
Abstract
Purpose: Ionizing radiation induces DNA damage, some of which are present in clusters, defined as two or more lesions within one to two helical turns of DNA by passage of a single radiation track. These clusters are thought to contribute to the detrimental effects of radiation, in part due to the compromised repair of clustered DNA damaged sites.
Materials and methods: The repair of three-lesion cluster present in oligonucleotides were determined in vitro using the hamster cell line CHO-K1 nuclear extract or purified proteins involved in base excision repair. The mutagenic potential of these clusters present in a plasmid was determined using an Escherichia coli reporter assay.
Results: We have shown that the repair of an abasic (AP) site within a three-lesion cluster, comprised of an AP site and bi-stranded 8-oxo-7,8-dihydroguanine (8-oxoG) lesions, is retarded compared to that of an isolated AP site in an in vitro base excision repair (BER) assay. Further, the mutation frequency of the clustered damaged site is up to three times greater than that of an isolated 8-oxoG lesion.
Conclusions: As a consequence of enhanced mutagenic potential of clusters, non-double-strand break (DSB) DNA damage may contribute to the detrimental effects of radiation, in addition to the effects of DSB.
Introduction
The spatial distribution of damage on DNA is responsible, in part, for the detrimental effects of ionizing radiation. Unlike damage resulting from oxygen metabolism and other DNA oxidizing agents (e.g., hydrogen peroxide), a percentage of the ionizing radiation damage formed is present in clusters on the DNA molecule. The clusters of damage, defined as two or more lesions within one to two helical turns of DNA induced by passage of a single radiation track, are formed due to the nature of the spatial distribution of ionization events along the radiation track. Low linear energy transfer (LET) radiation, such as X-rays and γ-radiation, deposits energy in sparsely ionizing radiation tracks. In contrast, high LET radiation tracks, such as those resulting from α-radiation, are densely ionizing and as a consequence several ionizing events may occur within a few nanometres, resulting in the DNA molecule being ‘hit’ several times within a small volume, typically within one to two helical turns, resulting in clusters of DNA lesions. As the LET of the irradiation increases and so the density of ionizing events increase, the yield and complexity of clustered damage increases, as predicted from biophysical and chemical modelling (Ward Citation1988, Goodhead Citation1994).
Double-strand breaks (DSB), thought to be the most harmful DNA damage as they can lead to chromosomal aberrations and cell death, are the most studied type of cluster damage (a cluster composed of two single-strand breaks [SSB]). However non-DSB clustered damage is also induced by ionizing radiation in yields 4–8 times greater than that of DSB in mammalian cells (Gulston et al. Citation2002, Sutherland et al. Citation2002). The chemical nature of the individual lesions that make up non-DSB clustered damage sites are the same as those induced endogenously and by other DNA oxidizing agents such as SSB, abasic (AP) sites and base lesions. These lesions tend to be processed by the base excision repair (BER)/single-strand break repair (SSBR) machinery.
Since low LET ionizing radiation-induced DNA damage sites are randomly distributed, it is difficult to study the repair of specific lesions or clusters within DNA in mammalian cells with the exception of SSB or DSB. Synthetic oligonucleotides containing site-specific lesions together with purified proteins or cell extracts have established that processing of bi-stranded lesions within clustered DNA damaged sites is compromised when compared to the processing of the same lesions when formed in isolation (reviewed in Eccles et al. Citation2011). To date, these biochemical studies have concentrated on two-lesion clusters although some recent studies have investigated three- and five-lesion clusters (Eot-Houllier et al. Citation2005, Citation2007, Eccles et al. Citation2010).
The processing of the lesions within the cluster is hierarchical, thus limiting the formation of DSB (Lomax et al. Citation2004a, Eot-Houllier et al. Citation2005, Citation2007, Eccles et al. Citation2010). However, two opposing lesions that can be rapidly cleaved to form a SSB, such as AP sites and 5-hydroxyuracil (hU) do lead to the formation of DSB (Chaudhry and Weinfeld Citation1997, Lomax et al. Citation2004b, Paap et al. Citation2008, Eccles et al. Citation2010, Sedletska et al. Citation2013). The efficiency of repair of clustered damaged sites depends on the number and types of lesions within the cluster, the distance between the lesions and the orientation of the lesions to each other. In general, AP sites and SSB confer retardation on the excision of base lesions and are repaired first (David-Cordonnier et al. Citation2000, Citation2002, Lomax et al. Citation2004a) although base lesions also impair the efficiency of repair of SSB (Lomax et al. Citation2004a, Eot-Houllier et al. Citation2005). In addition, the excision of base lesions follows a hierarchy with, hU, 5,6-dihydrothymine (DHT) and thymine glycol being excised initially in a cluster containing 8-oxo- 7,8-dihydroguanine (8-oxoG) and 5-formyluracil (fU) (Eot-Houllier et al. Citation2005, Shikazono et al. Citation2006, Bellon et al. Citation2009, Shikazono and O’Neill Citation2009).
Since the repair efficiency of DNA clustered damaged sites is reduced, the lifetime of the lesions within the cluster is extended and thus there is an increased chance of the lesions being present at replication. Studies in Escherichia coli and mammalian cells have shown that the mutagenic potential of DNA lesions within clustered sites is greater than that of the same lesions present in isolation (Malyarchuk et al. Citation2004, Pearson et al. Citation2004, Bellon et al. Citation2009). The mutation frequency of 8-oxoG in a bi-stranded cluster with uracil (which is rapidly converted to an AP site then to a SSB in vivo) is up to eight times greater than that of an isolated 8-oxoG in E. coli (Pearson et al. Citation2004). Some clusters containing bi-stranded uracils are processed to form DSB in vivo, seen as a reduced transformation efficiency of E. coli (D’Souza and Harrison Citation2003, Harrison et al. Citation2006, Shikazono and O’Neill, 2009, Eccles et al. Citation2010). In addition, two bi-stranded furans are converted into a DSB in mammalian cells together with deletions and mutations due to inaccurate repair (Malyarchuk et al. Citation2008).
8-oxoG is one of the most common lesions induced in cells by ionizing radiation and oxidative stress (Pouget et al. Citation2002). It can exist in two forms; the ‘anti’ form base pairs with cytosine (Oda et al. Citation1991) and the ‘syn’ form mis-pairs with adenine (Kouchakdjian et al. Citation1991). There are glycosylases that can remove both the 8-oxoG lesion and the adenine mis-paired with 8-oxoG (Michaels et al. Citation1992); however, the fact that 8-oxoG can mis-pair with adenine makes it a highly mutagenic lesion, giving rise to G:C to T:A transversions (Cheng et al. Citation1992). AP sites can be induced directly by ionizing radiation (Regulus et al. Citation2007) and are also an intermediate of the BER pathway (Boiteux and Guillet Citation2004). As they are non-coding they are mutagenic, often base pairing with adenine (Boiteux and Guillet Citation2004), and can also block replicative DNA polymerases (Boiteux and Guillet Citation2004).
We have previously shown (Eccles et al. Citation2010) that processing of three-lesion clusters containing two bi-stranded AP sites and an 8-oxoG site tends to induce DSB in vitro and in vivo. Additionally, we have shown that an AP site within 5 bases of 8-oxoG on the same strand can compromise repair of the lesions (Cunniffe et al. Citation2007). As few studies have investigated the processing of three-lesion clusters, we wanted to know whether the hierarchy of processing of these clusters is similar to that predicted from bi-stranded two-lesion clusters. We have used a three-lesion clustered-damaged site containing two bi-stranded 8-oxoG lesions with an AP site on the same strand (in tandem) as one of the 8-oxoG lesions. Using in vitro base excision repair assays and a plasmid based mutation assay using E. coli, the findings are consistent with the hierarchy of repair being similar to that predicted from previous knowledge derived from bi-stranded two-lesion clusters.
Materials and methods
Oligonucleotides
Forty base single-stranded oligonucleotides were purchased from Eurogentec (Seraing, Belgium), polyacrylamide gel electrophoresis (PAGE) purified. The lyophilized pellets were resuspended in Tris/ethylenediaminetetra acetic acid (TE) pH 8.0 (10 mM Tris, 1 mM ethylenediaminetetra acetic acid [EDTA]) to 1 mg/ml and aliquots were stored at − 20°C. The double-stranded oligonucleotide sequences containing two 8-oxoG lesions and a uracil (precursor to an AP site) are shown in . A previous similar study by our group showed that the efficiency of rejoining of a SSB resulting from the incision of an AP site is independent of the sequence context of the oligonucleotides (Lomax et al. Citation2004a).
Table I. Sequence of double-stranded oligonucleotides.
Preparation of 5’ labelled double-stranded substrates
A total of 0.2 μg of uracil containing oligonucleotides were 5’ end labelled with 32P, by incubating at 37°C for 30 min with 10 U T4 polynucleotide kinase (Invitrogen, Paisley, UK), 15 μCi of [γ-32P] adenosine triphosphate (ATP) (6000 Ci/mmol, 10 mCi/ml, Perkin Elmer, Waltham, MA, USA), in 20 μl of buffer (70 mM Tris-HCl pH 7.6, 10 mM MgCl2, 100 mM KCl, 1 mM 2-mercaptoethanol). The labelled oligonucleotide was purified using an illustra MicroSpin™ G-50 column (GE Healthcare, Little Chalfont, Buckinghamshire, UK) following manufacturer's instructions. The purified labelled oligonucleotides were hybridized to their complementary oligonucleotide by mixing with a 2-fold excess of complementary oligonucleotide, incubating at 95°C for 10 min and leaving to cool, slowly, to room temperature over 2–3 h. Hybridization was verified by running an aliquot of the double-stranded oligonucleotide alongside a single-stranded control on a 12% native polyacrylamide mini gel in 1 × Tris/borate/EDTA (TBE) buffer (89 mM Tris-HCl, 89 mM boric acid, 2 mM EDTA pH 8.3) at 120 V for 1 h 15 min.
To convert the uracil residues to AP sites, hybridized oligonucleotides were incubated with 2 U of uracil DNA glycosalyase (UDG) (Invitrogen) in 10 μl of buffer (10 mM Tris-HCl pH 7.5, 50 mM NaCl, 1 mM EDTA) at 37°C for 30 min. Complete AP site conversion was verified by treating an aliquot of the UDG-treated oligonucleotide with 10 ng of purified human AP endonuclease 1 (APE1) in 5 μl buffer (20 mM 4-(2-hydroxyethyl)1-1-piperazineethanesulphonic acid (HEPES) pH 7.9, 100 mM KCl, 0.2 mM EDTA 1 mM MgCl2) at 37°C for 30 min. The oligonucleotides were subjected to electrophoresis on a 12% denaturing polyacrylamide mini gel (8 M urea) in 1 × TBE at 300 V for 30 min. The activity of the oligonucleotides was measured in a scintillation counter and diluted to 10 000 counts per minute (cpm)/μl with TE pH 8.0.
Preparation of CHO-K1 nuclear extract
The nuclear extracts were prepared as previously described (David-Cordonnier et al. Citation2001a) from the hamster cell line, CHO-K1 cells. Briefly, the cells were grown in exponential phase in Hams F-12 media (Sigma Aldrich, Gillingham, Kent, UK) supplemented with 10% foetal bovine serum (FBS) (Mycoplex, PAA Laboratories, Teddington, Middlesex, UK), 100 U/ml penicillin (Gibco BRL, Gaithersburg, Maryland, USA) and 100 mg/ml streptomycin (Gibco BRL). The cells were harvested and the pelleted cells were resuspended in an equal volume of buffer (10 mM HEPES pH 7.9, 100 mM KCl, 1.5 mM MgCl2, 0.5 mM dithiothreitol [DTT]) and incubated on ice for 15 min. The cytoplasmic membranes were broken down by drawing the cell suspension into a 0.5 μm diameter needle 10 times. Following a 20 s centrifugation at 12,000 g, 4°C, the supernatant was removed and the nuclear pellet was resuspended in 2/3 volume high salt buffer (20 mM HEPES pH7.9, 420 mM NaCl, 25% glycerol, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 0.5 mM phenylmethanesulfonylfluoride [PMSF]) and incubated for 30 min with agitation, on ice. Following centrifugation for 10 min at 12,000 g, 4°C, the supernatant was dialysed twice over a total period of 16 h against 1 l of buffer (20 mM HEPES pH7.9, 100 mM KCl, 0.2 mM EDTA, 20% glycerol, 0.5 mM DTT, 0.5 mM PMSF). The protein concentration was determined using the Bradford colourimetric technique (Bio-Rad, Hercules, CA, USA) and aliquots were stored at − 80°C.
In vitro BER repair assays using nuclear extract
The double-stranded oligonucleotides (10,000 cpm/μl, 16 fmol) were incubated with 0.5 μg CHO-K1 nuclear extract (as determined by the Bradford colourimetric technique) in 5 μl repair buffer (70 mM Tris-HCl pH 7.5, 10 mM MgCl2, 10 mM DTT, 4 mM ATP, 40 mM phosphocreatine, 1.6 μg/ml phosphocreatine kinase, 0.4 mM deoxynucleotide (dNTP) and incubated at 37°C for 0, 1, 5, 15, 30, and 60 min. The concentration of extract was optimized from titration studies (data not shown). To stop the reactions 10 μl denaturing stop solution (98% formamide, 2 mM EDTA, 0.025% bromophenol blue, 0.025% xylene cyanol) was added. The samples were then subjected to electrophoresis on a 15% denaturing polyacrylamide gel containing 8 M urea in 1 × TBE (89 mM Tris-HCl, 89 mM boric acid, 2 mM EDTA pH 8.3) for 90 min at a constant power of 85 W. The dried gel was exposed to a Bio-Rad PhosphorImager screen (Bio-Rad, Molecular Imager FX) for visualization of repair products and quantified with Quantity One software (Bio-Rad). When following the time dependence of the repair of the AP site, the intensity of the bands representing the single-stranded DNA with base(s) added (before ligation) and ligated DNA (ligation of the SSB following addition of the missing base) are expressed as a percentage of the total intensities for all bands.
To determine if DSB had been formed, the in vitro repair reactions were stopped by adding 2 μl native loading dye (40% sucrose, 5 mM EDTA, 0.025% bromophenol, 0.025% xylene cyanol) and subjected to electrophoresis on a 15% native polyacrylamide gel in 1 × TBE, at a constant voltage of 300 V, for 3 h and the dried gel was analyzed using phosphorimaging technology as above.
In vitro BER repair assays using purified proteins
The double-stranded oligonucleotides (10,000 cpm/μl, 16 fmol) were incubated with the following BER proteins, depending on the experiment, in a 10 μl reaction solution (80 mM HEPES pH 7.9, 10 mM MgCl2, 2 mM DTT, 200 μM EDTA, 4 mM ATP, 800 μg/ml bovive serum albumin (BSA), 160 μM dNTP) at 37°C for 0, 1, 5, 15 and 30 min. All proteins were the human homologues of the proteins. Proteins used were: 0.5 ng APE1, 2 ng polymerase β, 4 ng flap endonuclease 1 (FEN1), 10 ng ligase 1, 3 ng oxogunaine glycosylase (OGG1). The reactions were stopped by the addition of 10 μl denaturing stop solution (98% formamide, 2 mM EDTA, 0.025% bromophenol blue, 0.025% xylene cyanol) and then subjected to electrophoresis on a 20% denaturing polyacrylamide gel containing 8 M urea in 1 × TBE (89 mM Tris-HCl, 89 mM boric acid, 2 mM EDTA pH 8.3) at a constant power of 85 W. The bands in the gel were quantified as described above.
Escherichia coli strains
Isogenic strain BH990 (fpg::KanR mutY::KanR) was a kind gift from Dr S. Boiteux (CNRS, France).
Determination of mutation frequency
The mutation frequency arising from the clustered DNA-damaged site was determined as described previously (Pearson et al. Citation2004). Briefly, the double-stranded oligonucleotides () were phosphorylated at their 5’ termini with 10 U of T4 polynucleotide kinase (Invitrogen, Paisley, UK) and 25 mM ATP in 20 μl of buffer (70 mM Tris-HCl pH 7.6, 10 mM MgCl2, 100 mM KCl, 1 mM β-2-mercaptoethanol) for 30 min at 37°C. pUC18 plasmid DNA was linearized with SmaI followed by treatment with 20 U calf intestinal phosphatase (New England Biolabs, Ipswich, MA, USA) for 15 min at 37°C, followed by a second incubation for 45 min at 55°C. The plasmid DNA was then gel purified using the Qiaquick gel purification system (Qiagen, Hilden, Germany) and the concentration determined spectrophotometrically. Phosphorylated double-stranded oligonucleotide (5 pmol) was ligated into 200 fmol linearized pUC18 DNA with 400 U T4 ligase (New England Biolabs) in 20 μl buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 10 mM DTT, 1 mM ATP, 25 μg/ml BSA) for 16 h at 16°C, followed by dialysis using 0.025 μm nitrocellulose filters (Merck Millipore, Billerica, MA, USA). 50 ng of the ligation product was transformed into 60 μl electro-competent bacteria using a Bio-Rad E. coli pulser set at 1.8 mV. Transformants were selected on Luria Bertani (LB) broth agar plates containing 100 μg/ml ampicillin for 16 h at 37°C. Single bacterial colonies were picked at random, transferred to a well of a 96-well plate containing LB agar with 100 μg ampicillin and incubated for 16 h at 37°C. The plasmid within the colonies was sequenced by Source BioScience LifeSciences (Nottingham, UK) using primer, 5’-CTTCGCTATT ACGCCAGCTG-3’. The primers amplify sequence across the site of damage in the cloned oligonucleotide. The sequencing data was analyzed using FinchTV software.
Results
In vitro repair of an AP site when in a three-lesion cluster with bi-stranded 8-oxoG lesions
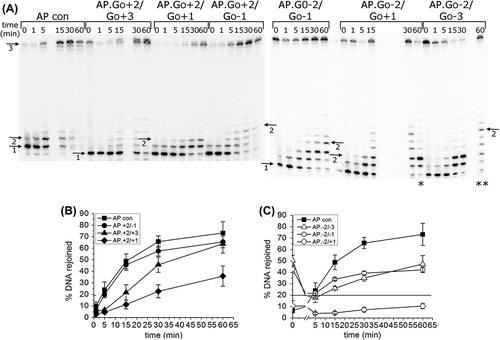
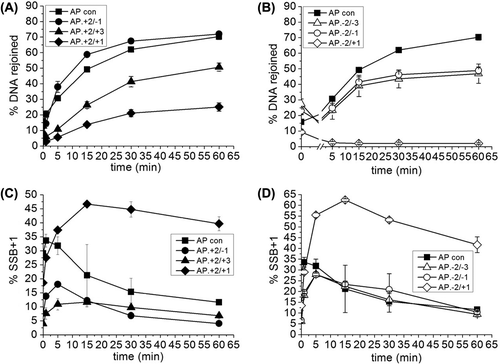
shows the repair of an AP site when it is in a cluster with bi-stranded 8-oxoG lesions (see for relative positions of 8-oxoG and the AP site in the cluster). The repair of the clustered AP site is compared to the repair of an AP site in isolation. The efficiency of repair of the cluster (+ 2/− 1) with the bistranded 8-oxoG lesions positioned 2 bases 5’ on the same strand and 1 base 3’ on the opposing strand to the AP site is similar to that of an isolated AP site (). In contrast, bi-stranded 8-oxoG lesions in any of the other positions tested (Figures 1B, 1C) reduce the efficiency of repair of the AP site to a greater or lesser degree. There is virtually no repair of the AP site when the 8-oxoG lesions are in position (− 2/+ 1). The efficiency of repair of the AP site is reduced by 3-fold and 2.3-fold, respectively, when 8-oxoG lesions are in positions (− 2/− 1) and (− 2/− 3), compared to the control (). When bi-stranded 8-oxoG lesions are positioned (− 2/− 1) and (− 2/− 3) to the AP site, the level of incision of the AP site at time zero is 50–60% and increases to 80% within the first minute, whereas in the control and in all other clusters the level of incision of the AP site is ≥ 90% at time zero (Figures 1B and 1C). Thus the efficiency of incision of the AP site is reduced in the AP.8-oxoG-2/8-oxoG-1 and AP.8-oxoG-2/8-oxoG-3 clusters. The horizontal line in indicates the base line from which the repair of the AP site in these two clusters is estimated. Restoration of the full length oligomer by BER, rather than chain elongation, has been confirmed in our previous studies (Lomax et al. Citation2004a, Citation2004b, Byrne et al. Citation2009), using dideoxy-version of dNTP that would be incorporated four bases downstream from the repair gap.
Figure 1. Repair of an AP site in a three-lesion cluster with bi-stranded 8-oxoG lesions. (A) Phosphorimaging scan of a polyacrylamide gel showing the repair products from an in vitro BER assay. Band 1; the SSB resulting from incision of the AP site. Band 2; the intermediate band at which the greatest number of nucleotides were incorporated. Band 3; the intact 40 mer band, either before incision of the AP site or following rejoining. Due to a loading error sample AP.8-oxoG-2/8-oxoG + 1 60 min is located in lane ** and sample AP.8-oxoG-2/8-oxoG-3 60 min is located in lane *. (B) and (C) Graphical representation of the rejoined band following repair of the AP site, over time. The horizontal line in C is to show the base line from which the repair of the clusters AP.8-oxoG-2/8-oxoG-1 and AP.8-oxoG-2/8-oxoG-3 were measured. The error bars represent standard deviations of three independent experiments.
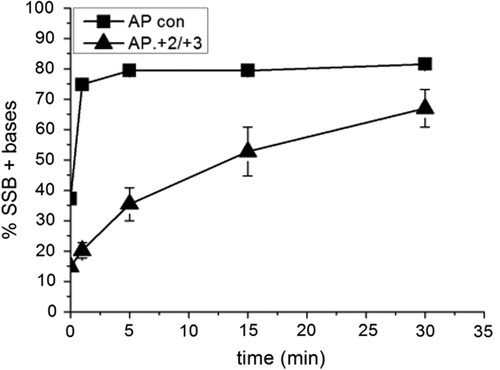
The phosphorimaging scan of the repair gel () shows a reduction in the number of repair intermediates with the (+ 2/+ 3) cluster, reflecting a reduction in the polymerase activity. Indeed no SSB + 1 base added band (band 2, ) can be seen before 5 min indicating a lag in repair () although the level of repair reaches to that of the control by 60 min. To investigate this further, in vitro repair assays were carried out using purified APE1 to incise the AP site and purified DNA polymerase β for repair synthesis. As demonstrates polymerase activity is reduced in the AP.8-oxoG + 2/8-oxoG + 3 cluster based on levels of SSB+ bases compared with that of the AP control.
Figure 2. Activity of polymerase β following incision of a control AP site and incision of an AP site in the AP.8-oxoG + 2/8-oxoG + 3 cluster. Polymerase β activity was measured by incorporation of nucleotides following incision of the AP site. The error bars represent standard deviations of three independent experiments.
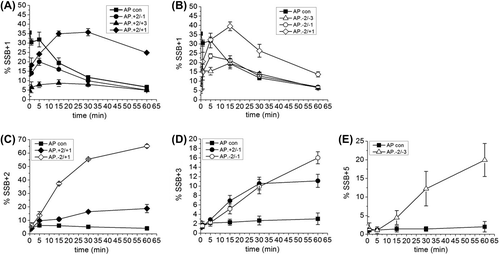
In all the other three-lesion clusters tested, repair intermediates are formed with > 1 base incorporated into the repair gap (Figures 1A and 3). Thus, long patch (LP) BER repair processes contribute to the repair of these clusters. The overall number of bases added in to the repair gap prior to ligation varies for the different clusters. The data in shows the accumulation of the first base (SSB + 1 band) incorporated in to the repair gap ( and ) whereas Figures 3C–E show the accumulation of the intermediates containing 2, 3 or 5 bases for each cluster. It should be noted that the repair of an isolated AP site occurs mainly via the short patch (SP) BER pathway as demonstrated by the lack of incorporation of more than one base into the repair gap prior to ligation (). In the AP site control, the SSB + 1 intermediate reaches a steady state level of ˜ 35% before decreasing as ligation occurs to give rejoined DNA rather than polymerase addition of further bases (Figures 3A, 3B). For example, the addition of more than one base into the gap in competition with ligation can be seen with the AP.8-oxoG + 2/8-oxoG-1, AP.8-oxoG-2/8-oxoG-3, AP.8-oxoG-2/8-oxoG-1, and AP.8-oxoG-2/8-oxoG + 1 clusters (Figures 3A, 3B) through the accumulation of additional intermediates (Figures 3C–E). This accumulation of additional intermediates indicates that SP the ligase step of repair is retarded. The AP.8-oxoG + 2/8-oxoG + 3 cluster does not accumulate SSB intermediates containing more than one base added (), presumably as the SSB + 1 intermediate is ligated as soon as it is formed, reflecting the low activity of polymerase on this cluster.
Figure 3. Intermediates of repair of an AP site in a three-lesion cluster with bi-stranded 8-oxoG lesions. (A) and (B) Graphical representation of the SSB + 1 intermediate, over time. (C), (D) and (E) Graphical representation of the intermediate with the greatest number of incorporated nucleotides for each of the three-lesion clusters, over time. The error bars represent standard deviations of three independent experiments.
The contribution of SP and LP repair processes was assessed in more detail through substitution of the second dNTP with the corresponding di-dNTP, which would be incorporated as the second base. As a consequence, additional bases would not be incorporated so that ligation products could only be formed following incorporation of the first base. Thus SP-BER would remain unaffected whereas LP-BER would be inhibited. and show that the level of DNA rejoining is not significantly affected by the inhibition of LP-BER. The level of accumulation of the SSB + 1 intermediate in the AP.8-oxoG + 2/8-oxoG + 1 and AP.oxoG-2/8-oxoG + 1 clusters (Figures 5C, 5D) is however higher when LP-BER is inhibited, reflecting poor rejoining of the AP site in these two clusters.
No evidence of DSB was seen during the processing of the three-lesion clusters () when the products of the in vitro repair assay were run on native polyacrylamide gels. However a higher molecular weight band was seen in some samples (). To determine if this was due to binding of OGG1 to the 8-oxoG lesions within the cluster, BER was reconstituted with purified proteins either with or without OGG1 protein. The mobility of the bands in the presence and absence of OGG1 are the same (), thus OGG1 binding to 8-oxoG within the clusters is not responsible for the higher mobility band seen in . The higher mobility band remains as yet unknown.
Figure 4. Repair of a three-lesion cluster with an AP site and bi-stranded 8-oxoG lesions. (A) Phosphorimaging scan of a native polyacrylamide gel showing the repair products from an in vitro BER assay with CHO-K1 nuclear extracts. Band 1; double-stranded 40 mer oligonucleotide. Band 2; expected site of shortened double-stranded oligonucleotide resulting from a DSB, if one was formed. (B) Phosphorimage scan of a native polyacrylamide gel showing the repair products from an in vitro BER assay reconstituted with purified BER proteins, in the absence (−) and presence (+) of OGG1 The repair assay was performed for 30 min. Band 1; double-stranded 40 mer oligonucleotide. Band 2; expected site of shortened double-stranded oligonucleotide resulting from a DSB, if one had been formed.
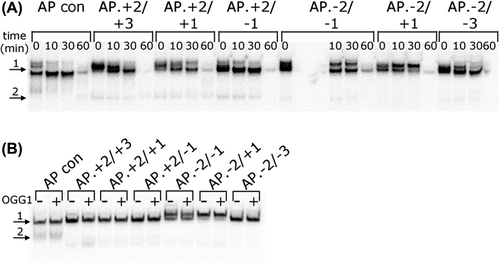
Figure 5. SP repair of an AP site in a three-lesion cluster with bi-stranded 8-oxoG lesions. Deoxynucleotides were substituted with di-deoxynucleotides such that only the SSB + 1 nucleotide could be incorporated, inhibiting LP repair. (A) and (B) Graphical representation of the rejoined band following repair of the AP site, over time. (C) and (D) Graphical representation of the SSB + 1 intermediate, over time. The error bars represent standard deviations of three independent experiments.
Mutagenic potential of a three-lesion cluster with bi-stranded 8-oxoG lesions and an AP site
An E. coli-based plasmid assay was used to measure the mutagenic potential of a single uracil, a single 8-oxoG and the three-lesion clusters of a uracil and bi-stranded 8-oxoG lesions. The uracil residue is rapidly converted in to an AP site and then subsequently to a SSB (Shikazono and O’Neill 2009). Unlike our previously developed E. coli-based plasmid assay (Pearson et al. Citation2004), the oligonucleotides used in this study did not have a restriction site that could be utilized as a pre-screen for those plasmids containing a mutation; therefore, mutations could only be detected by directly sequencing plasmid DNA rescued from E. coli. To optimize detection and quantification of mutations without pre-screening and knowing that the frequency of mutation is very low in wild-type E. coli but increases significantly in E. coli strains deficient in the glycosylases Fpg and MutY (Pearson et al. Citation2004, Shikazono et al. Citation2006, Bellon et al. Citation2009, Noguchi et al. Citation2012), the mutations were determined by sequencing of plasmids recovered from MutY/Fpg null E. coli. Secondly, as MutY is involved in post-replicative repair, the level of mutations determined more accurately reflect the level of clusters that have been present at replication relative to the levels of mutations for plasmids containing a single 8-oxoG.
A single uracil results in a mutation frequency (calculated by dividing the number of individual mutations found in the oligonucleotide sequence ligated in to pUC19 plasmid by the total number of oligonucleotides sequenced) of just 4.5% but only 20% (1 in 5) of the detected mutations involve the uracil residue. In contrast a single 8-oxoG lesion results in a mutation frequency of 22.6% () with over half of the mutations G:C to T:A transversions (). This frequency for a single 8-oxoG corresponds with previous determinations (Shikazono et al. Citation2006, Cunniffe et al. Citation2007).
Figure 6. Mutation frequency of a single uracil residue, a single 8-oxoG lesion and a three-lesion cluster of an AP site with bi-stranded 8-oxoG lesions in MutY/Fpg null E. coli. The mutation frequency was calculated by dividing the number of individual mutations found in the oligonucleotide sequence ligated in to pUC19 plasmid by the total number of oligonucleotides sequences sequenced. Some plasmids contained two oligonucleotide sequences. (A) The total number of mutations. The error bars represent standard deviations of three independent experiments. (B) Mutations at either of the 8-oxoG lesions or the uracil residue. (C) Mutations other than those at the 8-oxoG lesions or the uracil residue.
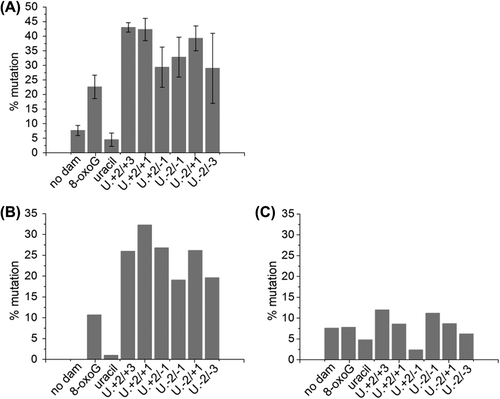
Table II. Mutations detected in oligonucleotides ligated into plasmids, recovered from E. coli.
When the uracil and 8-oxoG lesions are in a three-lesion cluster, no decrease in transformation efficiency is seen, indicating that the clusters are not significantly processed to form DSB. The mutation frequencies of the various clusters rise to between 29–43% (), reflecting the poor repair of the lesions within the cluster. The major mutation seen is a G:C to T:A transversion, accounting for 48.3–79.2% of the mutations, depending on the cluster (). Of the remaining mutations, 5–10% involve a mutation at an 8-oxoG lesion or a uracil residue and the remainder are small deletions (typically of one base) at sites other than the damage sites.
It was observed that the range of mutations was much broader than that seen in previous studies looking at two lesion clusters (Pearson et al. Citation2004, Shikazono et al. Citation2006, Cunniffe et al. Citation2007, Bellon et al. Citation2009, Shikazono and O’Neill 2009, Noguchi et al. Citation2012). For assessment of mutation frequency in the two-lesion clusters a pre-screen removes sequencing of any plasmids that have mutations outside of the cluster region. Therefore, it was of interest to see how the mutation frequency would be affected if mutations outside of the three-lesion cluster area were excluded from analysis of the mutation frequency. and , show that in oligonucleotides carrying the clusters, there is only an increase in mutation frequency (compared with that for single lesions) at the lesion sites, suggesting that an impairment of lesion repair results in an increase in mutation induction. The tendency is for the 8-oxoG on the DNA strand with a single 8-oxoG lesion to result in a greater number of mutations, than seen at the 8-oxoG/U strand ().
Discussion
The major findings are that (i) the repair of an AP site when in a cluster with bi-stranded 8-oxoG lesions is impaired compared to that of an isolated AP site, (ii) the mutagenic potential of the three-lesion cluster is increased in E. coli compared to the component lesions, and (iii) DSB formation was not observed. From these findings in E coli, it is inferred that the hierarchy of processing of these three-lesion-containing clusters is as predicted, in that attempted repair of the AP site occurs prior to cleavage of the 8-oxoG lesions, consistent with previous studies and the lack of DSB formation (Lomax et al. Citation2004a, Malyarchuk et al. Citation2004, Pearson et al. Citation2004, Eccles et al. Citation2010).
From the findings in and , it is inferred that the repair of the AP site within the three-lesion cluster is retarded due to reduced efficiencies of APE1, polymerase and ligation, although the level of reduction depends on the position of the AP site in relation to the bi-stranded 8-oxoG lesions. Repair of the AP site is least efficient when the 8-oxoG and AP site are generally on the same DNA strand in the − 2 position (compared to when they are in the + 2 position). One possibility is that during the repair of the SSB, resulting from the incision of the AP site, the polymerase is reading towards the tandem 8-oxoG lesion when the lesions are in the − 2 position, whereas when they are in the + 2 position the polymerase is reading away from the tandem 8-oxoG lesion. For the AP.8-oxoG-2/8-oxoG + 1 cluster there is little or no rejoining of the resulting SSB following incision of the AP site, even though nucleotides are incorporated in to the repair gap with similar efficiencies as the control AP site. It is inferred therefore that repair is impaired at the ligation stage. In contrast with the AP.8-oxoG + 2/8-oxoG + 1 cluster, not only the activity of the polymerase is reduced compared to the control, but also the efficiency of ligation (as seen by an accumulation of repair intermediates). Polymerase β activity is also reduced in the AP.8-oxoG + 2/8-oxoG + 3 cluster but ligation is unaffected. This is consistent with virtually no repair of an AP site if it is in the negative orientation to 8-oxoG in tandem (Cunniffe et al. Citation2007) or the reduced repair of an AP site when 8-oxoG is on the opposing DNA strand (Lomax et al. Citation2004b). Consistent with other studies on the excision of an 8-oxoG lesion in a bi-stranded 8-oxoG cluster (David-Cordonnier et al. Citation2001a, Citation2001b, Citation2001c), excision within the three-lesion cluster of either of the 8-oxoG lesions is prevented by the presence of the AP site. There is a contribution from the LP-BER pathway during the repair of the AP site in the clusters from the higher levels of the SSB + 1 (Figures 5A, 5B) when LP-BER is inhibited. However, when this sub-pathway is inhibited, the observed AP site repair by the SP-BER pathway occurs with similar efficiency. In in vitro repair assays, polymerase β continues to add nucleotides into the gap if left unchecked when ligase is absent in the repair reaction (data not shown). A possibility is that impairment of ligation in the clusters allows more nucleotides to be incorporated in to the repair gap by polymerase β, in competition with ligation. As a consequence of inhibition of LP-BER when polymerase β is not available, ligation by SP-BER occurs at a similar rate under these conditions.
To enhance the frequency of mutations MutY/Fpg null E. coli were used (Pearson et al. Citation2004) in the absence of pre-screening. The spectrum of mutations seen is much wider than those studies looking at two-lesion clusters, probably reflecting that all plasmids were sequenced even those that carried no mutations. Even for plasmids containing no damage, a higher mutation frequency was seen. Indeed if the mutations that involve 8-oxoG or uracil present in the clusters are removed from the calculation, then the mutation frequencies of all the plasmids, i.e., the no damage, the single lesion and the cluster containing plasmids, are similar (). shows that the mutation frequency affecting 8-oxoG and uracil are greatly increased in the cluster containing plasmids compared to the single-lesion controls.
Comparison of the mutation frequencies in E. coli of the three-lesion clusters with that of bi-stranded clusters of 8-oxoG lesions and AP sites (Malyarchuk et al. Citation2004, Pearson et al. Citation2004) indicate that the three-lesion clusters also have an increased mutagenic potential compared to the component lesions (). In contrast, clusters containing a tandem AP site and 8-oxoG have lower mutation frequencies than that of an isolated 8-oxoG in MutY/Fpg null E. coli (Cunniffe et al. Citation2007). It was proposed that when the AP site in tandem to an 8-oxoG lesion is incised, the repair is so slow that the lesion containing strand is lost before repair could take place, thus removing the 8-oxoG lesion before any mutagenic consequences (Cunniffe et al. Citation2007). Consistent with this latter observation is the lower frequency of mutations affecting the DNA strand containing the tandem 8-oxoG and AP site than that seen on the DNA strand containing a single 8-oxoG.
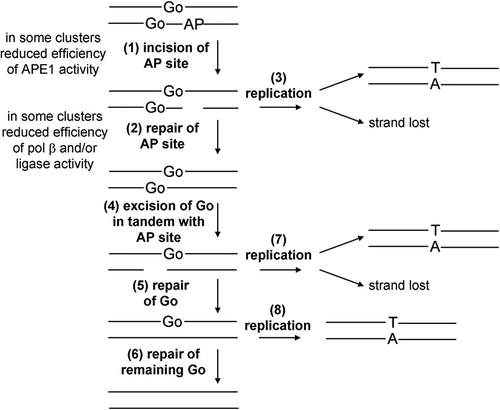
Based on the findings and the lack of DSB formation in the three-lesion clusters, a schematic representation of the processing of three-lesion clusters in E. coli is presented in . It is important to remember that the mutation frequencies were determined in a MutY/Fpg null background. The mutation frequency of both the single 8-oxoG lesion and the clusters is expected to be about 4–8 times lower in a wild-type background (Pearson et al. Citation2004, Shikazono et al. Citation2006, Bellon et al. Citation2009, Noguchi et al. Citation2012). The uracil is rapidly converted into an AP site and the repair of the AP site or the resulting SSB from AP endonuclease activity is retarded due to the proximity of the neighboring 8-oxoG sites. As a consequence the SSB, resulting from incision of the AP site, could remain unrepaired at replication, leading to loss of the DNA strand carrying the uracil and 8-oxoG in tandem (, pathways 1 and 3), as seen previously (Cunniffe et al. Citation2007). In this case, only the DNA strand with the single 8-oxoG lesion would be able to report for mutations (see , pathways 1 and 3), since this 8-oxoG lesion would still be present, as its repair would have been retarded due to the close proximity of the AP site (, pathways 1 and 3). That mutations on the DNA strand carrying the tandem lesions are formed (), suggests that if the AP site is repaired (or misrepaired, see ) then the tandem 8-oxoG lesion has the potential to be mutagenic. The cluster would behave as a bi-stranded 8-oxoG cluster, as previously reported, and either 8-oxoG may report as a mutation (, pathways 2, 4, 5 and 7). The increased level of mutations seen with these three-lesion clusters, compared to the component lesions, and lack of DSB formation is consistent with predictions when compared with processing of three-lesion clusters containing two bi-stranded AP sites and a 8-oxoG site, when DSB are induced and few mutations seen relating to 8-oxoG (Eccles et al. Citation2010).
Figure 7. Schematic representation of the repair of the three-lesion cluster in E. coli. Initially the AP site is cleaved (1) and then repaired (2). If replication (3) occurs before repair of the AP site then there is the potential for the SSB.8-oxoG containing strand to be lost or for mutations to arise from the single 8-oxoG containing strand (3). Following repair of the AP site the 8-oxoG lesions are repaired sequentially (4–6). Again if repair is not completed by replication (7 and 8) there is potential for strand loss or mutation induction.
In summary it has been shown that the repair of a three-lesion cluster comprised of 8-oxoG lesions and an AP site is compromised in an in vitro BER assay and the delayed repair leads to an increased potential for mutations to be formed in a plasmid-based E. coli assay, compared to that of the component lesions present in isolation. The hierarchy of processing these three-lesion clusters are as would be predicted from the present knowledge obtained with clusters containing bi-stranded lesions. Since non-DSB clustered lesions are induced in yields 4–8 times those of DSB (Gulston et al. Citation2002, Sutherland et al. Citation2002) more complex clustered damage sites may contribute to the detrimental effects of ionizing radiation, particularly mutation induction and ultimately tumourigenesis.
Acknowledgements
This study is dedicated to the memory of Professor Clemens von Sonntag. It was funded by Medical Research Council UK, grant number (MC_PC_12001).
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Bellon S, Shikazono N, Cunniffe S, Lomax M, O’Neill P. 2009. Processing of thymine glycol in a clustered DNA damage site: mutagenic or cytotoxic. Nucleic Acids Res 37:4430–4440.
- Boiteux S, Guillet M. 2004. Abasic sites in DNA: Repair and biological consequences in Saccharomyces cerevisiae. DNA Repair (Amst) 3:1–12.
- Byrne S, Cunniffe S, O’Neill P, Lomax ME. 2009. 5,6-Dihydrothymine impairs the base excision repair pathway of a closely opposed AP site or single-strand break. Radiat Res 172:537–549.
- Chaudhry MA, Weinfeld M. 1997. Reactivity of human apurinic/apyrimidinic endonuclease and Escherichia coli exonuclease III with bistranded abasic sites in DNA. J Biol Chem 272:15650–15655.
- Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 1992. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G to T and A to C substitutions. J Biol Chem 267:166–172.
- Cunniffe SM, Lomax ME, O’Neill P. 2007. An AP site can protect against the mutagenic potential of 8-oxoG when present within a tandem clustered site in E. coli. DNA Repair (Amst) 6:1839–1849.
- D’Souza DI, Harrison L. 2003. Repair of clustered uracil DNA damages in Escherichia coli. Nucleic Acids Res 31:4573–4581.
- David-Cordonnier MH, Boiteux S, O’Neill P. 2001a. Efficiency of excision of 8-oxo-guanine within DNA clustered damage by XRS5 nuclear extracts and purified human OGG1 protein. Biochemistry 40:11811–11818.
- David-Cordonnier MH, Boiteux S, O’Neill P. 2001b. Excision of 8-oxoguanine within clustered damage by the yeast OGG1 protein. Nucleic Acids Res 29:1107–1113.
- David-Cordonnier MH, Cunniffe SM, Hickson ID, O’Neill P. 2002. Efficiency of incision of an AP site within clustered DNA damage by the major human AP endonuclease. Biochemistry 41:634–642.
- David-Cordonnier MH, Laval J, O’Neill P. 2000. Clustered DNA damage, influence on damage excision by XRS5 nuclear extracts and Escherichia coli Nth and Fpg proteins. J Biol Chem 275:11865–11873.
- David-Cordonnier MH, Laval J, O’Neill P. 2001c. Recognition and kinetics for excision of a base lesion within clustered DNA damage by the Escherichia coli proteins Fpg and Nth. Biochemistry 40:5738–5746.
- Eccles LJ, Lomax ME, O’Neill P. 2010. Hierarchy of lesion processing governs the repair, double-strand break formation and mutability of three-lesion clustered DNA damage. Nucleic Acids Res 38: 1123–1134.
- Eccles LJ, O’Neill P, Lomax ME. 2011. Delayed repair of radiation induced clustered DNA damage: Friend or foe?Mutat Res 711: 134–141.
- Eot-Houllier G, Eon-Marchais S, Gasparutto D, Sage E. 2005. Processing of a complex multiply damaged DNA site by human cell extracts and purified repair proteins. Nucleic Acids Res 33:260–271.
- Eot-Houllier G, Gonera M, Gasparutto D, Giustranti C, Sage E. 2007. Interplay between DNA N-glycosylases/AP lyases at multiply damaged sites and biological consequences. Nucleic Acids Res 35: 3355–3366.
- Goodhead DT. 1994. Initial events in the cellular effects of ionizing radiations: Clustered damage in DNA. Int J Radiat Biol 65:7–17.
- Gulston M, Fulford J, Jenner T, de Lara C, O’Neill P. 2002. Clustered DNA damage induced by gamma radiation in human fibroblasts (HF19), hamster (V79-4) cells and plasmid DNA is revealed as Fpg and Nth sensitive sites. Nucleic Acids Res 30:3464–3472.
- Harrison L, Brame KL, Geltz LE, Landry AM. 2006. Closely opposed apurinic/apyrimidinic sites are converted to double strand breaks in Escherichia coli even in the absence of exonuclease III, endonuclease IV, nucleotide excision repair and AP lyase cleavage. DNA Repair (Amst) 5:324–335.
- Kouchakdjian M, Bodepudi V, Shibutani S, Eisenberg M, Johnson F, Grollman AP, Patel DJ. 1991. NMR structural studies of the ionizing radiation adduct 7-hydro-8-oxodeoxyguanosine (8-oxo-7H-dG) opposite deoxyadenosine in a DNA duplex. 8-Oxo-7H-dG(syn).dA(anti) alignment at lesion site. Biochemistry 30:1403–1412.
- Lomax ME, Cunniffe S, O’Neill P. 2004a. 8-OxoG retards the activity of the ligase III/XRCC1 complex during the repair of a single-strand break, when present within a clustered DNA damage site. DNA Repair (Amst) 3:289–299.
- Lomax ME, Cunniffe S, O’Neill P. 2004b. Efficiency of repair of an abasic site within DNA clustered damage sites by mammalian cell nuclear extracts. Biochemistry 43:11017–11026.
- Malyarchuk S, Brame KL, Youngblood R, Shi R, Harrison L. 2004. Two clustered 8-oxo-7,8-dihydroguanine (8-oxodG) lesions increase the point mutation frequency of 8-oxodG, but do not result in double strand breaks or deletions in Escherichia coli. Nucleic Acids Res 32:5721–5731.
- Malyarchuk S, Castore R, Harrison L. 2008. DNA repair of clustered lesions in mammalian cells: involvement of non-homologous end-joining. Nucleic Acids Res 36:4872–4882.
- Michaels ML, Cruz C, Grollman AP, Miller JH. 1992. Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc Natl Acad Sci USA 89:7022–7025.
- Noguchi M, Urushibara A, Yokoya A, O’Neill P, Shikazono N. 2012. The mutagenic potential of 8-oxoG/single strand break-containing clusters depends on their relative positions. Mutat Res 732:34–42.
- Oda Y, Uesugi S, Ikehara M, Nishimura S, Kawase Y, Ishikawa H, Inoue H, Ohtsuka E. 1991. NMR studies of a DNA containing 8-hydroxydeoxyguanosine. Nucleic Acids Res 19:1407–1412.
- Paap B, Wilson DM 3rd, Sutherland BM. 2008. Human abasic endonuclease action on multilesion abasic clusters: Implications for radiation-induced biological damage. Nucleic Acids Res 36: 2717–2727.
- Pearson CG, Shikazono N, Thacker J, O’Neill P. 2004. Enhanced mutagenic potential of 8-oxo-7,8-dihydroguanine when present within a clustered DNA damage site. Nucleic Acids Res 32:263–270.
- Pouget JP, Frelon S, Ravanat JL, Testard I, Odin F, Cadet J. 2002. Formation of modified DNA bases in cells exposed either to gamma radiation or to high-LET particles. Radiat Res 157:589–595.
- Regulus P, Duroux B, Bayle PA, Favier A, Cadet J, Ravanat JL. 2007. Oxidation of the sugar moiety of DNA by ionizing radiation or bleomycin could induce the formation of a cluster DNA lesion. Proc Natl Acad Sci USA 104:14032–14037.
- Sedletska Y, Radicella JP, Sage E. 2013. Replication fork collapse is a major cause of the high mutation frequency at three-base lesion clusters. Nucleic Acids Res 41:9339–9348.
- Shikazono N, O’Neill P. 2009. Biological consequences of potential repair intermediates of clustered base damage site in Escherichia coli. Mutat Res 669:162–168.
- Shikazono N, Pearson C, O’Neill P, Thacker J. 2006. The roles of specific glycosylases in determining the mutagenic consequences of clustered DNA base damage. Nucleic Acids Res 34:3722–3730.
- Sutherland BM, Bennett PV, Sutherland JC, Laval J. 2002. Clustered DNA damages induced by x rays in human cells. Radiat Res 157:611–616.
- Ward JF. 1988. DNA damage produced by ionizing radiation in mammalian cells: Identities, mechanisms of formation, and reparability. Prog Nucleic Acid Res Mol Biol 35:95–125.