitochondrial PV antibody; Cs, caspase; FasL, Fas ligand; FasR, Fas receptor; MRPVAg, mitochondria-related PV antigen; NO, nitric oxide; OPVAb, PV antibodies of other specificities, including anti-AChR and anti-PERP antibodies; OPVAg, other types of putative PV antigens; PVAb, PV antibody; TNFα, tumor necrosis factor α. Modified from Ref. Citation[37].](/cms/asset/874a7f25-cb0c-46d8-9264-cbe478211944/iaut_a_606444_f0007_b.gif)
Acantholysis and cell death (apoptosis/oncosis) are inseparable in PV because both processes are triggered by the same signal effectors activated due to PV IgG binding to keratinocytes and mediated by the same set of cell death enzymes. This is evident from the reports that inhibitors of Src, EGFRK, p38 MAPK, and mTOR block both acantholysis and apoptosis Citation[1,47,176,177,189,190,215,251] and caspase inhibitors prevent acantholysis both in vitro and in vivo Citation[190,225,246]. Moreover, it has been demonstrated that apoptotic enzymes cleave Dsg 1, 2, and 3 Citation[236,252,253].
For instance, when PV IgG was added to keratinocytes in the presence of anti-FasL neutralizing antibody, the cleavage of the intracellular portion of Dsg 3 and its degradation decreased Citation[1]. The fact that structural damage and death of keratinocytes in PV are mediated by the same set of enzymes has justified introduction of the new term “apoptolysis” to distinguish the unique mechanism of autoantibody-induced keratinocyte damage in PV from other known forms of cell death Citation[1]. The apoptolysis hypothesis links the basal cell shrinkage to suprabasal acantholysis and cell death, and emphasizes that apoptotic enzymes contribute to acantholysis in terms of both molecular events and chronologic sequence. It postulates that cell detachment and death in PV develop through the following five major steps ():
| • | Step 1: binding of pathogenic antibodies to keratinocytes via a receptor–ligand type of interaction sends an array of the agonist- and antagonist-like signals; | ||||
| • | Step 2: activation of Src, EGFRK, p38 MAPK and mTOR, and other signaling elements downstream of ligated PV antigens and elevation of intracellular Ca2+, altogether, initiate cell death enzymatic cascades; | ||||
| • | Step 3: suprabasal acantholysis starts when basal cells shrink due to reorganization of cortical actin filaments, collapse and retraction of the tonofilaments (TFs) cleaved by executioner caspases, as well as dissociation and internalization of intercellular adhesion complexes caused by phosphorylation of adhesion molecules and their cleavage by caspases; | ||||
| • | Step 4: acantholysis advances due to continued degradation and massive collapse of structural proteins by the same cell death enzymes, leading to separation and rending of preexisting desmosomes from the CM by shear forces, thus separating the collapsing cells and stimulating production of secondary (scavenging) antibodies; | ||||
| • | Step 5: rounding up and death of acantholytic cells in the lower epidermal compartment follows irreversible damage of mitochondrial and nuclear proteins. | ||||
Figure 8. Hypothetical scheme of keratinocyte apoptolysis in PV. Step 1: apoptolysis is triggered by binding of autoantibodies to the PV antigens capable of transducing apopotolytic signals from the keratinocyte plasma membrane, such as PERP and AChRs. Step 2: outside-in signaling from ligated antigens launches the cell death cascades. Step 3: collapse and retraction of the TFs cleaved by executioner caspases and dissociation of interdesmosomal adhesion complexes caused by phosphorylation of adhesion molecules result in basal cell shrinkage, most of desmosomes remain intact. Step 4: massive cleavage of cellular proteins by activated cell death enzymes leads to collapse of the cytoskeleton and tearing off desmosomes from the CM with subsequent production of scavenging (i.e. secondary) autoantibodies mainly to sloughed adhesion molecules. Step 5: suprabasal acantholytic cells die rendering a tombstone appearance to surviving basal cells. Modified from Ref. Citation[1].
![Figure 8. Hypothetical scheme of keratinocyte apoptolysis in PV. Step 1: apoptolysis is triggered by binding of autoantibodies to the PV antigens capable of transducing apopotolytic signals from the keratinocyte plasma membrane, such as PERP and AChRs. Step 2: outside-in signaling from ligated antigens launches the cell death cascades. Step 3: collapse and retraction of the TFs cleaved by executioner caspases and dissociation of interdesmosomal adhesion complexes caused by phosphorylation of adhesion molecules result in basal cell shrinkage, most of desmosomes remain intact. Step 4: massive cleavage of cellular proteins by activated cell death enzymes leads to collapse of the cytoskeleton and tearing off desmosomes from the CM with subsequent production of scavenging (i.e. secondary) autoantibodies mainly to sloughed adhesion molecules. Step 5: suprabasal acantholytic cells die rendering a tombstone appearance to surviving basal cells. Modified from Ref. Citation[1].](/cms/asset/ba45849b-fdba-4c36-8afa-fdd5d501d81b/iaut_a_606444_f0008_b.gif)
In conclusion, the mechanism of apoptolysis in PV encompasses several tiers of events triggered through distinct antigen–antibody systems. Because apoptolysis develops in a stepwise fashion, late morphologic features of apoptosis, such as rounding up, nuclear fragmentation, and plasma membrane blebbing, do not become apparent until the keratinocyte death in Step 5. Signaling mechanisms may vary from patient to patient because a unique composition of the pool of autoantibodies determines the principal pathway. The Dsg 3-dependent late peak of p38 MAPK activity and activation of JNK represent the pathways mediating processing and utilization of internalized Dsg 3, rather than a primary downstream signaling emanating from the CM. The apoptolysis hypothesis has several important implications: (1) it links together a number of apparently unrelated, and previously held contradictory, observations on the events surrounding acantholysis; (2) it opens new avenues of investigation into the pathomechanism of pemphigus; and (3) it creates new approaches to the treatment of pemphigus based on interfering with or blocking the signaling pathways and enzymatic processes that lead to blistering.
Treatment of pemphigus
The high-dose, long-term systemic glucocorticoid therapy remains the mainstay of current therapy of PV and PF patients. Corticosteroid hormones (usually prednisone tablets) are essential to establish control of PV during the acute stage Citation[2,254,255]. Optimal dosing proved to be variable and could not be predicted at the outset in any given patient Citation[4]. Some patients respond rapidly and completely to treatment with moderate doses of oral prednisone (1 mg/kg/day), others are rather refractory and require much higher doses. If there is no response after 5–7 days, the dose is increased by 50–100% Citation[256,257]. Once the control of the disease is achieved (lack of new lesions, epithelialization of existing erosions and negative Nikolskiy sign Citation[258,259]), the prednisone dosage is decreased in a “logarithmic fashion”, i.e. by 10–20% every 7–15 days. Chronic PV patients with disease exacerbation are treated exactly the same way as new patients, i.e. prednisone dose is increased until disease control is achieved and than tapered Citation[256,257].
The first report of administration of glucocorticoids to a pemphigus patient is dated to 1940 Citation[260], i.e. some 25 years earlier than pemphigus antibodies were discovered. Adrenocortical extract was tried for treatment, because it had been noticed that pemphigus is associated with changes in patients' blood chemistry characteristic of abnormal (deficient) function of the adrenal gland producing cortisone. The synthetic cortisone was introduced to the treatment of pemphigus approximately 10 years later Citation[261]. Prior to the introduction of therapy with oral corticosteroids in the 1950s, the disease had a dismal natural course with a 50% mortality rate at 2 years and 100% mortality rate by 5 years after the onset of the disease. Although there has been a significant decrease in mortality nowadays Citation[262], it remains at a relatively high level of approximately 12% Citation[263], with death being almost invariably related to complications of therapy.
The early adverse effects of systemic glucocorticosteroids that are essentially unavoidable include enhanced appetite, fluid, and salt retention leading to weight gain and neuropsychiatric disorders such as emotional lability, insomnia, irritability, anxiety, depression, euphoria, hyperactivity, and manic episodes. Delayed and insidious adverse effects that depend on cumulative dose include the Cushingoid appearance, hypothalamic–pituitary–adrenal suppression, menstrual disorders, hyperlipidemias, atherosclerosis, cardiovascular events, fatty liver, cataracts, growth retardation, osteoporosis, osteonecrosis, myopathy, muscle cramps and weakness, and skin bruising and thinning Citation[264,265]. Rare and unpredictable adverse effects are glaucoma, pancreatitis and pseudotumor cerebri. Treatment of pemphigus patients with corticosteroids can also unmask or aggravate concomitant acne vulgaris, diabetes mellitus hypertension, and peptic ulcer disease.
The goal of pemphigus research is to develop an effective treatment modality that would allow to achieve and maintain clinical remission without the need to use systemic corticosteroids. Although this goal has not been reached yet, a substantial progress was made toward development of steroid-sparing regimens. Based on assumption that glucocorticoids treat pemphigus owing to their immunosuppressive properties, most of the clinical research has been focused on the immunosuppressive therapy. The following is a chronological sequence of the reports of steroid-sparing drugs and therapeutic modalities in the treatment of pemphigus. Immunosuppressive cytotoxic drugs: methotrexate Citation[266], azathioprine Citation[267], cyclophosphamide Citation[268], chlorambucil Citation[269], and mycophenolate mofetil Citation[270]. Immunomodulators: heparin Citation[271], cyclosporine Citation[272], T-cell immunocorrection via photophoresis Citation[273], high-dose intravenous γglobulin, i.e. IVIg Citation[274], rituximab Citation[275] and daclizumab Citation[276]. Anti-inflammatory drugs: gold Citation[277], dapsone Citation[278], doxycycline Citation[279], tetracycline Citation[280], minocycline Citation[281], tranilast Citation[282], and thalidomide Citation[283]. Extracorporeal autoantibody eliminations: plasmapheresis Citation[284], plasma exchange Citation[285], hemocarbofiltration Citation[243], and protein A immunoadsorption Citation[286].
Unfortunately, these therapies do not allow reliable control of acute pemphigus without systemic glucocorticosteroids, indicating that in addition to immunosuppression the therapeutic action of corticosteroids in pemphigus includes other mechanisms, such as direct anti-acantholytic effect on keratinocytes. Moreover, although there is a bulk of evidence that PV is predominantly a Th2-type autoimmune disease, at least with regard to the anti-Dsg 3 antibody production Citation[113,144,159,287], the data on the mechanisms of immunomodulatory action of glucocorticosteroids counterintuitively demonstrate that these drugs foster Th2 polarization of CD4+ T cells Citation[288–290].
Direct anti-acantholytic effects of the corticosteroids methylprednisolone and hydrocortisone on keratinocytes were discovered in in vitro experiments, in which high doses of these drugs blocked PV IgG-induced acantholysis Citation[291,292]. Because antibody-producing cells were not present in cultures, these drugs could not exhibit their anti-acantholytic effects by way of acting upon lymphocytes. Subsequent in vivo experiments demonstrated that administration of methylprednisolone significantly decreased the extent of acantholysis in the epidermis of 3–5-day-old nude mice injected with PV IgG Citation[214]. This was in keeping with the clinical observations that blistering in pemphigus patients stops within 24–48 hrs after initiation of a high dose, “pulse” therapy with methylprednisolone or dexamethasone Citation[293–296], while the major decline in autoantibody titers occurs 3–4 weeks after initiation of glucocorticoid therapy Citation[297]. It is well known that pemphigus therapy improves disease earlier than decreasing the antibody titers Citation[298].
Also, local administration of a 0.05% clobetasol propionate cream can initially control cutaneous lesions in mild cases of PV Citation[299]. The direct effects of corticosteroids that can protect keratinocytes from PV IgG may include alterations in gene expression, as revealed by a DNA microarray assay Citation[214]. PV IgG downregulated and methylprednisolone upregulated expression of the genes encoding the keratinocyte adhesion molecules Dsg 3 and periplakin, regulators of cell cycle progression and apoptosis, differentiation markers, protein kinases and phosphatases, serine proteases and their inhibitors, and some other genes. Furthermore, methylprednisolone blocked phosphorylation of Dsg 3, E-cadherin, and β- and γ-catenins induced by PV IgG Citation[214].
These pharmacologic effects of methylprednisolone help explain the dose-dependent therapeutic action of corticosteroids in pemphigus patients. It is well known that extremely high corticosteroid doses are sometime required to attain control of acantholysis in the acute stage of disease. Thus, in addition to their immunosuppressive and anti-inflammatory actions in pemphigus, glucocorticoids can also regulate adhesion and viability of keratinocytes through a combination of their genomic and non-genomic effects.
Historically, the earliest attempt to treat pemphigus patients by protecting the target cells (keratinocytes) from the PV IgG-induced damage was made using the proteinase inhibitors aprotinin (Contrykal) and ε-aminocaproic acid Citation[242]. A few years later, Dobrev et al. Citation[300] administered p-aminomethylbenzoic acid. In both studies, the addition of protease inhibitors allowed to achieve therapeutic effects at lower doses of systemic corticosteroids, thus decreasing the risk for adverse reactions. More recently, TNFα inhibitors have been tried.
Clinical benefit in PV was reported for etanercept (Enbrel) Citation[301–303] and infliximab (Remicade) Citation[304,305], and for adalimumab (Humira) in IgA pemphigus Citation[306]. The p38 MAPK inhibitor KC-706 was used in a multicenter, open-label trial that had to be aborted due to severe adverse reactions. According to Dr Rubenstein's report at the JC Bystryn pemphigus & pemphigoid Meeting Citation[307], KC-706 had been administered orally to 15 patients with PV. One half of the patients experienced a partial response to treatment, while the remaining patients either failed to improve or experienced worsened disease.
Oral nicotinamide (niacinamide) is often used to treat pemphigus patients Citation[280,281,308,309]. Although the exact mechanism of its therapeutic action in pemphigus remains unknown, the efficacy of 4% nicotinamide gel in the treatment of cutaneous erosions of PV patients in a double-blind, placebo-controlled study Citation[310] suggests that it exerts a pharmacologic effect on target cells. In lesional skin, nicotinamide can stimulate keratinocyte adhesion and facilitate epithelialization through its cholinomimetic action Citation[311] that includes both stimulation of ACh release Citation[312] and inhibition of ACh degradation by acetylcholinesterase Citation[313].
The importance of nicotinergic stimulation for the treatment of pemphigus was first suggested by the report of a PV patient whose disease worsened when he stopped smoking and improved shortly after he resumed smoking Citation[314]. The epidemiologic studies confirmed the beneficial effect of smoking on pemphigus. Brenner et al. Citation[315] reported that 25.9% of 126 patients were smokers vs. 48.5% of controls. According to Sullivan et al. Citation[316], only 15.3% of 59 patients were current or former smokers, compared to 47.4% in the general population. These findings have recently been validated in a study involving 199 patients with PV, 11 with PF, and 205 control subjects Citation[317]. Also, it has been reported that smokers with PV achieve partial remission more frequently than non-smokers at the end of the 1st year of treatment, and that the number of patients in remission at the end of the 2nd year of therapy is significantly higher for smokers than for non-smokers Citation[318]. The cigarette smoke contains the nicotinergic agent nicotine that not only upregulates epithelialization in vitro Citation[131] but also facilitates healing of skin erosions Citation[319]. In addition to stimulation of epithelialization, nicotine may exhibit its therapeutic effect in pemphigus by affecting the immune system.
Experimental studies demonstrated that activation of nAChRs suppresses B-cell activation Citation[320], abrogates phytohemagglutinin-dependent upregulation of TNFα and IFNγ receptors in T cells Citation[321], inhibits expression of the TNFα, IL-6 and IL-18 genes, and upregulates IL-10 production in macrophages Citation[322]. Furthermore, the nicotinergic signaling facilitates T-cell polarization toward Th1 lineage, inhibits Th17 differentiation, and upregulates Tregs Citation[323,324]. Altogether, these immunopharmacologic effects of nicotine may be able to correct the immune dysregulation characteristic of pemphigus ().
The perspective for the development of steroid-sparing therapy employing cholinergic drugs is very promising, because cholinergic agonists of both nicotinic and muscarinic classes have already shown their therapeutic activities in pemphigus patients. The nAChRs in keratinocytes can be directly activated by pyridostigmine bromide (Mestinon) Citation[325]. Besides its underappreciated nicotinergic action, pyridostigmine bromide is reversible acetylcholinesterase inhibitor Citation[326] that can elevate tissue levels of auto/paracrine ACh, thus augmenting signaling through both muscarinic and nicotinic pathways in the cells secreting ACh, like human keratinocytes Citation[327]. Pyridostigmine bromide has been shown to antagonize the effects of PV antibodies in both in vitro Citation[328] and in vivo experiments Citation[329]. Most importantly, a clinical trial of Mestinon in the treatment of eight pemphigus patients brought encouraging results Citation[330]. Three patients showed a very good response, and five patients did not show any significant improvement. One patient was able to discontinue glucocorticosteroids and immunosuppressive medications, and control the disease using Mestinon only.
In contrast to glucocorticosteroids and protease inhibitors that can only block but not reverse acantholysis Citation[291,331], muscarinic agonists both prevented cell detachment and restored the integrity of keratinocyte monolayers exposed to PV IgGs, the serine proteinase trypsin or the calcium chelator EDTA Citation[130]. These observations indicate that the muscarinic effects stem from activation of the epithelialization program that comprises both the adhesive and the migratory functions of keratinocytes (reviewed in Citation[55,198]). Consistent with the ability of the cholinomimetic carbachol to prevent acantholysis and skin blistering in the neonatal athymic nude mice with passively transferred PV IgGs Citation[329], a double-blind, placebo-controlled study showed therapeutic activity of the muscarinic agonist pilocarpine 4% gel applied to skin erosions of PV patients Citation[332].
Pilocarpine preferentially binds to and activate the M1 molecular subtype of keratinocyte mAChRs Citation[333] that has been recently found to be specifically targeted by PV antibodies Citation[49]. Pilocarpine blocks PV IgG-induced phosphorylation of p120- and β-catenins in keratinocytes, because it elevates both serine/threonine and tyrosine phosphatase activities Citation[215]. Furthermore, the anti-acantholytic activity of pilocarpine synergizes with that of the α7 nAChR agonist AR-R17779 that both activates tyrosine phosphatase and inhibits Src Citation[215]. Taken together, these findings identify novel paradigm of regulation of signaling kinases associated with cholinergic receptors and provide mechanistic explanation of therapeutic activity of cholinomimetics in PV patients.
A large variety of steroid-sparing effects reported thus far in the literature suggests that it should be possible to replace corticosteroids by combining the steroid-sparing drugs and/or treatment modalities that can provide for simultaneous inhibition of antibody production and protection of keratinocytes from autoantibody action. Unfortunately, such putative combination has not been devised yet, though a combination of rituximab and IVIg allows to treat certain PV patients without corticosteroids Citation[334]. Most recently, it has been reported that the mTOR inhibitor sirolimus (Rapamune, Rapamycin®) combined with IVIg allowed rapid and complete withdrawal of systemic glucocorticosteroids in a PV patient who developed disease exacerbation that could not be controlled with 40 mg/day of prednisone Citation[120]. Sirolimus is a naturally occurring lipophilic microcyclic lactone isolated from Streptomyces hygroscopicus discovered at Rapa Nui (Easter Island). It binds to immunophilin and FK binding protein-12.
The sirolimus–FKBP-12 complex targets the 290 kD serine–threonine kinase of the phosphoinositide 3-kinases/Akt pathway termed mTOR Citation[335]. Sirolimus exhibits potent immunosuppressive activity due to suppression of T- and B-cell activation and IL-2- and IL-4-dependent proliferation via inhibition of new ribosomal protein synthesis and arrest of the G1–S phase of the cell cycle Citation[336]. In PV, the immunosuppressive action of sirolimus may become therapeutic when it is taken for a period of time. The rapid healing of the skin lesion in the reported case of PV patient in the acute stage of disease Citation[120], however, suggests that it had a direct effect on keratinocytes that protected them from autoantibody action.
As already mentioned, the mTOR pathway is activated in keratinocytes exposed to PV IgG and mTOR inhibition prevents acantholysis in the murine passive transfer model of PV IgG Citation[190]. Additionally, sirolimus may prevent damage of keratinocytes and enforce their adhesive function by inhibiting reorganization of the actin cytoskeleton and phosphorylation of focal adhesion proteins, and upregulating E-cadherin expression Citation[337,338]. The clinical trial of sirolimus in pemphigus patients is currently underway at University of California Irvine (http://clinicaltrials.gov/ct2/show/NCT01313923).
In conclusion, corticosteroids remain an essential component of pemphigus treatment. Early and aggressive use of corticosteroids is required to decrease the duration of treatment and avoid relapses. Adjuvant drugs allow a decrease in the total dose of corticosteroids. The natural course of pemphigus has improved with new therapies. Cholinomimetics can achieve a steroid-sparing effect in pemphigus patients by both stimulating epithelialization and inhibiting autoimmune aggression. The dualistic pharmacologic action of sirolimus that affects both effectors of autoimmunity and target cells apparently mediates its therapeutic effect in pemphigus. Further elucidation of the molecular mechanisms mediating aberrant signaling along the mTOR pathway in PV should improve our understanding of the pathogenesis and lead to novel therapeutic approaches for the development of steroid-free treatment of pemphigus.
Summary
| (1) | Recent advances of knowledge on pemphigus autoimmunity scrutinize old dogmas, resolve controversies, and open novel perspectives for treatment (). | ||||
| (2) | The initial insult is sustained by the autoantibodies to the cell-membrane receptor antigens triggering the intracellular signaling by Src, EGFRK, PKC, phospholipase C, mTOR, p38 MAPK, other tyrosine kinases, and calmodulin that cause basal cell shrinkage and ripping desmosomes off the CM. | ||||
| (3) | Autoantibodies synergize with the effectors of apoptotic and oncotic pathways, serine proteases and inflammatory cytokines to overcome the natural resistance and activate the cell death program in keratinocytes. | ||||
| (4) | The process of keratinocyte shrinkage/detachment and death via apoptosis/oncosis has been termed apoptolysis to emphasize that it is triggered by the same signal effectors and mediated by the same cell death enzymes. | ||||
| (5) | Although a high-dose, long-term systemic glucocorticoid therapy remains the mainstay of current treatment of patients with PV or PF, causing severe adverse effects, a substantial progress has been made toward development of steroid-sparing therapies combining the immunosuppressive and direct anti-acantholytic effects. | ||||
| (6) | The onset of acantholysis in drug-induced pemphigus in patients taking angiotensin-converting enzyme inhibitors, such as captopril, apparently involves a drop in the concentration of auto/paracrine ACh that sustains keratinocyte shape and cohesiveness, due to a strong upregulation of the ACh degrading enzyme acetylcholinesterase Citation[351]. | ||||
Table II. Hypotheses and realities in the knowledge of pemphigus.
Declaration of Interest: The author reports no conflicts of interest. The author alone is responsible for the content and writing of the paper.
Related Research Data
References
- Grando SA, Bystryn JC, Chernyavsky AI, Frusic-Zlotkin M, Gniadecki R, Lotti R, Milner Y, Pittelkow MR, Pincelli C. Apoptolysis: A novel mechanism of skin blistering in pemphigus vulgaris linking the apoptotic pathways to basal cell shrinkage and suprabasal acantholysis. Exp Dermatol. 2009; 18:764–770.
- Carson PJ, Hameed A, Ahmed AR. Influence of treatment on the clinical course of Pemphigus vulgaris. J Am Acad Dermatol. 1996; 34:645–652.
- Ahmed AR, Moy R. Death in Pemphigus. J Am Acad Dermatol. 1982; 7:221–228.
- Rosenberg FR, Sanders S, Nelson CT. Pemphigus: A 20-year review of 107 patients treated with corticosteroids. Arch Dermatol. 1976; 112:962–970.
- Anhalt GJ, Kim SC, Stanley JR, Korman NJ, Jabs DA, Kory M, Izumi H3rd, Ratrie H, Mutasim D, Ariss-Abdo L, Labib RS. Paraneoplastic Pemphigus. An autoimmune mucocutaneous disease associated with neoplasia. N Engl J Med. 1990; 323:1729–1735.
- Kaplan RP, Callen JP. Pemphigus associated diseases and induced Pemphigus. Clin Dermatol. 1983; 1:42–71.
- Armin A, Nadimi H, Robinson J. Pemphigus vulgaris and malignancy. Int J Oral Surg. 1985; 14:376–380.
- Younus J, Ahmed AR. The relationship of Pemphigus to neoplasia. J Am Acad Dermatol. 1990; 23:498–502.
- Ohyama M, Amagai M, Hashimoto T, Nousari HC, Anhalt GJ, Nishikawa T. Clinical phenotype and anti-desmoglein autoantibody profile in paraneoplastic Pemphigus. J Am Acad Dermatol. 2001; 44:593–598.
- Nguyen VT, Ndoye A, Bassler KD, Shultz LD, Shields MC, Ruben BS, Webber RJ, Pittelkow MR, Lynch PJ, Grando SA. Classification, clinical manifestations, and immunopathological mechanisms of the epithelial variant of paraneoplastic autoimmune multiorgan syndrome: A reappraisal of paraneoplastic Pemphigus. Arch Dermatol. 2001; 137:193–206.
- Nousari HC, Deterding R, Wojtczack H, Aho S, Uitto J, Hashimoto T, Anhalt GJ. The mechanism of respiratory failure in paraneoplastic Pemphigus. N Engl J Med. 1999; 340:1406–1410.
- Czernik A, Camilleri M, Pittelkow MR, Grando SA. Paraneoplastic autoimmune multiorgan syndrome: Twenty years after. Int J Dermatol. 2011; 50 8: 905–914.
- Billet SE, Grando SA, Pittelkow MR. Paraneoplastic autoimmune multiorgan syndrome: Review of the literature and support for a cytotoxic role in pathogenesis. Autoimmunity. 2006; 39:617–630.
- Beutner E, Jordon R. Demonstration of skin antibodies in sera of Pemphigus vulgaris patients by indirect immunofluorescent staining. Proc Soc Exp Biol Med. 1964; 117:505–510.
- Beutner EH, Prigenzi LS, Hale W, Leme Cde A, Bier OG. Immunofluorescent studies of autoantibodies to intercellular areas of epithelia in Brazilian Pemphigus foliaceus. Proc Soc Exp Biol Med. 1968; 127:81–86.
- Stanley JR, Yaar M, Hawley-Nelson P, Katz SI. Pemphigus antibodies identify a cell surface glycoprotein synthesized by human and mouse keratinocytes. J Clin Invest. 1982; 70:281–288.
- Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in Pemphigus vulgaris, a disease of cell adhesion. Cell. 1991; 67:869–877.
- Nguyen VT, Ndoye A, Shultz LD, Pittelkow MR, Grando SA. Antibodies against keratinocyte antigens other than desmogleins 1 and 3 can induce Pemphigus vulgaris-like lesions. J Clin Invest. 2000; 106:1467–1479.
- Ablin RJ, Bronson P, Beutner EH. Immunochemical characterization of epithelial antigen(s) reactive with Pemphigus-like antibodies of rabbit and human Pemphigus autoantibodies. J Hyg Epidemiol Microbiol Immunol. 1969; 13:321–329.
- Shu SY, Beutner EH. Isolation and characterization of antigens reactive with Pemphigus antibodies. J Invest Dermatol. 1973; 61:270–276.
- Miyagawa S, Hojo T, Ishii H, Yoshioka J, Sakamoto K. Isolation and characterization of soluble epidermal antigens reactive with Pemphigus antibodies. Acta Derm Venereol. 1977; 57:7–13.
- Calvanico NJ, Swartz SJ. A non-desmoglein component of bovine epidermis reactive with Pemphigus foliaceus sera. J Autoimmun. 1994; 7:231–242.
- Calvanico NJ, Martins CR, Diaz LA. Characterization of Pemphigus foliaceus antigen from human epidermis. J Invest Dermatol. 1991; 96:815–821.
- Diaz LA, Patel H, Calvanico NJ. Isolation of Pemphigus antigen from human saliva. J Immunol. 1980; 124:760–765.
- Diaz LA, Sampaio SA, Martins CR, Rivitti EA, Macca ML, Roscoe JT, . An autoantibody in Pemphigus serum, specific for the 59 kD keratin, selectively binds the surface of keratinocytes: Evidence for an extracellular keratin domain. J Invest Dermatol. 1987; 89:287–295.
- Murahata RI, Ahmed AR. Partial purification and characterization of Pemphigus-like antigens in urine. Arch Derm Res. 1983; 275:118–123.
- Peterson LL, Wuepper KD. Isolation and purification of a Pemphigus vulgaris antigen from human epidermis. J Clin Invest. 1984; 73:1113–1120.
- Acosta E, Ivanyi L. Identification of Pemphigus-like antigens expressed by SCaBER cells. Br J Dermatol. 1985; 112:157–164.
- Stanley JR, Koulu L, Thivolet C. Distinction between epidermal antigens binding Pemphigus vulgaris and Pemphigus foliaceus autoantibodies. J Clin Invest. 1984; 74:313–320.
- Gilbert D, Joly P, Jouen F, Thibout A, Delpech A, Thomine E, Lauret P, Tron F. Production of a human monoclonal anti-epithelial cell surface antibody derived from a patient with Pemphigus vulgaris. J Autoimmun. 1992; 5:173–182.
- Lyubimov H, Goldshmit D, Michel B, Oron Y, Milner Y. Pemphigus—identifying the autoantigen and its possible induction of epidermal acantholysis via Ca2+ signalling. Israel J Med Sci. 1995; 31:42–48.
- Kiss M, Husz S, Molnar K, Dobozy A. Identification of different circulating autoantibodies in patients with bullous Pemphigoid and Pemphigus vulgaris by means of immunoblotting. Acta Microbiol Immunol Hung. 1996; 43:115–123.
- Ding X, Aoki V, Mascaro JMJr, Lopez-Swiderski A, Diaz LA, Fairley JA. Mucosal and mucocutaneous (generalized) Pemphigus vulgaris show distinct autoantibody profiles. J Invest Dermatol. 1997; 109:592–596.
- Joly P, Gilbert D, Thomine E, Zitouni M, Ghohestani R, Delpech A, Lauret P, Tron F. Identification of a new antibody population directed against a desmosomal plaque antigen in Pemphigus vulgaris and Pemphigus foliaceus. J Invest Dermatol. 1997; 108:469–475.
- Eyre RW, Stanley JR. Identification of Pemphigus vulgaris antigen extracted from normal human epidermis and comparison with Pemphigus foliaceus antigen. J Clin Invest. 1988; 81:807–812.
- Nguyen VT, Ndoye A, Grando SA. Novel human α9 acetylcholine receptor regulating keratinocyte adhesion is targeted by Pemphigus vulgaris autoimmunity. Am J Pathol. 2000; 157:1377–1391.
- Marchenko S, Chernyavsky AI, Arredondo J, Gindi V, Grando SA. Antimitochondrial autoantibodies in Pemphigus vulgaris: A missing link in disease pathophysiology. J Biol Chem. 2010; 285:3695–3704.
- Cirillo N, Gombos F, Lanza A. Pemphigus vulgaris immunoglobulin G can recognize a 130 000 MW antigen other than desmoglein 3 on peripheral blood mononuclear cell surface. Immunology. 2007; 121:377–382.
- Mejri K, Abida O, Kallel-Sellami M, Haddouk S, Laadhar L, Zarraa I, Ben Ayed M, Zitouni M, Mokni M, Lahmar H, Fezaa B, Turki H, Tron F, Masmoudi H, Makni S. Spectrum of autoantibodies other than anti-desmoglein in Pemphigus patients. J Eur Acad Dermatol Venereol. 2010; 25 7: 774–781.
- Stanley JR, Koulu L, Klaus-Kovtun V, Steinberg MS. A monoclonal antibody to the desmosomal glycoprotein desmoglein I binds the same polypeptide as human autoantibodies in Pemphigus foliaceus. J Immunol. 1986; 136:1227–1230.
- Kurzen H, Brenner S. Significance of autoimmunity to non-desmoglein targets in Pemphigus. Autoimmunity. 2006; 39:549–556.
- Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR. Explanations for the clinical and microscopic localization of lesions in Pemphigus foliaceus and vulgaris. J Clin Invest. 1999; 103:461–468.
- Chen J, Den Z, Koch PJ. Loss of desmocollin 3 in mice leads to epidermal blistering. J Cell Sci. 2008; 121:2844–2849.
- Amagai M, Karpati S, Klaus-Kovtun V, Udey MC, Stanley JR. Extracellular domain of Pemphigus vulgaris antigen (Desmoglein 3) mediates weak homophilic adhesion. J Invest Dermatol. 1994; 102:402–408.
- Green KJ, Simpson CL. Desmosomes: New perspectives on a classic. J Invest Dermatol. 2007; 127:2499–2515.
- Desai BV, Harmon RM, Green KJ. Desmosomes at a glance. J Cell Sci. 2009; 122:4401–4407.
- Chernyavsky AI, Arredondo J, Kitajima Y, Sato-Nagai M, Grando SA. Desmoglein versus non-desmoglein signaling in Pemphigus acantholysis: Characterization of novel signaling pathways downstream of Pemphigus vulgaris antigens. J Biol Chem. 2007; 282:13804–13812.
- Nguyen VT, Lee TX, Ndoye A, Shultz LD, Pittelkow MR, Dahl MV, Lynch PJ, Grando SA. The pathophysiological significance of non-desmoglein targets of Pemphigus autoimmunity. Pemphigus vulgaris and foliaceus patients develop antibodies against keratinocyte cholinergic receptors. Arch Dermatol. 1998; 134:971–980.
- Kalantari-Dehaghi M, Molina DM, Farhadieh M, Morrow WJ, Liang X, Felgner PL, Grando SA. New targets of Pemphigus vulgaris antibodies identified by protein array technology. Exp Dermatol. 2011; 20:154–156.
- Kalantari-Dehaghi M, Anhalt G, Camilleri M, Leiferman KM, Marchenko S, Pittelkow M, Zone JJ, Grando SA. Pemphigus vulgaris antibodies target PERP and several other keratinocyte membrane and mitochondrial proteins. J Invest Dermatol. 2011; 131 Suppl. 1: S11.
- Ihrie RA, Marques MR, Nguyen BT, Horner JS, Papazoglu C, Bronson RT, Mills AA, Attardi LD. Perp is a p63-regulated gene essential for epithelial integrity. Cell. 2005; 120:843–856.
- Bektas M, Rubenstein DS. Perp and Pemphigus: A disease of desmosome destabilization. J Invest Dermatol. 2009; 129:1606–1608.
- Ihrie RA, Reczek E, Horner JS, Khachatrian L, Sage J, Jacks T, Attardi LD. Perp is a mediator of p53-dependent apoptosis in diverse cell types. Curr Biol. 2003; 13:1985–1990.
- Davies L, Gray D, Spiller D, White MR, Damato B, Grierson I, Paraoan L. P53 apoptosis mediator PERP: Localization, function and caspase activation in uveal melanoma. J Cell Mol Med. 2009; 13:1995–2007.
- Grando SA. Cholinergic control of epidermal cohesion in norm and pathology. Exp Dermatol. 2006; 15:265–282.
- Grando SA, Kurzen H. 2009. Cholinergic control of keratinocyte cohesion. In: Cirillo N, Lanza A, Gombo F. editors. Pathophysiology of the desmosome104 Research Signpost, Managing Editor Dr. S.G. Pandalai, Trivandrum, Kerala, India.
- Grando SA, George PM, Dahl MV, Conti-Tronconi BM. Antibody against keratinocyte nicotinic acetylcholine receptor in patient with coexistent Pemphigus foliaceus, myasthenia gravis and thymoma. J Invest Dermatol. 1994; 102:609 (Abstr.).
- Craft J, Fatenejad S. Self antigens and epitope spreading in systemic autoimmunity. Arthritis Rheum. 1997; 40:1374–1382.
- Lindstrom J. Autoimmune diseases involving nicotinic receptors. J Neurobiol. 2002; 53:656–665.
- Kalantari M, Molina DM, Farhadieh M, Vigil A, Morrow WJ, Liang X, Felgner PL, Grando SA. Partial evaluation of Pemphigus vulgaris autoantibody profile using the protein array technology. J Invest Dermatol. 2010; 130 Suppl. 1: S21.
- Patel M, Furstenberg G, Hazelton J, Tiagi S, Seiffert-Sinha K, Haab BB, Robinson WH, Sinha AA. Development of protein microarrays to investigate autoantibody profiles in Pemphigus vul-garis (abstr. #111). J Invest Dermatol. 2010; 130 Suppl. 1: s19.
- Nguyen VT, Ndoye A, Grando SA. Pemphigus vulgaris antibody identifies pemphaxin—a novel keratinocyte annexin-like molecule binding acetylcholine. J Biol Chem. 2000; 275:29466–29476.
- Bastian BC, Nuss B, Romisch J, Kraus M, Brocker EB. Autoantibodies to annexins: A diagnostic marker for cutaneous disorders?. J Dermatol Sci. 1994; 8:194–202.
- Abreu Velez AM, Yi H, Gao W, Smoller BR, Grossniklaus HE, Howard MS. Antibodies to pilosebaceous units along their neurovascular supply routes in a new variant of endemic Pemphigus foliaceus in Colombia, South America. Eur J Dermatol. 2011; 21 3: 371–375.
- Echigo T, Hasegawa M, Inaoki M, Yamazaki M, Sato S, Takehara K. Antiphospholipid antibodies in patients with autoimmune blistering disease. J Am Acad Dermatol. 2007; 57:397–400.
- Blondin DA, Zhang Z, Shideler KK, Hou H, Fritzler MJ, Mydlarski PR. Prevalence of non-organ-specific autoantibodies in patients with Pemphigus vulgaris. J Cutan Med Surg. 2009; 13:82–87.
- Amagai M, Karpati S, Prussick R, Klaus-Kovtun V, Stanley JR. Autoantibodies against the amino-terminal cadherin-like binding domain of Pemphigus vulgaris antigen are pathogenic. J Clin Invest. 1992; 90:919–926.
- Amagai M, Hashimoto T, Shimizu N, Nishikawa T. Absorption of pathogenic autoantibodies by the extracellular domain of Pemphigus vulgaris antigen (Dsg3) produced by baculovirus. J Clin Invest. 1994; 94:59–67.
- Amagai M, Hashimoto T, Green KJ, Shimizu N, Nishikawa T. Antigen-specific immunoadsorption of pathogenic autoantibodies in Pemphigus foliaceus. J Invest Dermatol. 1995; 104:895–901.
- Amagai M, Nishikawa T, Nousari HC, Anhalt GJ, Hashimoto T. Antibodies against desmoglein 3 (Pemphigus vulgaris antigen) are present in sera from patients with paraneoplastic Pemphigus and cause acantholysis in vivo in neonatal mice. J Clin Invest. 1998; 102:775–782.
- Moller NP. Fc-mediated immune precipitation. I. A new role of the Fc-portion of IgG. Immunology. 1979; 38:631–640.
- Moller NP, Steensgaard J. Fc-mediated immune precipitation. II. Analysis of precipitating immune complexes by rate-zonal ultracentrifugation. Immunology. 1979; 38:641–648.
- Kolenko P, Dohnalek J, Duskova J, Skalova T, Collard R, Hasek J. New insights into intra- and intermolecular interactions of immunoglobulins: Crystal structure of mouse IgG2b-Fc at 2.1-A resolution. Immunology. 2009; 126:378–385.
- Kaithamana S, Fan JL, Memar O, Li K, Jutto J, Seetharamaiah GS, Prabhakar BS. Relevance of differential immunogenicity of human and mouse recombinant desmoglein-3 for the induction of acantholytic autoantibodies in mice. Clin Exp Immunol. 2003; 132:16–23.
- Angelini G, Bonamonte D, Lin MS, Lucchese A, Mittelman A, Serpico R, Simone S, Sinha AA, Kanduc D. Characterization of polyclonal antibodies raised against a linear peptide determinant of desmoglein-3. J Exp Ther Oncol. 2005; 5:1–7.
- Nishimura H, Strominger JL. Involvement of a tissue-specific autoantibody in skin disorders of murine systemic lupus erythematosus and autoinflammatory diseases. Proc Natl Acad Sci USA. 2006; 103:3292–3297.
- Amagai M, Tsunoda K, Suzuki H, Nishifuji K, Koyasu S, Nishikawam T. Use of autoantigen-knockout mice in developing an active autoimmune disease model for Pemphigus. J Clin Invest. 2000; 105:625–631.
- Aoki-Ota M, Tsunoda K, Ota T, Iwasaki T, Koyasu S, Amagai M, Nishikawa T. A mouse model of Pemphigus vulgaris by adoptive transfer of naive splenocytes from desmoglein 3 knockout mice. Br J Dermatol. 2004; 151:346–354.
- Amagai M. Pemphigus vulgaris and its active disease mouse model. Curr Dir Autoimmun. 2008; 10:167–181.
- Holm TL, Markholst H. Confirmation of a disease model of Pemphigus vulgaris: Characterization and correlation between disease parameters in 90 mice. Exp Dermatol. 2010; 19:e158–e165.
- Ishii K, Harada R, Matsuo I, Shirakata Y, Hashimoto K, Amagai M. In vitro keratinocyte dissociation assay for evaluation of the pathogenicity of anti-desmoglein 3 IgG autoantibodies in Pemphigus vulgaris. J Invest Dermatol. 2005; 124:939–946.
- Bhol KC, Ahmed AR. Production of non-pathogenic human monoclonal antibodies to desmoglein 3 from Pemphigus vulgaris patient. Autoimmunity. 2002; 35:87–91.
- Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, Tsunoda K, Amagai M, Stanley JR, Siegel DL. Genetic and functional characterization of human Pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest. 2005; 115:888–899.
- Shimizu A, Ishiko A, Ota T, Tsunoda K, Amagai M, Nishikawa T. IgG binds to desmoglein 3 in desmosomes and causes a desmosomal split without keratin retraction in a Pemphigus mouse model. J Invest Dermatol. 2004; 122:1145–1153.
- Shimizu A, Ishiko A, Ota T, Saito H, Oka H, Tsunoda K, Amagai M, Nishikawa T. In vivo ultrastructural localization of the desmoglein 3 adhesive interface to the desmosome mid-line. J Invest Dermatol. 2005; 124:984–989.
- Lever WF. Pemphigus. Medicine. 1953; 32:2–123.
- Wilgram GF, Caulfield JB, Lever WF. An electron microscopic study of acantholysis in Pemphigus vulgaris. J Invest Dermatol. 1961; 36:373–382.
- Hu CH, Michel B, Schiltz JR. Epidermal acantholysis induced in vitro by Pemphigus autoantibody. An ultrastructural study. Am J Pathol. 1978; 90:345–361.
- Barlow Y, Wray D. Ultrastructural alterations associated with in vivo and in vitro bound Pemphigus antibodies in cultured oral epithelial cells. J Oral Pathol Med. 1991; 20:241–244.
- Diercks GF, Pas HH, Jonkman MF. The ultrastructure of acantholysis in Pemphigus vulgaris. Br J Dermatol. 2009; 160:450–474.
- Lambert LL, Spriet E, Vandewiele A, Naeyaert J. Desmoglein 1 and 3 IgG auto-antibody titers do not correlate with Pemphigus disease activity in a prospective study. J Invest Dermatol. 2006; 126 Suppl. 1: 11 (Abstract 65).
- Abasq C, Mouquet H, Gilbert D, Tron F, Grassi V, Musette P, Joly P. ELISA testing of anti-desmoglein 1 and 3 antibodies in the management of Pemphigus. Arch Dermatol. 2009; 145:529–535.
- Akman A, Uzun S, Alpsoy E. Immunopathologic features of Pemphigus in the east Mediterranean region of Turkey: A prospective study. Skinmed. 2010; 8:12–16.
- Arin MJ, Engert A, Krieg T, Hunzelmann N. Anti-CD20 monoclonal antibody (rituximab) in the treatment of Pemphigus. Br J Dermatol. 2005; 153:620–625.
- Kwon EJ, Yamagami J, Nishikawa T, Amagai M. Anti-desmoglein IgG autoantibodies in patients with Pemphigus in remission. J Eur Acad Dermatol Venereol. 2008; 22:1070–1075.
- Belloni-Fortina A, Faggion D, Pigozzi B, Peserico A, Bordignon M, Baldo V, Alaibac M. Detection of autoantibodies against recombinant desmoglein 1 and 3 molecules in patients with Pemphigus vulgaris: Correlation with disease extent at the time of diagnosis and during follow-up. Clin Dev Immunol. 2009; 2009:187864.
- Brandsen R, Frusic-Zlotkin M, Lyubimov H, Yunes F, Michel B, Tamir A, Milner Y, Brenner S. Circulating Pemphigus IgG in families of patients with Pemphigus: Comparison of indirect immunofluorescence, direct immunofluorescence, and immunoblotting. J Am Acad Dermatol. 1997; 36:44–52.
- Warren SJ, Lin MS, Giudice GJ, Hoffmann RG, Hans-Filho G, Aoki V, Rivitti EA, Santos V, Diaz LA. The prevalence of antibodies against desmoglein 1 in endemic Pemphigus foliaceus in Brazil. Cooperative Group on Fogo Selvagem Research. N Engl J Med. 2000; 343:23–30.
- Kricheli D, David M, Frusic-Zlotkin M, Goldsmith D, Rabinov M, Sulkes J, Milner Y. The distribution of Pemphigus vulgaris-IgG subclasses and their reactivity with desmoglein 3 and 1 in Pemphigus patients and their first-degree relatives. Br J Dermatol. 2000; 143:337–342.
- Torzecka JD, Narbutt J, Sysa-Jedrzejowska A, Waszczykowska E, Lukamowicz J, Pas HH. Detection of Pemphigus autoantibodies by IIF and ELISA tests in patients with Pemphigus vulgaris and foliaceus and in healthy relatives. Med Sci Monit. 2003; 9:CR528–CR533.
- Torzecka JD, Wozniak K, Kowalewski C, Waszczykowska E, Sysa-Jedrzejowska A, Pas HH, Narbutt J. Circulating Pemphigus autoantibodies in healthy relatives of Pemphigus patients: Coincidental phenomenon with a risk of disease development?. Arch Dermatol Res. 2007; 299:239–243.
- Hilario-Vargas J, Dasher DA, Li N, Aoki V, Hans-Filho G, Dos Santos V, Qaqish BF, Rivitti EA, Diaz LA. Prevalence of anti-desmoglein-3 antibodies in endemic regions of Fogo Selvagem in Brazil. J Invest Dermatol. 2006; 126:2044–2048.
- Ortega Loayza AG, Ramos W, Elgart G, Bouman P, Jimenez G, Avila J, Rojas I, Vilcarromero M, Hurtado J, Lindo G, Galarza C. Antibodies against desmoglein 1 in healthy subjects in endemic and nonendemic areas of Pemphigus foliaceus (fogo selvagem) in Peru. Int J Dermatol. 2006; 45:538–542.
- Diaz LA, Arteaga LA, Hilario-Vargas J, Valenzuela JG, Li N, Warren S, Aoki V, Hans-Filho G, Eaton D, dos Santos V, Nutman TB, de Mayolo AA, Qaqish BF, Sampaio SA, Rivitti EA. Anti-desmoglein-1 antibodies in onchocerciasis, leishmaniasis and Chagas disease suggest a possible etiological link to Fogo selvagem. J Invest Dermatol. 2004; 123:1045–1051.
- Yoshimura T, Seishima M, Nakashima K, Yasuhara Y, Adachi S, Kawaguchi M, Minoura N, Nakao T, Kobayashi J, Yamazaki F. Increased antibody levels to desmogleins 1 and 3 after administration of carbamazepine. Clin Exp Dermatol. 2001; 26:441–445.
- Gallo R, Massone C, Parodi A, Guarrera M. Allergic contact dermatitis from thiurams with Pemphigus-like autoantibodies. Contact Dermatitis. 2002; 46:364–365.
- Cozzani E, Rosa GM, Drosera M, Intra C, Barsotti A, Parodi A. ACE inhibitors can induce circulating antibodies directed to antigens of the superficial epidermal cells. Arch Dermatol Res. 2010; 303 5: 327–332.
- Khandpur S, Sharma VK, Sharma A, Pathria G, Satyam A. Comparison of enzyme-linked immunosorbent assay test with immunoblot assay in the diagnosis of Pemphigus in Indian patients. Indian J Dermatol Venereol Leprol. 2010; 76:27–32.
- Empinotti JC, Aoki V, Filgueira A, Sampaio SA, Rivitti EA, Sanches JAJr, Li N, Hilario-Vargas J, Diaz LA. Clinical and serological follow-up studies of endemic Pemphigus foliaceus (fogo selvagem) in Western Parana, Brazil (2001–2002). Br J Dermatol. 2006; 155:446–450.
- Bhol K, Mohimen A, Ahmed AR. Correlation of subclasses of IgG with disease activity in Pemphigus vulgaris. Dermatology. 1994; 189 Suppl 1: 85–89.
- Bhol K, Natarajan K, Nagarwalla N, Mohimen A, Aoki V, Ahmed AR. Correlation of peptide specificity and IgG subclass with pathogenic and nonpathogenic autoantibodies in Pemphigus vulgaris: A model for autoimmunity. Proc Natl Acad Sci USA. 1995; 92:5239–5243.
- Ayatollahi M, Joubeh S, Mortazavi H, Jefferis R, Ghaderi A. IgG4 as the predominant autoantibody in sera from patients with active state of Pemphigus vulgaris. J Eur Acad Dermatol Venereol. 2004; 18:241–242.
- Nagel A, Lang A, Engel D, Podstawa E, Hunzelmann N, de Pita O, Borradori L, Uter W, Hertl M. Clinical activity of Pemphigus vulgaris relates to IgE autoantibodies against desmoglein 3. Clin Immunol. 2010; 134:320–330.
- Mentink LF, de Jong MC, Kloosterhuis GJ, Zuiderveen J, Jonkman MF, Pas HH. Coexistence of IgA antibodies to desmogleins 1 and 3 in Pemphigus vulgaris, Pemphigus foliaceus and paraneoplastic Pemphigus. Br J Dermatol. 2007; 156:635–641.
- Amagai M, Tsunoda K, Zillikens D, Nagai T, Nishikawa T. The clinical phenotype of Pemphigus is defined by the anti-desmoglein autoantibody profile. J Am Acad Dermatol. 1999; 40:167–170.
- Arteaga LA, Prisayanh PS, Warren SJ, Liu Z, Diaz LA, Lin MS. A subset of Pemphigus foliaceus patients exhibits pathogenic autoantibodies against both desmoglein-1 and desmoglein-3. J Invest Dermatol. 2002; 118:806–811.
- Cunha PR, Bystryn JC, Medeiros EP, de Oliveira JR. Sensitivity of indirect immunofluorescence and ELISA in detecting intercellular antibodies in endemic Pemphigus foliaceus (Fogo Selvagem). Int J Dermatol. 2006; 45:914–918.
- Zagorodniuk I, Weltfriend S, Shtruminger L, Sprecher E, Kogan O, Pollack S, Bergman R. A comparison of anti-desmoglein antibodies and indirect immunofluorescence in the serodiagnosis of Pemphigus vulgaris. Int J Dermatol. 2005; 44:541–544.
- Sharma VK, Prasad HR, Khandpur S, Kumar A. Evaluation of desmoglein enzyme-linked immunosorbent assay (ELISA) in Indian patients with Pemphigus vulgaris. Int J Dermatol. 2006; 45:518–522.
- Grando SA, Laquer VT, Le HM. Sirolimus for acute Pemphigus vulgaris: A case report and discussion of dualistic action providing for both immunosuppression and keratinocyte protection. J Am Acad Dermatol. 2011; 65 3: 684–686.
- Amagai M, Ahmed AR, Kitajima Y, Bystryn JC, Milner Y, Gniadecki R, Hertl M, Pincelli C, Fridkis-Hareli M, Aoyama Y, Frusic-Zlotkin M, Muller E, David M, Mimouni D, Vind-Kezunovic D, Michel B, Mahoney M, Grando S. Are desmoglein autoantibodies essential for the immunopathogenesis of Pemphigus vulgaris, or just “witnesses of disease”?. Exp Dermatol. 2006; 15:815–831.
- Ohata Y, Amagai M, Ishii K, Hashimoto T. Immunoreactivity against intracellular domains of desmogleins in Pemphigus. J Dermatol Sci. 2001; 25:64–71.
- Lanza A, Femiano F, De Rosa A, Cammarota M, Lanza M, Cirillo N. The N-terminal fraction of desmoglein 3 encompassing its immunodominant domain is present in human serum: Implications for Pemphigus vulgaris autoimmunity. Int J Immunopathol Pharmacol. 2006; 19:399–407.
- Dmochowski M, Hashimoto T, Garrod DR, Nishikawa T. Desmocollins I and II are recognized by certain sera from patients with various types of Pemphigus particularly Brazilian Pemphigus foliaceus. J Invest Dermatol. 1993; 100:380–384.
- Dmochowski M, Hashimoto T, Chidgey MAJ, Yue KKM, Wilkinson RW, Nishikawa T, Garrod DR. Demonstration of antibodies to bovine desmocollin isoforms in certain Pemphigus sera. Br J Dermatol. 1995; 133:519–525.
- Hisamatsu Y, Amagai M, Garrod DR, Kanzaki T, Hashimoto T. The detection of IgG and IgA autoantibodies to desmocollins 1–3 by enzyme-linked immunosorbent assays using baculovirus-expressed proteins, in atypical Pemphigus but not in typical Pemphigus. Br J Dermatol. 2004; 151:73–83.
- Muller R, Heber B, Hashimoto T, Messer G, Mullegger R, Niedermeier A, Hertl M. Autoantibodies against desmocollins in European patients with Pemphigus. Clin Exp Dermatol. 2009; 34:898–903.
- Spindler V, Heupel WM, Efthymiadis A, Schmidt E, Eming R, Rankl C, Hinterdorfer P, Muller T, Drenckhahn D, Waschke J. Desmocollin 3-mediated binding is crucial for keratinocyte cohesion and is impaired in Pemphigus. J Biol Chem. 2009; 284:30556–30564.
- Mao X, Nagler AR, Farber SA, Choi EJ, Jackson LH, Leiferman KM, Ishii N, Hashimoto T, Amagai M, Zone JJ, Payne AS. Autoimmunity to desmocollin 3 in Pemphigus vulgaris. Am J Pathol. 2010; 177:2724–2730.
- Grando SA, Dahl MV. Activation of keratinocyte muscarinic acetylcholine receptors reverses Pemphigus acantholysis. J Eur Acad Dermatol Venereol. 1993; 2:72–86.
- Grando SA, Horton RM, Pereira EFR, Diethelm-Okita BM, George PM, Albuquerque EX, Conti-Fine BM. A nicotinic acetylcholine receptor regulating cell adhesion and motility is expressed in human keratinocytes. J Invest Dermatol. 1995; 105:774–781.
- Nguyen VT, Chernyavsky AI, Arredondo J, Bercovich D, Orr-Urtreger A, Vetter DE, Wess J, Beaudet AL, Kitajima Y, Grando SA. Synergistic control of keratinocyte adhesion through muscarinic and nicotinic acetylcholine receptor subtypes. Exp Cell Res. 2004; 294:534–549.
- Kurzen H, Henrich C, Booken D, Poenitz N, Gratchev A, Klemke CD, Engstner M, Goerdt S, Maas-Szabowski N. Functional characterization of the epidermal cholinergic system in vitro. J Invest Dermatol. 2006; 126:2458–2472.
- Grando SA. Autoimmunity to keratinocyte acetylcholine receptors in Pemphigus. Dermatology. 2000; 201:290–295.
- Dick SE, Werth VP. Pemphigus: A treatment update. Autoimmunity. 2006; 39:591–600.
- Brickman CM, Shoenfeld Y. The mosaic of autoimmunity. Scand J Clin Lab Invest Suppl. 2001; 235:3–15.
- Tron F, Gilbert D, Joly P, Mouquet H, Drouot L, Ayed MB, Sellami M, Masmoudi H, Makni S. Immunogenetics of Pemphigus: An update. Autoimmunity. 2006; 39:531–540.
- Saha M, Harman K, Mortimer NJ, Binda V, Black MM, Kondeatis E, Vaughan R, Groves RW. Pemphigus vulgaris in White Europeans is linked with HLA Class II allele HLA DRB1*1454 but not DRB1*1401. J Invest Dermatol. 2010; 130:311–314.
- Ahmed AR, Wagner R, Khatri K, Notani G, Awdeh Z, Alper CA, Yunis EJ. Major histocompatibility complex haplotypes and class II genes in non-Jewish patients with Pemphigus vulgaris. Proc Natl Acad Sci USA. 1991; 88:5056–5060.
- Niizeki H, Inoko H, Narimatsu H, Takata H, Sonoda A, Tadakuma T, Ando A, Tsuji K, Hashimoto T, Nishikawa T. HLA class II antigens are associated with Japanese Pemphigus patients. Hum Immunol. 1991; 31:246–250.
- Niizeki H, Inoko H, Mizuki N, Inamoto N, Watababe K, Hashimoto T, Nishikawa T. HLA-DQA1, -DQB1 and -DRB1 genotyping in Japanese Pemphigus vulgaris patients by the PCR-RFLP method. Tissue Antigens. 1994; 44:248–251.
- Miyagawa S, Amagai M, Niizeki H, Yamashina Y, Kaneshige T, Nishikawa T, Shirai T, Inoko H. HLA-DRB1 polymorphisms and autoimmune responses to desmogleins in Japanese patients with Pemphigus. Tissue Antigens. 1999; 54:333–340.
- Hertl M, Karr RW, Amagai M, Katz SI. Heterogeneous MHCII restriction pattern of autoreactive desmoglein 3 specific T cell responses in Pemphigus vulgaris patients and normals. J Invest Dermatol. 1998; 110:388–392.
- Lin MS, Swartz SJ, Lopez A, Ding X, Fernandez-Vina MA, Stastny P, Fairley JA, Diaz LA. Development and characterization of desmoglein-3 specific T cells from patients with Pemphigus vulgaris. J Clin Invest. 1997; 99:3–40.
- Riechers R, Grotzinger J, Hertl M. HLA class II restriction of autoreactive T cell responses in Pemphigus vulgaris: Review of the literature and potential applications for the development of a specific immunotherapy. Autoimmunity. 1999; 30:183–196.
- Veldman C, Stauber A, Wassmuth R, Uter W, Schuler G, Hertl M. Dichotomy of autoreactive Th1 and Th2 cell responses to desmoglein 3 in patients with Pemphigus vulgaris (PV) and healthy carriers of PV-associated HLA class II alleles. J Immunol. 2003; 170:635–642.
- Nishifuji K, Amagai M, Kuwana M, Iwasaki T, Nishikawa T. Detection of antigen-specific B cells in patients with Pemphigus vulgaris by enzyme-linked immunospot assay: Requirement of T cell collaboration for autoantibody production. J Invest Dermatol. 2000; 114:88–94.
- Tsunoda K, Ota T, Suzuki H, Ohyama M, Nagai T, Nishikawa T, Amagai M, Koyasu S. Pathogenic autoantibody production requires loss of tolerance against desmoglein 3 in both T and B cells in experimental Pemphigus vulgaris. Eur J Immunol. 2002; 32:627–633.
- Takahashi H, Amagai M, Nishikawa T, Fujii Y, Kawakami Y, Kuwana M. Novel system evaluating in vivo pathogenicity of desmoglein 3-reactive T cell clones using murine Pemphigus vulgaris. J Immunol. 2008; 181:1526–1535.
- Aoki-Ota M, Kinoshita M, Ota T, Tsunoda K, Iwasaki T, Tanaka S, Koyasu S, Nishikawa T, Amagai M. Tolerance induction by the blockade of CD40/CD154 interaction in Pemphigus vulgaris mouse model. J Invest Dermatol. 2006; 126:105–113.
- Hertl M, Amagai M, Sundaram H, Stanley J, Ishii K, Katz SI. Recognition of desmoglein 3 by autoreactive T cells in Pemphigus vulgaris patients and normals. J Invest Dermatol. 1998; 110:62–66.
- Grando SA, Glukhenky BT, Drannik GN, Kostromin AP, Boiko Y, Senyuk OF. Autoreactive cytotoxic T lymphocytes in Pemphigus and Pemphigoid. Autoimmunity. 1989; 3:247–260.
- Veldman CM, Gebhard KL, Uter W, Wassmuth R, Grotzinger J, Schultz E, Hertl M. T cell recognition of desmoglein 3 peptides in patients with Pemphigus vulgaris and healthy individuals. J Immunol. 2004; 172:3883–3892.
- Ahmed AR, Mohimen A, Yunis EJ, Mirza NM, Kumar V, Beutner EH, Alper CA. Linkage of Pemphigus vulgaris antibody to the major histocompatibility complex in healthy relatives of patients. J Exp Med. 1993; 177:419–424.
- Gebhard KL, Veldman CM, Wassmuth R, Schultz E, Schuler G, Hertl M. Ex vivo analysis of desmoglein 1-responsive T-helper (Th) 1 and Th2 cells in patients with Pemphigus foliaceus and healthy individuals. Exp Dermatol. 2005; 14:586–592.
- Costantino CM, Baecher-Allan CM, Hafler DA. Human regulatory T cells and autoimmunity. Eur J Immunol. 2008; 38:921–924.
- Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 2000; 164:183–190.
- Yamaguchi T, Sakaguchi S. Regulatory T cells in immune surveillance and treatment of cancer. Semin Cancer Biol. 2006; 16:115–123.
- Hertl M, Eming R, Veldman C. T cell control in autoimmune bullous skin disorders. J Clin Invest. 2006; 116:1159–1166.
- Sugiyama H, Matsue H, Nagasaka A, Nakamura Y, Tsukamoto K, Shibagaki N, Kawamura T, Kitamura R, Ando N, Shimada S. CD4+CD25 high regulatory T cells are markedly decreased in blood of patients with Pemphigus vulgaris. Dermatology. 2007; 214:210–220.
- Veldman C, Hohne A, Dieckmann D, Schuler G, Hertl M. Type I regulatory T cells specific for desmoglein 3 are more frequently detected in healthy individuals than in patients with Pemphigus vulgaris. J Immunol. 2004; 172:6468–6475.
- Arakawa M, Dainichi T, Yasumoto S, Hashimoto T. Lesional Th17 cells in Pemphigus vulgaris and Pemphigus foliaceus. J Dermatol Sci. 2009; 53:228–231.
- Sebaratnam DF, Martin LK, Rubin AI, Tran K, Pas HH, Marr PJ, Edmonds J, Murrell DF. Reversible relapse of Pemphigus foliaceus triggered by topical imiquimod suggests that Toll-like receptor 7 inhibitors may be useful treatments for Pemphigus. Clin Exp Dermatol. 2011; 36:91–93.
- Shlomchik MJ. Activating systemic autoimmunity: B's, T's, and tolls. Curr Opin Immunol. 2009; 21:626–633.
- Amagai M. Autoimmunity against desmosomal cadherins in Pemphigus. J Dermatol Sci. 1999; 20:92–102.
- Udey MC, Stanley JR. Pemphigus—diseases of antidesmosomal autoimmunity. J Am Med Assoc. 1999; 282:572–576.
- Karpati S, Amagai M, Prussick R, Cehrs K, Stanley JR. Pemphigus vulgaris antigen, a desmoglein type of cadherin, is localized within keratinocyte desmosomes. J Cell Biol. 1993; 122:409–415.
- Iwatsuki K, Han GW, Fukuti R, Ohtsuka M, Kikuchi S, Akiba H, Kaneko F. Internalization of constitutive desmogleins with the subsequent induction of desmoglein 2 in Pemphigus lesions. Br J Dermatol. 1999; 140:35–43.
- Bedane C, Prost C, Thomine E, Intrator L, Joly P, Caux F, Blecker M, Bernard P, Leboutet MJ, Tron F, Lauret P, Bonnetblanc JM, Dubertret L. Binding of autoantibodies is not restricted to desmosomes in Pemphigus vulgaris: Comparison of 14 cases of Pemphigus vulgaris and 10 cases of Pemphigus foliaceus studied by western immunoblot and immunoelectron microscopy. Arch Dermatol Res. 1996; 288:343–352.
- Waschke J, Bruggeman P, Baumgartner W, Zillikens D, Drenckhahn D. Pemphigus foliaceus IgG causes dissociation of desmoglein 1-containing junctions without blocking desmoglein 1 transinteraction. J Clin Invest. 2005; 115:3157–3165.
- Muller EJ, Williamson L, Kolly C, Suter MM. Outside-in signaling through integrins and cadherins: A central mechanism to control epidermal growth and differentiation?. J Invest Dermatol. 2008; 128:501–516.
- Sharma P, Mao X, Payne AS. Beyond steric hindrance: The role of adhesion signaling pathways in the pathogenesis of Pemphigus. J Dermatol Sci. 2007; 48:1–14.
- Bektas M, Runager K, Petersen JS, Rubenstein DS. Advances in Pemphigus research, signaling, and acantholysis. G Ital Dermatol Venereol. 2010; 145:675–688.
- Berkowitz P, Hu P, Liu Z, Diaz LA, Enghild JJ, Chua MP, Rubenstein DS. Desmosome signaling: Inhibition of p38MAPK prevents Pemphigus vulgaris IgG induced cytoskeleton reorganization. J Biol Chem. 2005; 280:23778–23784.
- Rubenstein DS, Diaz LA. Pemphigus antibody induced phosphorylation of keratinocyte proteins. Autoimmunity. 2006; 39:577–586.
- Lee HE, Berkowitz P, Jolly PS, Diaz LA, Chua MP, Rubenstein DS. Biphasic activation of p38MAPK suggests that apoptosis is a downstream event in Pemphigus acantholysis. J Biol Chem. 2009; 284:12524–12532.
- Jolly PS, Berkowitz P, Bektas M, Lee HE, Chua M, Diaz LA, Rubenstein DS. p38MAPK signaling and desmoglein-3 internalization are linked events in Pemphigus acantholysis. J Biol Chem. 2010; 285:8936–8941.
- de Bruin A, Caldelari R, Williamson L, Suter MM, Hunziker T, Wyder M, Muller EJ. Plakoglobin-dependent disruption of the desmosomal plaque in Pemphigus vulgaris. Exp Dermatol. 2007; 16:468–475.
- Muller EJ, Hunziker T, Suter MM. Keratin intermediate filament retraction is linked to plakoglobin-dependent signaling in Pemphigus vulgaris. J Am Acad Dermatol. 2007; 56:890–891 author reply 891–892.
- Williamson L, Raess NA, Caldelari R, Zakher A, de Bruin A, Posthaus H, Bolli R, Hunziker T, Suter MM, Muller EJ. Pemphigus vulgaris identifies plakoglobin as key suppressor of c-Myc in the skin. EMBO J. 2006; 25:3298–3309.
- Caldelari R, de Bruin A, Baumann D, Suter MM, Bierkamp C, Balmer V, Muller E. A central role for the armadillo protein plakoglobin in the autoimmune disease Pemphigus vulgaris. J Cell Biol. 2001; 153:823–834.
- Calkins CC, Setzer SV, Jennings JM, Summers S, Tsunoda K, Amagai Mq, Kowalczyk AP. Desmoglein endocytosis and desmosome disassembly are coordinated responses to Pemphigus autoantibodies. J Biol Chem. 2006; 281:7623–7634.
- Delva E, Jennings JM, Calkins CC, Kottke MD, Faundez V, Kowalczyk AP. Pemphigus vulgaris IgG-induced desmoglein-3 endocytosis and desmosomal disassembly are mediated by a clathrin- and dynamin-independent mechanism. J Biol Chem. 2008; 283:18303–18313.
- Jennings JM, Tucker DK, Kottke MD, Saito M, Delva E, Hanakawa Y, Amagai M, Kowalczyk AP. Desmosome disassembly in response to Pemphigus Vulgaris IgG occurs in distinct phases and can be reversed by expression of exogenous Dsg3. J Invest Dermatol. 2011; 131 3: 706–718.
- Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, Koyasu S, Nishikawa T, Amagai M. Induction of Pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol. 2003; 170:2170–2178.
- Kawasaki Y, Aoyama Y, Tsunoda K, Amagai M, Kitajima Y. Pathogenic monoclonal antibody against desmoglein 3 augments phosphorylation of desmoglein 3 and p38 MAPK in a human squamous carcinoma cell line. Autoimmunity. 2006; 39:587–590.
- Nguyen B, Dusek RL, Beaudry VG, Marinkovich MP, Attardi LD. Loss of the desmosomal protein perp enhances the phenotypic effects of Pemphigus vulgaris autoantibodies. J Invest Dermatol. 2009; 129:1710–1718.
- Sanchez-Carpintero I, Espana A, Pelacho B, Lopez Moratalla N, Rubenstein DS, Diaz LA, Lopez-Zabalza MJ. In vivo blockade of Pemphigus vulgaris acantholysis by inhibition of intracellular signal transduction cascades. Br J Dermatol. 2004; 151:565–570.
- Frusic-Zlotkin M, Raichenberg D, Wang X, David M, Michel B, Milner Y. Apoptotic mechanism in Pemphigus autoimmunoglobulins-induced acantholysis: Possible involvement of the EGF receptor. Autoimmunity. 2006; 39:563–575.
- Pretel M, España A, Marquina M, Pelacho B, Lopez-Picazo J, Lopez-Zabalza M. An imbalance in Akt/mTOR is involved in the apoptotic and acantholytic processes in a mouse model of Pemphigus vulgaris. Exp Dermatol. 2009; 18:771–780.
- Spindler V, Vielmuth F, Schmidt E, Rubenstein DS, Waschke J. Protective endogenous cyclic adenosine 5′-monophosphate signaling triggered by Pemphigus autoantibodies. J Immunol. 2010; 185:6831–6838.
- Seishima M, Iwasaki-Bessho Y, Itoh Y, Nozawa Y, Amagai M, Kitajima Y. Phosphatidylcholine-specific phospholipase C, but not phospholipase D, is involved in Pemphigus IgG-induced signal transduction. Arch Dermatol Res. 1999; 291:606–613.
- Cheng H, Kartenbeck J, Kabsch K, Mao X, Marques M, Alonso A. Stress kinase p38 mediates EGFR transactivation by hyperosmolar concentrations of sorbitol. J Cell Physiol. 2002; 192:234–243.
- Kippenberger S, Bernd A, Loitsch S, Guschel M, Muller J, Bereiter-Hahn J, Kaufmann R. Signaling of mechanical stretch in human keratinocytes via MAP kinases. J Invest Dermatol. 2000; 114:408–412.
- D'Alessandro M, Russell D, Morley SM, Davies AM, Lane EB. Keratin mutations of epidermolysis bullosa simplex alter the kinetics of stress response to osmotic shock. J Cell Sci. 2002; 115:4341–4351.
- Schmidt E, Waschke J. Apoptosis in Pemphigus. Autoimmun Rev. 2009; 8:533–537.
- Mao X, Sano Y, Park JM, Payne AS. p38 mitogen activated protein kinase (MAPK) activation is downstream of the loss of intercellular adhesion in Pemphigus vulgaris. J Biol Chem. 2010; 286 2: 1283–1291.
- Grando SA, Pittelkow MR, Schallreuter KU. Adrenergic and cholinergic control in the biology of epidermis: Physiological and clinical significance. J Invest Dermatol. 2006; 126:1948–1965.
- Calautti E, Grossi M, Mammucari C, Aoyama Y, Pirro M, Ono Y, Li J, Dotto GP. Fyn tyrosine kinase is a downstream mediator of Rho/PRK2 function in keratinocyte cell–cell adhesion. J Cell Biol. 2002; 156:137–148.
- Serres M, Filhol O, Lickert H, Grangeasse C, Chambaz EM, Stappert J, Vincent C, Schmitt D. The disruption of adherens junctions is associated with a decrease of E-cadherin phosphorylation by protein kinase CK2. Exp Cell Res. 2000; 257:255–264.
- Moon HS, Choi EA, Park HY, Choi JY, Chung HW, Kim JI, Park WI. Expression and tyrosine phosphorylation of E-cadherin, beta- and gamma- catenin, and epidermal growth factor receptor in cervical cancer cells. Gynecol Oncol. 2001; 81:355–359.
- Hu P, O'Keefe EJ, Rubenstein DS. Tyrosine phosphorylation of human keratinocyte beta-catenin and plakoglobin reversibly regulates their binding to E-cadherin and alpha- catenin. J Invest Dermatol. 2001; 117:1059–1067.
- Lilien J, Balsamo J, Arregui C, Xu G. Turn-off, drop-out: Functional state switching of cadherins. Dev Dyn. 2002; 224:18–29.
- Volberg T, Zick Y, Dror R, Sabanay I, Gilon C, Levitzki A, Geiger B. The effect of tyrosine-specific protein phosphorylation on the assembly of adherens-type junctions. EMBO J. 1992; 11:1733–1742.
- Behrens J, Vakaet L, Friis R, Winterhager E, Van Roy F, Mareel MM, Birchmeier W. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J Cell Biol. 1993; 120:757–766.
- Potter MD, Barbero S, Cheresh DA. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem. 2005; 280:31906–31912.
- Yin T, Getsios S, Caldelari R, Godsel LM, Kowalczyk AP, Muller EJ, Green KJ. Mechanisms of plakoglobin-dependent adhesion: Desmosome-specific functions in assembly and regulation by epidermal growth factor receptor. J Biol Chem. 2005; 280:40355–40363.
- Amar LS, Shabana AH, Oboeuf M, Martin N, Forest N. Involvement of desmoplakin phosphorylation in the regulation of desmosomes by protein kinase C, in HeLa cells. Cell Adhes Commun. 1999; 7:125–138.
- Mueller H, Franke WW. Biochemical and immunological characterization of desmoplakins I and II, the major polypeptides of the desmosomal plaque. J Mol Biol. 1983; 163:647–671.
- Boyer B, Tucker GC, Valles AM, Franke WW, Thiery JP. Rearrangements of desmosomal and cytoskeletal proteins during the transition from epithelial to fibroblastoid organization in cultured rat bladder carcinoma cells. J Cell Biol. 1989; 109:1495–1509.
- Pasdar M, Li Z, Chan H. Desmosome assembly and disassembly are regulated by reversible protein phosphorylation in cultured epithelial cells. Cell Motil Cytoskeleton. 1995; 30:108–121.
- Aoyama Y, Owada MK, Kitajima Y. A pathogenic autoantibody. Pemphigus vulgaris-IgG, induces phosphorylation of desmoglein 3, and its dissociation from plakoglobin in cultured keratinocytes. Eur J Immunol. 1999; 29:2233–2240.
- Aoyama Y, Kitajima Y. Pemphigus vulgaris-IgG causes a rapid depletion of desmoglein 3 (Dsg3) from the triton X-100 soluble pools, leading to the formation of Dsg3-depleted desmosomes in a human squamous carcinoma cell line, DJM-1 cells. J Invest Dermatol. 1999; 112:67–71.
- Nguyen VT, Arredondo J, Chernyavsky AI, Kitajima Y, Pittelkow M, Grando SA. Pemphigus vulgaris IgG and methylprednisolone exhibit reciprocal effects on keratinocytes. J Biol Chem. 2004; 279:2135–2146.
- Chernyavsky AI, Arredondo J, Piser T, Karlsson E, Grando SA. Differential coupling of M1 muscarinic and α7 nicotinic receptors to inhibition of Pemphigus acantholysis. J Biol Chem. 2008; 283:3401–3408.
- Kitajima Y, Inoue S, Yaoita H. Effects of Pemphigus antibody on the organization of microtubules and keratin-intermediate filaments in cultured human keratinocytes. Br J Dermatol. 1986; 114:171–179.
- Jinbu Y, Kitajima Y, Koto S, Akasaka Y, Yaoita H. Different effects of Pemphigus antibody and plasmin on the distribution of keratin intermediate filaments and desmoplakins between cultured oral and epidermal keratinocytes. J Dermatol Sci. 1992; 3:6–12.
- Baroni A, Buommino E, Paoletti I, Orlando M, Ruocco E, Ruocco V. Pemphigus serum and captopril induce heat shock protein 70 and inducible nitric oxide synthase overexpression, triggering apoptosis in human keratinocytes. Br J Dermatol. 2004; 150:1070–1080.
- Hashimoto K, Lever WF. An electron microscopic study on Pemphigus vulgaris of the mouth and the skin with special reference to the intercellular cement. J Invest Dermatol. 1967; 48:540–552.
- Hashimoto K, Lever WF. The intercellular cement in Pemphigus vulgaris, an electron microscopic study. Dermatologica. 1967; 135:27–34.
- Sams WMJr, Gammon WR. Mechanism of lesion production in Pemphigus and Pemphigoid. J Am Acad Dermatol. 1982; 6:431–452.
- Takahashi Y, Patel HP, Labib RS, Diaz LA, Anhalt GJ. Experimentally induced Pemphigus vulgaris in neonatal BALB/c mice: A time-course study of clinical, immunologic, ultrastructural, and cytochemical changes. J Invest Dermatol. 1985; 84:41–46.
- Bystryn J-C, Grando SA. A novel explanation for acantholysis in Pemphigus vulgaris—the “basal cell shrinkage” hypothesis. J Am Acad Dermatol. 2006; 54:513–516.
- Hotti A, Jarvinen K, Siivola P, Holtta E. Caspases and mitochondria in c-Myc-induced apoptosis: Identification of ATM as a new target of caspases. Oncogene. 2000; 19:2354–2362.
- Arredondo J, Chernyavsky AI, Karaouni A, Grando SA. Novel mechanisms of target cell death and survival and of therapeutic action of IVIg in Pemphigus. Am J Pathol. 2005; 167:1531–1544.
- Deb-Basu D, Aleem E, Kaldis P, Felsher DW. CDK2 is required by MYC to induce apoptosis. Cell Cycle. 2006; 5:1342–1347.
- Lanza A, Cirillo N, Rossiello R, Rienzo M, Cutillo L, Casamassimi A, de Nigris F, Schiano C, Rossiello L, Femiano F, Gombos F, Napoli C. Evidence of key role of Cdk2 overexpression in Pemphigus vulgaris. J Biol Chem. 2008; 283:8736–8745.
- Gera JF, Mellinghoff IK, Shi Y, Rettig MB, Tran C, Hsu JH, Sawyers CL, Lichtenstein AK. AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. J Biol Chem. 2004; 279:2737–2746.
- Gorshtein A, Rubinfeld H, Kendler E, Theodoropoulou M, Cerovac V, Stalla GK, Cohen ZR, Hadani M, Shimon I. Mammalian target of rapamycin inhibitors rapamycin and RAD001 (everolimus) induce anti-proliferative effects in GH-secreting pituitary tumor cells in vitro. Endocr Relat Cancer. 2009; 16:1017–1027.
- Gniadecki R, Jemec GB, Thomsen BM, Hansen M. Relationship between keratinocyte adhesion and death: Anoikis in acantholytic diseases. Arch Dermatol Res. 1998; 290:528–532.
- Puviani M, Marconi A, Cozzani E, Pincelli C. Fas ligand in Pemphigus sera induces keratinocyte apoptosis through the activation of caspase-8. J Invest Dermatol. 2003; 120:164–167.
- Pelacho B, Natal C, Espana A, Sanchez-Carpintero I, Iraburu MJ, Lopez-Zabalza MJ. Pemphigus vulgaris autoantibodies induce apoptosis in HaCaT keratinocytes. FEBS Lett. 2004; 566:6–10.
- Milner Y, Metezeau P, Kiefer H, Finemesser M, Bregegere F, Zlotkin M, Wang X, Michel B. 1999. Pemphigus an autoimmune disease of the skin: Cell–cell separation versus membrane signaling and apoptosis in acantholysis. In: Shoenfeld Y. editors. The decade of autoimmunityElsevier Science B.V.606–615.
- Frusic-Zlotkin M, Pergamentz R, Michel B, David M, Mimouni D, Bregegere F, Milner Y. The interaction of Pemphigus autoimmunoglobulins with epidermal cells: Activation of the fas apoptotic pathway and the use of caspase activity for pathogenicity tests of Pemphigus patients. Ann N Y Acad Sci. 2005; 1050:371–379.
- Pacheco-Tovar MG, Avalos-Diaz E, Vega-Memije E, Bollain-y-Goytia JJ, Lopez-Robles E, Hojyo-Tomoka MT, Dominguez-Soto L, Herrera-Esparza R. The final destiny of acantholytic cells in Pemphigus is Fas mediated. J Eur Acad Dermatol Venereol. 2009; 23:697–701.
- Lotti R, Shu E, Truzzi F, Palazzo E, Marconi A, Kitajima Y, Pincelli C. Apoptosis precedes desmoglein cleavage and keratinocyte dissociation in Pemphigus: Anti-Fas Ligand neutralizing antibodies prevent acantholysis both in vitro and in vivo. J Invest Dermatol. 2009; 129 Suppl: S14 (Abstr. #80).
- Feliciani C, Toto P, Amerio P, Pour SM, Coscione G, Shivji G, Wang B, Sauder DN. In vitro and in vivo expression of interleukin-1alpha and tumor necrosis factor-alpha mRNA in Pemphigus vulgaris: Interleukin-1alpha and tumor necrosis factor-alpha are involved in acantholysis. J Invest Dermatol. 2000; 114:71–77.
- Lopez-Robles E, Avalos-Diaz E, Vega-Memije E, Hojyo-Tomoka R, Villalobos S, Fraire L, Domiguez-Soto L, Herrera-Esparza R. TNFalpha and IL-6 are mediators in the blistering process of Pemphigus. Int J Dermatol. 2001; 40:185–188.
- D'Auria L, Bonifati C, Mussi A, D'Agosto G, De Simone C, Giacalone B, Ferraro C, Ameglio F. Cytokines in the sera of patients with Pemphigus vulgaris: Interleukin-6 and tumour necrosis factor-alpha levels are significantly increased as compared to healthy subjects and correlate with disease activity. Eur Cytokine Netw. 1997; 8:383–387.
- Ameglio F, D'Auria L, Cordiali-Fei P, Trento E, D'Agosto G, Mastroianni A, Giannetti A, Giacalone B. Anti-intercellular substance antibody log titres are correlated with serum concentrations of interleukin-6, interleukin-15 and tumor necrosis factor-alpha in patients with Pemphigus vulgaris relationships with peripheral blood neutrophil counts, disease severity and duration and patients' age. J Biol Regul Homeost Agents. 1999; 13:220–224.
- Orlov MD, Chernyavsky AI, Arredondo J, Grando SA. Synergistic actions of Pemphigus vulgaris IgG. Fas-ligand and tumor necrosis factor-alpha during induction of basal cell shrinkage and acantholysis. Autoimmunity. 2006; 39:557–562.
- Grando SA. Decompensation in proteinase-inhibitor system and application of proteinase inhibitors in Pemphigus and Pemphigoid. J Dermatol Sci. 1992; 4:95–97.
- Grando SA, Drannik GN, Glukhenky BT, Kostromin AP, Romanenko AB, Chayun OA, Chernyavsky AI. Clinical and laboratory evaluation of hemocarboadsorption in autoimmune bullous dermatoses. Int J Artif Organs. 1990; 3:181–188.
- Keskin DB, Stern JN, Fridkis-Hareli M, Ahmed AR. Cytokine profiles in Pemphigus vulgaris patients treated with intravenous immunoglobulins as compared to conventional immunosuppressive therapy. Cytokine. 2008; 41:315–321.
- Cirillo N, Lanza M, Femiano F, Gaeta GM, De Rosa A, Gombos F, Lanza A. If Pemphigus vulgaris IgG are the cause of acantholysis, new IgG-independent mechanisms are the concause. J Cell Physiol. 2007; 212:563–567.
- Wang X, Bregegere F, Frusic-Zlotkin M, Feinmesser M, Michel B, Milner Y. Possible apoptotic mechanism in epidermal cell acantholysis induced by Pemphigus vulgaris autoimmunoglobulins. Apoptosis. 2004; 9:131–143.
- Wang X, Bregegere F, Soroka Y, Frusic-Zlotkin M, Milner Y. Replicative senescence enhances apoptosis induced by Pemphigus autoimmune antibodies in human keratinocytes. FEBS Lett. 2004; 567:281–286.
- Grando SA, Glukhenky BT, Drannik GN, Kostromin AP, Chernyavsky AI. Cytotoxic proteases in blister fluid of Pemphigus and Pemphigoid patients. Int J Tissue React. 1989; 11:195–201.
- Grando SA, Glukhenky BT, Drannik GN, Epshtein EV, Kostromin AP, Korostash TA. Mediators of inflammation in blister fluids from patients with Pemphigus vulgaris and bullous Pemphigoid. Arch Dermatol. 1989; 125:925–930.
- Rico MJ, Benning C, Weingart ES, Streilein RD, Hall RPIII. Characterization of skin cytokines in bullous Pemphigoid and Pemphigus vulgaris. Br J Dermatol. 1999; 140:1079–1086.
- Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS. p38MAPK inhibition prevents disease in Pemphigus vulgaris mice. Proc Natl Acad Sci USA. 2006; 103:12855–12860.
- Lanza A, Cirillo N. Caspase-dependent cleavage of desmoglein 1 depends on the apoptotic stimulus. Br J Dermatol. 2007; 156:400–402.
- Cirillo N, Lanza M, De Rosa A, Cammarota M, La Gatta A, Gombos F, Lanza A. The most widespread desmosomal cadherin, desmoglein 2, is a novel target of caspase 3-mediated apoptotic machinery. J Cell Biochem. 2008; 103:598–606.
- Holubar K, Fellner MJ. 1986. Pemphigus and related diseases. In: Rook A, Parish LC, Beare JM. editors. Practical management of the dermatologic patient. Philadelphia: Lippincott Company153–155.
- Muller S, Stanley JR. 1990. Pemphigus: Pemphigus vulgaris and Pemphigus foliaceus. In: Wojnarowska F, Briggaman RA. editors. Management of blistering diseases. London: Chapman and Hall Medical43–61.
- Lever WF. 1965. Pemphigus and pemphigoidCharles C. Thomas, Springfield.
- Lever WF, Schaumburg-Lever G. Treatment of Pemphigus vulgaris. Results obtained in 84 patients between 1961 and 1982. Arch Dermatol. 1984; 120:44–47.
- Murrell DF, Dick S, Ahmed AR, Amagai M, Barnadas MA, Borradori L, Bystryn JC, Cianchini G, Diaz L, Fivenson D, Hall R, Harman KE, Hashimoto T, Hertl M, Hunzelmann N, Iranzo P, Joly P, Jonkman MF, Kitajima Y, Korman NJ, Martin LK, Mimouni D, Pandya AG, Payne AS, Rubenstein D, Shimizu H, Sinha AA, Sirois D, Zillikens D, Werth VP. Consensus statement on definitions of disease, end points, and therapeutic response for Pemphigus. J Am Acad Dermatol. 2008; 58:1043–1046.
- Grando SA, Grando AA, Glukhenky BT, Doguzov V, Nguyen VT, Holubar K. History and clinical significance of mechanical symptoms in blistering dermatoses: A reappraisal. J Am Acad Dermatol. 2003; 48:86–92.
- Talbott JH, Lever WF, Consolazio WV. Metabolic studies on patients with Pemphigus. J Invest Dermatol. 1940; 3:31.
- Thorn GW, Forsham PH, Frawley TF, Hill SRJr, Roche M, Staehelin D, Wilson DL. Clinical usefulness of ACTH and cortisone. New Eng J Med. 1950; 242:783, 824, 865.
- Risser J, Lewis K, Weinstock MA. Mortality of bullous skin disorders from 1979 through 2002 in the United States. Arch Dermatol. 2009; 145:1005–1008.
- Alexandroff AB, Harman KE. Blistering skin disorders: An evidence-based update. Conference report. Br J Dermatol. 2009; 160:502–504.
- Curtis JR, Westfall AO, Allison J, Bijlsma JW, Freeman A, George V, Kovac SH, Spettell CH, Saag KG. Population-based assessment of adverse events associated with long-term glucocorticoid use. Arthritis Rheum. 2006; 55:420–426.
- McDonough AK, Curtis JR, Saag KG. The epidemiology of glucocorticoid-associated adverse events. Curr Opin Rheumatol. 2008; 20:131–137.
- Lever WF, Goldberg HS. Treatment of Pemphigus vulgaris with methotrexate. Arch Dermatol. 1969; 100:70–78.
- Wolff K, Schreiner E. Immunosuppressive therapy of Pemphigus vulgaris. Preliminary results of azathioprine (Imuran) treatment (in German). Arch Klin Exp Dermatol. 1969; 235:63–77.
- Krain LS, Landau JW, Newcomer VD. Cyclophosphamide in the treatment of Pemphigus vulgaris and bullous Pemphigoid. Arch Dermatol. 1982; 106:657–661.
- Piamphongsant T, Ophaswongse S. Treatment of Pemphigus. Int J Dermatol. 1991; 30:139–146.
- Enk AH, Knop J. Treatment of Pemphigus vulgaris with mycophenolate mofetil. Lancet. 1997; 350:494.
- Mashkilleyson NA. Heparin action in Pemphigus vulgaris: Clinical and immunologic studies. Acta Derm Venereol. 1985; 65:545–547.
- Balda BR, Rosenzweig D. Cyclosporin A in the treatment of Pemphigus foliaceus and Pemphigus erythematosus (In German). Hautarzt. 1986; 37:454–457.
- Knobler RM. Photopheresis—extracorporeal irradiation of 8-MOP containing blood—a new therapeutic modality. Blut. 1987; 54:247–250.
- Humbert P, Derancourt C, Aubin F, Agache P. Effects of intravenous gamma-globulin in Pemphigus. J Am Acad Dermatol. 1990; 22:326.
- Salopek TG, Logsetty S, Tredget EE. Anti-CD20 chimeric monoclonal antibody (rituximab) for the treatment of recalcitrant, life-threatening Pemphigus vulgaris with implications in the pathogenesis of the disorder. J Am Acad Dermatol. 2002; 47:785–788.
- Renkl A, Mockenhaupt M, Technau K, Herouy Y, Norgauer J. A novel therapeutic option in Pemphigus vulgaris: Humanized monoclonal anti-CD25 antibody. Br J Dermatol. 2004; 150:1220–1222.
- Penneys NS, Eaglstein WH, Indgin S, Frost P. Gold sodium thiomalate treatment of Pemphigus. Arch Dermatol. 1973; 108:56–60.
- Haim S, Friedman-Birnbaum R. Dapsone in the treatment of Pemphigus vulgaris. Dermatologica. 1978; 156:120–123.
- Grando SA. Combined immunosuppressive therapy of autoimmune bullous dermatoses (In Russian). Sov Med. 1988; 2:113–115.
- Chaffins ML, Collison D, Fivenson DP. Treatment of Pemphigus and linear IgA dermatosis with nicotinamide and tetracycline: A review of 13 cases. J Am Acad Dermatol. 1993; 28:998–1000.
- Sawai T, Kitazawa K, Danno K, Sugie T, Machizuki T, Sugiura H, Uehara M. Pemphigus vegetans with oesophageal involvement: Successful treatment with minocycline and nicotinamide. Br J Dermatol. 1995; 132:668–670.
- Miyamoto H, Takahashi I. Successful treatment of Pemphigus vulgaris with prednisolone and tranilast. Acta Derm Venereol. 1997; 77:87–88.
- Cunha PR, de Oliveira JR, Salles MR, Jamora J, Bystryn JC. Pemphigus vulgaris with involvement of the cervix treated using thalidomide therapy. Int J Dermatol. 2004; 43:682–684.
- Ruocco V, Rossi A, Argenziano G, Aatarita C, Alviggi L, Farzati B, Papaleo G. Pathogenicity of the intercellular antibodies of Pemphigus and their periodic removal from the circulation by plasmapheresis. Br J Dermatol. 1978; 98:237–241.
- Cotterill JA, Barker DJ, Millard LG. Plasma exchange in the treatment of Pemphigus vulgaris. Br J Dermatol. 1978; 98:243.
- Schmidt E, Klinker E, Opitz A, Herzog S, Sitaru C, Goebeler M, Mansouri Taleghoni B, Brocker EB, Zillikens D. Protein A immunoadsorption: A novel and effective adjuvant treatment of severe Pemphigus. Br J Dermatol. 2003; 148:1222–1229.
- Satyam A, Khandpur S, Sharma VK, Sharma A. Involvement of T(H)1/T(H)2 cytokines in the pathogenesis of autoimmune skin disease-Pemphigus vulgaris. Immunol Invest. 2000; 38:498–509.
- DeKruyff RH, Fang Y, Umetsu DT. Corticosteroids enhance the capacity of macrophages to induce Th2 cytokine synthesis in CD4+ lymphocytes by inhibiting IL-12 production. J Immunol. 1998; 160:2231–2237.
- Mainali ES, Kikuchi T, Tew JG. Dexamethasone inhibits maturation and alters function of monocyte-derived dendritic cells from cord blood. Pediatr Res. 2005; 58:125–131.
- Miyaura H, Iwata M. Direct and indirect inhibition of Th1 development by progesterone and glucocorticoids. J Immunol. 2002; 168:1087–1094.
- Swanson DL, Dahl MV. Methylprednisolone inhibits Pemphigus acantholysis in skin cultures. J Invest Dermatol. 1983; 81:258–260.
- Jeffes EW, Kaplan RP, Ahmed AR. Acantholysis produced in vitro with Pemphigus serum: Hydrocortisone inhibits acantholysis, while dapsone and 6-mercaptopurine do not inhibit acantholysis. J Clin Lab Immunol. 1984; 4:359–363.
- Fine JD, Appell ML, Green LK, Sams WMJr. Pemphigus vulgaris. Combined treatment with intravenous corticosteroid pulse therapy, plasmapheresis, and azathioprine. Arch Dermatol. 1988; 124:236–239.
- Werth VP. Treatment of Pemphigus vulgaris with brief, high-dose intravenous glucocorticoids. Arch Dermatol. 1996; 132:1435–1439.
- Roujeau JC. Pulse glucocorticoid therapy. The “big shot” revisited. Arch Dermatol. 1996; 132:1499–1502.
- Chryssomallis F, Dimitriades A, Chaidemenos GC, Panagiotides D, Karakatsanis G. Steroid-pulse therapy in Pemphigus vulgaris long term follow-up. Int J Dermatol. 1995; 34:438–442.
- Saxon A, Stevens RH, Ramer SJ, Clements PJ, Yu DT. Glucocorticoids administered in vivo inhibit human suppressor T lymphocyte function and diminish B lymphocyte responsiveness in in vitro immunoglobulin synthesis. J Clin Invest. 1978; 61:922–930.
- Aksu D, Peksari Y, Arica IE, Gurgey E. Assessing the autoantibody levels in relation to disease severity and therapy response in Pemphigus patients. Indian J Dermatol. 2010; 55:342–347.
- Dumas V, Roujeau JC, Wolkenstein P, Revuz J, Cosnes A. The treatment of mild Pemphigus vulgaris and Pemphigus foliaceus with a topical corticosteroid. Br J Dermatol. 1999; 140:1127–1129.
- Dobrev H, Popova L, Vlashev D. Proteinase inhibitors and Pemphigus vulgaris. An in vitro and in vivo study. Arch Derm Res. 1996; 288:648–655.
- Berookhim B, Fischer HD, Weinberg JM. Treatment of recalcitrant Pemphigus vulgaris with the tumor necrosis factor alpha antagonist etanercept. Cutis. 2004; 74:245–247.
- Lin MH, Hsu CK, Lee JY. Successful treatment of recalcitrant Pemphigus vulgaris and Pemphigus vegetans with etanercept and carbon dioxide laser. Arch Dermatol. 2005; 141:680–682.
- Shetty A, Marcum CB, Glass LF, Carter JD. Successful treatment of Pemphigus vulgaris with etanercept in four patients. J Drugs Dermatol. 2009; 8:940–943.
- Pardo J, Mercader P, Mahiques L, Sanchez-Carazo JL, Oliver V, Fortea JM. Infliximab in the management of severe Pemphigus vulgaris. Br J Dermatol. 2005; 153:222–223.
- Jacobi A, Shuler G, Hertl M. Rapid control of therapy-refractory Pemphigus vulgaris by treatment with the tumour necrosis factor-alpha inhibitor infliximab. Br J Dermatol. 2005; 153:448–449.
- Howell SM, Bessinger GT, Altman CE, Belnap CM. Rapid response of IgA Pemphigus of the subcorneal pustular dermatosis subtype to treatment with adalimumab and mycophenolate mofetil. J Am Acad Dermatol. 2005; 53:541–543.
- Schultz HY, Diaz LA, Sirois DA, Werth VP, Grando SA. Generating Consensus Research Goals and Treatment Strategies for Pemphigus and Pemphigoid: The 2010 JC Bystryn Pemphigus and Pemphigoid Meeting. J Invest Dermatol. 2011; 131 7: 1395–1399.
- Alpsoy E, Yilmaz E, Basaran E, Yazar S, Cetin L. Is the combination of tetracycline and nicotinamide therapy alone effective in Pemphigus?. Arch Dermatol. 1995; 131:1339–1340.
- Hausermann P, Gutersohn T, Beltraminelli H, Schiller P, Buchner SA, Rufli T. [Oral Pemphigus vulgaris. Successful treatment with minocycline and nicotinamide]. Hautarzt. 2002; 53:813–815.
- Iraji F, Banan L. The efficacy of nicotinamide gel 4% as an adjuvant therapy in the treatment of cutaneous erosions of Pemphigus vulgaris. Dermatol Ther. 2010; 23:308–311.
- Romanenko AV. The action of nicotinamide on neuromuscular transmission. Fiziologicheskii Zhurnal (Kiev). 1987; 33:51–56.
- Koeppen A, Klein J, Erb C, Loeffelholz K. Acetylcholine release and choline availability in rat hippocampus: Effects of exogenous choline and nicotinamide. J Pharmacol Exp Ther. 1997; 282:1139–1145.
- Stoytcheva M, Zlatev R. Bioelectrocatalytical studies of the effect of some pharmaceuticals on the acetylcholinesterase activity. Electroanalysis. 1996; 8:676–679.
- Mehta JN, Martin AG. A case of Pemphigus vulgaris improved by cigarette smoking. Arch Dermatol. 2000; 136:15–17.
- Brenner S, Tur E, Shapiro J, Ruocco V, D'Avino M, Ruocco E, Tsankov N, Vassileva S, Drenovska K, Brezoev P, Barnadas MA, Gonzalez MJ, Anhalt G, Nousari H, Silva MR, Pinto KT, Miranda MF. Pemphigus vulgaris: Environmental factors. Occupational, behavioral, medical, and qualitative food frequency questionnaire. Int J Dermatol. 2001; 40:562–569.
- Sullivan TP, Elgart GW, Kirsner RS. Pemphigus and smoking. Int J Dermatol. 2002; 41:528–530.
- Valikhani M, Kavusi S, Chams-Davatchi C, Daneshpazhooh M, Barzegari M, Ghiasi M, Abedini R. Pemphigus and associated environmental factors: A case-control study. Clin Exp Dermatol. 2007; 32:256–260.
- Valikhani M, Kavusi S, Chams-Davatchi C, Hallaji Z, Esmaili N, Ghandi N, Farahani F, Lajevardi V. Impact of smoking on Pemphigus. Int J Dermatol. 2008; 47:567–570.
- Morimoto N, Takemoto S, Kawazoe T, Suzuki S. Nicotine at a low concentration promotes wound healing. J Surg Res. 2008; 145:199–204.
- Arredondo J, Omelchenko DM, Chernyavsky AI, Qian J, Skok M, Grando SA. Functional role of the nicotinic arm of the acetylcholine regulatory axis in human B-cell lines. J Exp Pharmacol. 2009; 1:1–7.
- Chernyavsky AI, Arredondo J, Galitovskiy V, Qian J, Grando SA. Structure and function of the nicotinic arm of acetylcholine regulatory axis in human leukemic T cells. Int J Immunopathol Pharmacol. 2009; 22:461–472.
- Chernyavsky AI, Arredondo J, Skok M, Grando SA. Auto/paracrine control of inflammatory cytokines by acetylcholine in macrophage-like U937 cells through nicotinic receptors. Int Immunopharmacol. 2010; 10:308–315.
- Wang DW, Zhou RB, Yao YM, Zhu XM, Yin YM, Zhao GJ, Dong N, Sheng ZY. Stimulation of {alpha}7 nicotinic acetylcholine receptor by nicotine increases suppressive capacity of naturally occurring CD4+CD25+ regulatory T cells in mice in vitro. J Pharmacol Exp Ther. 2010; 335:553–561.
- Qian J, Galitovskiy V, Chernyavsky AI, Marchenko S, Grando SA. Plasticity of the murine spleen T-cell cholinergic receptors and their role in in vitro differentiation of naive CD4 T cells toward the Th1. Th2 and Th17 lineages. Genes Immun. 2011; 12:222–230.
- Akaike A, Ikeda SR, Brookes N, Pascuzzo GJ, Rickett DL, Albuquerque EX. The nature of the interactions of pyridostigmine with the nicotinic acetylcholine receptor-ionic channel complex. II. Patch clamp studies. Mol Pharmacol. 1984; 25:102–112.
- Taylor P. 1985. Anticholinesterase agents. In: Gilman AG, Goodman LS, Rall TW, Murad F. editors. Goodman and Gilman's pharmacological basis of therapeutics. New York: Macmillan110–127.
- Grando SA, Kist DA, Qi M, Dahl MV. Human keratinocytes synthesize, secrete and degrade acetylcholine. J Invest Dermatol. 1993; 101:32–36.
- Lanza A, Stellavato A, Heulfe I, Landi C, Gombos F, Cirillo N. Serum of patients with oral Pemphigus vulgaris impairs keratinocyte wound repair in vitro: A time-lapse study on the efficacy of methylprednisolone and pyridostigmine bromide. Oral Dis. 2009; 15:478–483.
- Nguyen VT, Arredondo J, Chernyavsky AI, Pittelkow MR, Kitajima Y, Grando SA. Pemphigus vulgaris acantholysis ameliorated by cholinergic agonists. Arch Dermatol. 2004; 140:327–334.
- Grando SA. New approaches to the treatment of Pemphigus. J Invest Dermatol Symp Proc. 2004; 9:84–91.
- Nait K, Morioka S, Nakajima S, Ogawa H. Proteinase inhibitors block formation of Pemphigus acantholysis in experimental models of neonatal mice and skin explants: Effects of synthetic and plasma proteinase inhibitors on Pemphigus acantholysis. J Invest Dermatol. 1989; 93:173–177.
- Iraji F, Yoosefi A. Healing effect of Pilocarpine gel 4% on skin lesions of Pemphigus vulgaris. Int J Dermatol. 2006; 45:743–746.
- . 1994. The RBI handbook of receptor classificationKebabian JW, Neumeyer JL. Natick: Research Biochemicals International.
- Ahmed AR, Spigelman Z, Cavacini LA, Posner MR. Treatment of Pemphigus vulgaris with rituximab and intravenous immune globulin. N Engl J Med. 2006; 355:1772–1779.
- Morath C, Arns W, Schwenger V, Mehrabi A, Fonouni H, Schmidt J, Zeier M. Sirolimus in renal transplantation. Nephrol Dial Transplant. 2007; 22 Suppl 8: viii61–viii65.
- Sehgal SN. Sirolimus: Its discovery, biological properties, and mechanism of action. Transplant Proc. 2003; 35:7S–14S.
- Luan FL, Hojo M, Maluccio M, Yamaji K, Suthanthiran M. Rapamycin blocks tumor progression: Unlinking immunosuppression from antitumor efficacy. Transplantation. 2002; 73:1565–1572.
- Liu L, Chen L, Chung J, Huang S. Rapamycin inhibits F-actin reorganization and phosphorylation of focal adhesion proteins. Oncogene. 2008; 27:4998–5010.
- Hertl M, Veldman C. T-cellular autoimmunity against desmogleins in Pemphigus, an autoantibody-mediated bullous disorder of the skin. Autoimmun Rev. 2003; 2:278–283.
- Schumann H, Baetge J, Tasanen K, Wojnarowska F, Schacke H, Zillikens D, Bruckner-Tuderman L. The shed ectodomain of collagen XVII/BP180 is targeted by autoantibodies in different blistering skin diseases. Am J Pathol. 2000; 156:685–695.
- Simpson A, Levin D, Kazerounian S, Uitto J, Aho S, Mahoney MG. Desmoglein 2 is a new antigenic target of Pemphigus autoantibodies (abstr. #23). J Invest Dermatol. 2001; 117:393.
- Kljuic A, Bazzi H, Sundberg JP, Martinez-Mir A, O'Shaughnessy R, Mahoney MG, Levy M, Montagutelli X, Ahmad W, Aita VM, Gordon D, Uitto J, Whiting D, Ott J, Fischer S, Gilliam TC, Jahoda CA, Morris RJ, Panteleyev AA, Nguyen VT, Christiano AM. Desmoglein 4 in hair follicle differentiation and epidermal adhesion. Evidence from inherited hypotrichosis and acquired Pemphigus vulgaris. Cell. 2003; 113:249–260.
- Kim SC, Chung YL, Kim J, Cho NJ, Amagai M. Pemphigus vulgaris with autoantibodies to desmoplakin. Br J Dermatol. 2001; 145:838–840.
- Evangelista F, Dasher DA, Diaz LA, Prisayanh PS, Li N. E-cadherin is an additional immunological target for Pemphigus autoantibodies. J Invest Dermatol. 2008; 128:1710–1718.
- Korman NJ, Eyre RW, Klaus-Kovtun V, Stanley JR. Demonstration of an adhering-junction molecule (plakoglobin) in the autoantigens of Pemphigus foliaceus and Pemphigus vulgaris. N Engl J Med. 1989; 321:631–635.
- Lambert J, Bracke S, van Roy F, Pas HH, Bonne S, De Schepper S. Serum plakophilin-3 autoreactivity in paraneoplastic Pemphigus. Br J Dermatol. 2010; 163:630–632.
- Fiebiger E, Hammerschmid F, Stingl G, Maurer D. Anti-FcεRIα autoantibodies in autoimmune-mediated disorders. Identification of a structure–function relationship. J Clin Invest. 1998; 101:243–251.
- Grando SA. Pemphigus in the XXI Century: New life to an old story. Autoimmunity. 2006; 39:521–530.
- Pitoia F, Moncet D, Glorio R, Graciela Diaz A, Rodriguez Costa G, Carbia S, Cabrera H, Niepomniszcze H. Prevalence of thyroid autoimmunity in patients with Pemphigus vulgaris. Medicina (B Aires). 2005; 65:307–310.
- Rickman L, Simrak D, Stevens HP, Hunt DM, King IA, Bryant SP, Eady RA, Leigh IM, Arnemann J, Magee AI, Kelsell DP, Buxton RS. N-terminal deletion in a desmosomal cadherin causes the autosomal dominant skin disease striate palmoplantar keratoderma. Hum Mol Genet. 1999; 8:971–976.
- Baroni A, Buommino E, Ruocco E, Petrazzuolo M, De Filippis A, Satriano RA, Ruocco V, . Captopril modulates acetylcholinesterase in human keratinocytes. Arch Dermatol Res 2011; 303:491–497.