Abstract
We have completed the first comprehensive transmembrane topology determination for a member of the ubiquitous and important SulP/SLC26 family of coupled anion transporters found in eukaryotes and prokaryotes. The prokaryotic member that we have mapped, namely BicA from Synechococcus PCC7002, is an important Na+-dependent bicarbonate transporter that is likely to play a major role in global primary productivity via the CO2 concentrating mechanism in cyanobacteria. We experimentally determined the topology based on phoA-lacZ topology mapping combined with reference to a range of predictive models based on hydropathy analysis and positive charge distribution. The 12-TMH structure for BicA is characterized by tight turns between several pairs of TMH and it features a prominent cytoplasmically-located STAS domain that is characteristic of the SulP family. A key difference from previous predicted models is that we identify a cytoplasmic loop between helices 8 and 9 where previous models suggested a TMH. This region includes a highly conserved motif that defines the SulP family. The identification of this region as cytoplasmic, rather than transmembrane, has implications for the function and perhaps regulation of SulP family members. This finding is used to reinterpret mutagenesis data relating to highly conserved residues in this region from both plant and human SulP transporters.
Introduction
BicA is a low/medium affinity, Na+-dependent bicarbonate transporter that is ubiquitous in a wide range of cyanobacterial families and some proteobacteria [Citation1–3]. It was first discovered and characterized in the marine coastal cyanobacterium, Synechococcus PCC7002; in addition, two other cyanobacterial orthologs were identified and characterized by gain-of-function analysis and many more homologs have been identified by bioinformatics [Citation1]. In cyanobacteria, HCO3- transporters form an integral part of the CO2 concentrating mechanism (CCM) where, in conjunction with active CO2 uptake systems, HCO3- is actively accumulated inside the cell, and then used within carboxysome micro-compartments to support optimal rates of photosynthetic CO2 fixation via the primary carboxylase, ribulose bisphosphate carboxylase-oxygenase or Rubisco [Citation3]. A functional CCM is an obligate growth requirement for all species of cyanobacteria so far examined and it is arguable that the cyanobacterial CCM contributes to primary productivity, especially in oceanic regions where cyanobacteria are estimated to contribute as much as 25% to annual global primary productivity [Citation4,Citation5]. The archetypal BicA transporter from Synechococcus PCC7002 is predicted to be 566 amino acids in length (59.6 kD) and topology predictions indicate it could have up to 10–12 transmembrane domains. The BicA transporter, like all the active CO2 and HCO3- uptake systems in cyanobacteria, is believed to be inactive in the dark, a process that potentially involves protein phosphorylation events [Citation3]. As a prelude to understanding the post-translational or allosteric activation of BicA we have sought to topology-map the structural domains of BicA so that putative cytoplasmic loops involved in regulation can be identified.
Interestingly, BicA is member of the large SulP family that includes the SLC26 family in mammals. SulP members include a wide range of anion transporters or exchangers with diverse substrates and physiological functions in prokaryotic and eukaryotic species. These include sulphate transport into and within plants [Citation6], chloride/HCO3- exchange in mammalian epithelial cells, iodide transport into the thyroid and sulphate uptake into fibroblasts [Citation7]. Structural and topological information for this family is extremely limited, with no structure determined for any member and topological predictions suggesting between 10 and 14 transmembrane helices (TMH). Although the sequence identity across the family is low, the distribution of hydrophobic regions is conserved, suggesting a similar structure for all family members. For several of the mammalian members, it has been established that the N- and C-termini are intracellular, indicating that there is an even number of transmembrane helices [Citation8–12]. A characteristic of the SulP family is a conserved C-terminal domain, the STAS domain (Sulfate Transporter Anti-Sigma factor domain) which has been suggested to be involved in interactions with other proteins and to have a role in regulation of transport activity. This is consistent with an intracellular location and for some transporters, interacting partners have been identified [Citation8,Citation9].
It is important to determine topology experimentally as a first step in understanding structure and function of a membrane protein. Identification of intracellular regions paves the way for further studies on potential regulatory regions, which may not be restricted to the STAS domain. One advantage of working with bacterial transporters is the availability of reliable fusion reporter systems for the determination of topology. One such system is the phoA/lacZ reporter system. This dual construct is fused to successive C-terminal truncations of the transporter gene and the activity of both enzymes determined [Citation13,Citation14]. Alkaline phosphatase is active only in the periplasm while β-galactosidase is active only in the cytoplasm so the relative activity indicates the topological orientation of the construct. An advantage of the dual reporter system is that the ratio of the two activities can be calculated and this corrects for any differences in the expression level of different fusion constructs [Citation13]. Fusion protein topology mapping has been used for several transporters for which the structure was determined subsequently and in these cases the topology was confirmed by the structure [Citation14,Citation15]. We therefore aimed to use the phoA/lacZ system to determine the topology of BicA and then fitted our data to a range of predictive models for a best fit. Prior to the present report, no member of the SulP family had been topology-mapped and so the present report includes basic structural information that may be applicable to other members of the SulP (SLC26) family.
Materials and methods
Bacterial strains and growth conditions
Escherichia coli JM109 was used as plasmid host in this study and strains were grown aerobically at 37°C in Luria Broth (LB). Dual indicator plates [Citation13] contained 1% Bacto-tryptone, 0.5% yeast extract, 0.5% NaCl, 1.5% Bacto-agar, 80 mM K2HPO4 (pH 7.0), 1 mM IPTG, 80 μg/ml X-phos (5-bromo-4-chloro-3-indolyl phosphate, disodium salt, Research Organics), 100 μg ml-1 Red-Gal (6-chloro-3-indolyl-β-D-galactoside, Research Organics) and 30 μg ml-1 of chloramphenicol.
Cloning of BicA into the dual reporter construct
Plasmid pNV1216 [Citation16] harbouring the phoA-lacZ fusion was digested with Bam HI and Xba I to remove the gtrII gene and replaced with the 1.7 kb bicA gene from Synechococcus PCC7002 that was PCR amplified from genomic DNA with forward primer BicA(7002)BamH1(F), and reverse primer BicA(7002)XbaI(R); tcatcacggatccgaaaaatgc and cgcagatcatctagataacccatctc, respectively. The phoA-lacZ reporter fusion was constructed according to Alexeyev and Winkler [Citation13].
Construction of BicA-phoA/lacZ fusions using Exonuclease-III
BicA(7002)/pNV1216 was linearized with the unique restriction sites, NsiI and XbaI. The Promega Erase-a-base kit was used to create a series of BicA-phoA/lacZ fusions according to the manufacturer's recommendations. After ligation, plasmid DNA was transformed into JM109 cells and plated onto dual indicator plates. Plasmid DNA was purified from blue, red and purple colonies, digested with Bam HI and Sac I to confirm deletion size, and sequenced with the PhoSeq primer (cgctaagagaatcacgcaga). Big-dye Sequencing was carried out following the manufacturer's protocol (Applied Biosystems).
Construction of BicA-phoA/lacZ fusions using PCR
PCR was used to generate a number of BicA-phoA/lacZ fusions at desired locations to create BicA-phoA/lacZ C-terminal fusions G59, T121, L340, A369 and I393 (amino acid position of last residue before reporter). Primers were designed with an XbaI restriction site 3′ of the fusion site to allow in-frame directional cloning into pNV1216. BicA (7002) was excised from pNV1216 using double digests with BamHI and XbaI. The following primer combinations were used for PCR reactions: BicA(7002)BamH1(F) and one of BicA(G59)XbaI(R), BicA(T121)XbaI(R), BicA(L340)XbaI(R), BicA(A369)XbaI(R) or BicA(I393)XbaI(R); tcctctagacccaaagagggcggcaaagaa, cattctagaggtgacgtatttgccgagtt, ccgtctagataaactagccgccccaag, cgttctagaggcccgttttaggaaactc, or gactctagaaatcaagtcaactaagaccgtca, respectively. PCR products were ligated into BamHI/XbaI digested pNV1216 and transformed into JM109 cells, sequenced and then colonies were analysed on dual indicator plates.
Fluorometric assays of alkaline phosphatase and β-galactosidase activities
E. coli JM109 cells containing BicA-phoA/lacZ fusion constructs were grown overnight, diluted 1:10 in fresh LB containing chloramphenicol and induced with 1 mM IPTG. Cells were grown for a further 2–3 h until A600 ∼ 0.5, then harvested by centrifugation and permeabilized as described previously [Citation17]. For the β-galactosidase assay, cells were harvested as above, except prior to permeabilization, cells were resuspended in the following buffer (25 mM Tris-HCl pH 7.5, 125 mM NaCl, 2 mM MgCl2 and 12 mM 2-mercaptoethanol). Stock solutions of 4-methylumbelliferyl β-D-galactopyranoside (4-MUG) and 4-methylumbelliferyl phosphate (4-MUP) were prepared by dissolving in sterile water and DMSO, respectively. The substrates were diluted in reaction buffer immediately prior to experiments. The reaction buffer for alkaline phosphatase assay was: 1 M Tris- HCl pH 8.0, 0.1 mM ZnCl2 and 100 μM 4-MUP, and for the β-galactosidase assay: 25 mM Tris-HCl pH 7.5, 125 mM NaCl, 2 mM MgCl2,12 mM 2-mercaptoethanol and 100 μM 4-MUG. Assays were performed using the TKO 100 Mini Fluorometer (Hoefer, Inc., Holliston, MA, USA) following the manufacturers' protocol. Reactions were stopped with 5% trichloroacetic acid (TCA) and the solution clarified by centrifugation. Glycine carbonate reagent (133 mM glycine, 83 mM Na2CO3, pH 10.7) was added and fluorescence was measured. Excitation occurs at 380 nm, and 4-MU emits at a wavelength of 460 nm. At least three independent cultures for each construct were assayed. A standard curve was generated with 4-methylumbelliferone (4-MU) at concentrations between 0 and 500 nM allowing the activity of samples to be calculated.
Topology prediction analysis
A number of computer programs available on the Internet, were used to examine the BicA protein sequence for predicted membrane topology. The TOPCONS website (http://topcons.net/index.php) was used to give a consensus prediction of membrane protein topology using several different algorithms: SCAMPI (single sequence mode) [Citation18], SCAMPI (multiple sequence mode) [Citation18], OCTOPUS and TMHMM [Citation19]. A number of other programs were used including MEMSAT3 [Citation20] and TopPred 2 [Citation21].
Results
PhoA-LacZ mapping of BicA from Synechococcus PCC7002
A total of 47 random in-frame fusions were initially identified on dual reporter plates and scored as red (putative cytoplasmic locations), blue (putative periplasmic locations) or purple colonies (putative membrane spanning domains; ). Several different topology prediction programs were used to predict topologies for BicA () and the locations indicated for the initial fusions were compared to the models generated. The results of different programs suggested 10–12 transmembrane helices (TMH). Some predictions (for example those with a periplasmic location for the STAS domain) could be immediately excluded based on the initial data. To distinguish between the remaining predicted topologies, a further five targeted C-terminal fusions of BicA were generated using PCR (see Methods). DNA sequencing was used to verify the location of all fusions generated by both methods.
Table I. Enzyme activity of BicA dual reporter fusions.
Table II. Predicted topologies for BicA using available prediction programs.
A collection of 41 fusion constructs was quantitatively assessed for alkaline phosphatase and β-galactosidase activity using a fluorescence enzyme assay (). Assay data were normalized and expressed as ratios to allow for any differences in the expression of fusion constructs (). In theory, a ratio of alkaline phosphatase to β-galactosidase activity greater than 1 should indicate a periplasmic location, while a ratio less than 1 would be consistent with a cytoplasmic location. In practice there are two complications. Firstly, some fusions may undergo cytosolic proteolysis which results in abnormally high β-galactosidase activity when a normally periplasmic C-terminus is released in the cytoplasm [Citation14]. However, the converse does not occur so a high alkaline phosphatase activity is a better indicator of a periplasmic location than β-galactosidase is of a cytoplasmic location. Secondly, TMH-located fusions may be targeted to both the periplast and cytoplast, and this can depend on the protein sequence and on the nature of the fusion protein [Citation14]. These two factors were taken into account to give the scoring parameters described in footnotes, which include colony colour and alkaline phosphatase activity as well as the ratio of alkaline phosphatase to β-galactosidase activities.
Data obtained from the activity ratios of the C-terminal fusions suggested a topology model of 12 transmembrane helices with the N- and C-termini in the cytoplasm (). Most predictive models used also take into account the distribution of positive charges, and as shown in our topology model conforms well to the positive-inside-rule [Citation22]. Five cytoplasmic loops could be clearly identified, with each containing at least one fusion where the activity ratio was less than 0.1 and the colony was red (; and ). Six periplasmic loops were also identified, where at least one fusion resulted from a blue colony with an activity ratio of greater than 1.0 (). Five of these fusions had significant alkaline phosphatase activity (> 30% of the maximum activity). In the remaining periplasmic loop, between putative helices 3 and 4, the only fusion that had an activity ratio greater than 1.0 (V100) appeared to be poorly expressed, as indicated by the low activity of both enzymes. However, the two preceding fusions had high alkaline phosphatase activity (G93 and A97) which is inconsistent with this region forming a cytoplasmic loop and therefore supports the existence of helices 3 and 4 in this location.
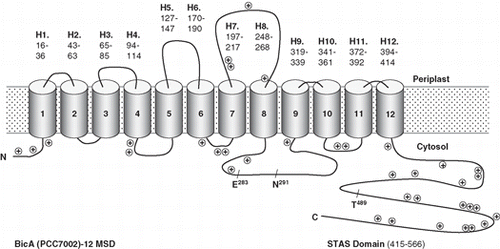
Figure 1. The topology map for the Synechococcus PCC7002 Na+-dependent HCO3- transporter, BicA. Experimentally determined phoA/lacZ mapping positions () are shown as black (cytoplasmic), mid-grey (TMH located) or light grey (periplasmic); in the online version these are shown as blue, purple and red, respectively. The inset shows a WebLogo (weblogo.berkeley.edu) representation of the consensus of the ungapped alignment of the 50 residue loop between TMH 8 and 9; all the SulP/SLC26A members used to generate the phylogenetic tree shown in were used and BicA numbering is shown. This region corresponds to the second signature sequence identified by Saier et al. [Citation23] for the SulP family; the most highly conserved subset, NSNKELIGQGLGN (279-291), is highlighted. This Figure is reproduced in colour in the online version of Molecular Membrane Biology.
![Figure 1. The topology map for the Synechococcus PCC7002 Na+-dependent HCO3- transporter, BicA. Experimentally determined phoA/lacZ mapping positions (Table I) are shown as black (cytoplasmic), mid-grey (TMH located) or light grey (periplasmic); in the online version these are shown as blue, purple and red, respectively. The inset shows a WebLogo (weblogo.berkeley.edu) representation of the consensus of the ungapped alignment of the 50 residue loop between TMH 8 and 9; all the SulP/SLC26A members used to generate the phylogenetic tree shown in Figure 3 were used and BicA numbering is shown. This region corresponds to the second signature sequence identified by Saier et al. [Citation23] for the SulP family; the most highly conserved subset, NSNKELIGQGLGN (279-291), is highlighted. This Figure is reproduced in colour in the online version of Molecular Membrane Biology.](/cms/asset/5131875b-2944-4eaf-b3a6-ac676594d9bb/imbc_a_440190_f0001_b.jpg)
Figure 2. A simplified version of the topology map for BicA (Synechococcus PCC7002) showing the positions of positive charged residues and three highly conserved residues that are discussed in the text, E283 and N291, that are implicated in human disease and T489, which is a putative phosphorylation site.
Implications of the BicA topology for the SulP family
Bacterial transporters have frequently been found to be good structural models for eukaryotic homologs so we examined other members of the SulP family in the light of the BicA topology. The SulP family is defined by conserved motifs in helices 1 and 2 and in the cytoplasmic region between helices 8 and 9 [Citation23]. Conserved amino acid residues in helices 1 and 2 have been identified as functionally important in the plant SulP transporter, SHST1 [Citation24] but the other conserved region has not been well studied. One feature of BicA that makes correct prediction of its topology difficult is the extremely tight turns between helices. While eukaryotic members of the family are generally 100–200 amino acids longer than bacterial members, at least some of the tight turns, as indicated by spacing on topology predictions, appear to be conserved. We therefore examined different topology predictions (see Methods) for BicA to determine which ones predicted a topology that agrees with the phoA/lacZ fusion data and compared these to predictions for other members of the family. As was found for BicA, a range of topologies were predicted for several eukaryotic members of the SulP family, usually showing 10–12 TMH, depending on the algorithm used.
The only programs that accurately predicted the topology of BicA were SCAMPI-msa and the latest version of MEMSAT3 (). Other programs were generally unable to distinguish some of the most closely connected helices (helices 1–3 and 9–12) and many also predicted a TMH for the region between helices 8 and 9 that we have shown is cytoplasmic. SCAMPI-msa and MEMSAT3 differ from other programs in that they both use a multiple sequence alignment as a basis for prediction. The entered query sequence is first used by each program to generate a multiple alignment, which is then used for topology prediction. This is thought to improve the accuracy of prediction as it takes into account sequence conservation as well as hydrophobicity [Citation20].
A phylogenetic tree was generated for a wide range of prokaryotic and eukaryotic SulP transporters ().
Figure 3. Phylogenetic tree showing the relationship of the cyanobacterial BicA family (30 members) to the Arabidopsis SulP sulphate transporter family (13 members), the human SLC26A family (9 members) and a selection of bacterial BicA-like proteins (13 members). Protein sequences were aligned using ClustalW and nearest neighbour-joining routines in MEGA4 software (www.megasoftware.net). The scale marker represents 0.2 substitutions per residue.
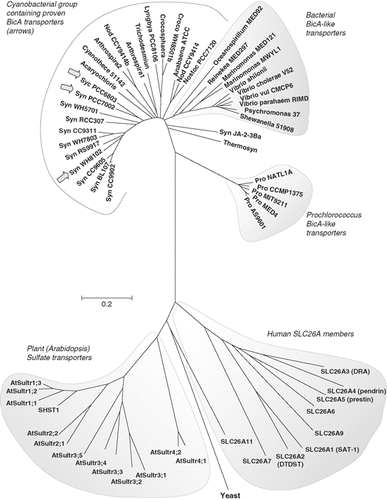
Figure 4. BicA (Synechococcus PCC7002) aligned to an extensively investigated member of the plant sulphate transporter family (SHST1, Stylosanthes hamata) and a member of the human SLC26A family (DTDST, SLC26A2) that is linked to the disease, diastrophic dysplasia. SCAMPI-msa predicted TMH regions are shaded grey based on separate queries for each protein. The two disease-causing residues, E283 and N291, and the putative phosphorylation site T489, are shown.

One of the most striking differences between the different topology models is the location of the region that we have shown forms a relatively large cytoplasmic loop (50 residues from V269 to R318) between helices 8 and 9 of BicA ( and ). All programs other than SCAMPI-msa and MEMSAT3 predicted a TMH in the middle of this region (). The experimentally determined topology for BicA is consistent with a cytoplasmic loop in this region (), although its hydrophobicity suggests that it could form a partially re-entrant loop. Such a loop is unlikely to be detected by a fusion approach because part of the loop will be replaced by the fusion protein. A re-entrant loop might only enter the membrane as a folded structure which would require the presence of the entire loop sequence. This region also includes a number of highly conserved residues, as shown on the WebLogo (weblogo.berkeley.edu) representation (, inset) derived from the aligned members shown in .
Discussion
We have shown that the cyanobacterial Na+-dependent HCO3- transporter, BicA, has 12 transmembrane helices identified by phoA/lacZ fusion mapping ( and ). The structure and function of BicA is of interest for several reasons. Firstly, BicA has an important physiological role in the uptake of bicarbonate for photosynthesis in cyanobacteria. As a transporter with a high flux rate that contributes to the cyanobacterial carbon concentrating mechanism and which has homologs in all major clades of cyanobacteria (), it is likely to have a significant role in photosynthetic productivity on a global scale. Secondly, BicA is a member of a larger family including eukaryotic as well as prokaryotic members – the SulP or SLC26 family. BicA is the first member of this family for which a comprehensive topological analysis has been done and our bioinformatic analyses suggest that the proposed 12 TMH topology may apply across the family.
The physiological role of BicA in the cyanobacterial CO2 concentrating mechanism has been well characterized but little is known about its regulation. Two types of allosteric regulation appear to exist, although both may involve protein phosphorylation [Citation3]. The first involves inactivation of HCO3- transporters (and the CO2 uptake systems) on the transition from light to dark, presumably to avoid futile cycling (pump and leak) in the dark, while the second, known as ‘fast induction’ manifests as a rapid activation of transporter affinity on exposure to severe HCO3-/CO2 limitation, and is known to be blocked by Ser/Thr protein kinase inhibitors [Citation25]. To gain a greater understanding of these processes, it is important to identify which parts of the protein are intracellular and may, therefore, contain phosphorylation or other regulatory sites. Topological analysis provides this information and therefore acts as a starting point for the identification of putative regulatory regions. An unexpected finding of this study is the existence of a 50 residue intracellular loop between helices 8 and 9. In this respect, our experimentally determined topology differs from predictions from most programs, which have been used to suggest this region formed a transmembrane helix. Our new topology has implications for the functioning and regulation of BicA and for eukaryotic members of the family, where there exists experimental data relating to the function of this region of the protein (discussed below).
An advantage of studying topology in bacterial transporters is the availability of effective dual reporter methods such as the phoA-lacZ system used here. Dual reporter systems have been shown to provide accurate topological information by comparing results with subsequent structures as they become available [Citation15]. A recent study tested the reliability of reporter fusion topology mapping by using this approach to determine the topology of the E. coli ClcA H+/Cl- exchange transporter, for which the structure is known [Citation15]. These authors used phoA and GFP fusions and found that for normal membrane-spanning helices, the results were highly reliable. It was concluded that the dual reporter accurately predicted ‘normal’ TMH although it did not detect the two shorter, and less hydrophobic, helical hairpins seen in the ClcA structure. Of interest to the present study, is that two TMH linked by a tight turn in ClcA which could not be clearly distinguished on the hydropathy plot were distinguished by the fusion data. In BicA, the first three helices and the pairs, 9 and 10, and 11 and 12, are linked by tight turns. The fusion data is consistent with the existence of these 7 helices, even though they are not predicted by all topology prediction programs (). It is possible that some of these helices may include short sections that are non-helical, as has been found for several transporters [Citation26]. This would serve to lengthen these helices, in addition to introducing a less hydrophobic region which may be important for transport function.
Different topologies with 10–14 transmembrane helices have been proposed for different members of the SulP family. Our results confirm the intracellular location of the N- and C-termini found for other members of the family [Citation9,Citation12,Citation27]. There have been some studies providing limited topological information for eukaryotic members of the SulP family, including several that examined the functionality of conserved glycosylation sites that occur after the third predicted transmembrane region. Two studies indicated that these are glycosylated [Citation28,Citation29] and therefore outside the cell (consistent with the BicA topology) but a third found no evidence for glycosylation [Citation30]. Epitope tagging was used to suggest a ten helix structure for prestin in which most TMH were inverted compared to other predictions [Citation30]. In the light of these conflicting data, it is hard to draw definitive conclusions. This is partly because these studies were done on different members of the family, using different techniques, in contrast to our comprehensive analysis of BicA.
Although a topology of 12 transmembrane helices has been predicted for many members of the SulP family, our experimentally determined topology differs from most of these in the positions of helices 9–12. Interestingly, the SCAMPI-msa topology prediction, which is based on a multiple sequence alignment, consistently predicts this structure, regardless of which SulP member is used as the query sequence. However, other topology predictions based on individual transporter sequences commonly miss one or more helices and also often predict a TMH in the cytoplasmic loop region between helices 8 and 9. The sequence previously thought to form helix 9 is clearly identified as cytoplasmic in BicA by multiple fusions in this region (). The predicted helices 10 and 11 become 9 and 10 in our model and are therefore inverted relative to earlier predictions. The final region of hydrophobicity is identified by our fusion data as a tight helical hairpin, bringing the C-terminal STAS domain into the cytoplasm, consistent with earlier studies. The orientation of these final four helices in BicA is more consistent with the positive-inside-rule than earlier predicted SulP family topologies ().
The sequence of the intracellular region between helices 8 and 9 is highly conserved and forms part of the SulP consensus motif [Citation23]. It is also quite hydrophobic, raising the possibility that it may form a partial re-entrant loop, which would not have been detected by our fusion analysis. Mutational analysis of amino acid residues in this region is consistent with it being involved in a protein/protein interaction, rather than forming an unstructured cytoplasmic loop. Mutations in the human SulP members, SLC26A2 and SLC26A4 are responsible for the diseases diastrophic dysplasia and Pendred syndrome, respectively [Citation7]. Some mutations affect conserved amino acid residues in the sequence between helices 8 and 9 [Citation31–33]. Mutations in some of the homologous residues have been introduced into the plant SulP member, SHST1, and primarily affected folding and trafficking of the mutant transporters in a yeast expression system [Citation34]. The two mutations tested either introduced or removed a charge (affecting the highly conserved residues, E283 and N291, BicA numbering, and ) so it was suggested that they affected interaction with other helices and hence folding. These results are more surprising for a cytoplasmic region of the protein but could be accounted for if the loop is involved in protein/protein interactions. This loop may therefore be functionally important and is a good candidate for an intracellular regulatory region.
In the light of these results, we now hypothesize that this loop region interacts with another part of the BicA protein, either within the membrane, or with the C-terminal STAS domain, which is homologous to a protein/protein interaction domain of the sporulation regulator, SpoIIA [Citation35]. Recent work on ammonium transporters from the Amt family has found that a C-terminal domain interacts with cytoplasmic loops to regulate transporter function [Citation36,Citation37]. This interaction is controlled by phosphorylation of the C-terminal domain. BicA, like most SulP transporters, contains a putative phosphorylation site in the STAS domain at T489 (). Our topology for BicA therefore raises the possibility that a similar mode of regulation to that found in the Amt family exists. Future mutagenesis studies could test this possibility as well as examining the potential functional and regulatory roles of the loop between helices 8 and 9 more generally. Our experimentally determined topology has provided the basis for a revised understanding of existing data for this important transporter family as well as suggesting new directions.
Acknowledgements
This work was supported by an Australian Research Council Discovery grant, number DP0984773, to GDP; we thank Loraine Tucker, Warren Hudson-Taylor, Sara Milward and Soumi Bala for occasional technical support. We thank Dr Fiona Leves for helpful discussions and Naresh Verma's lab for the gift of the pNV1216 plasmid.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Price GD, Woodger FJ, Badger MR, Howitt SM, Tucker L. 2004. Identification of a SulP-type bicarbonate transporter in marine cyanobacteria. Proc Nat Acad Sci USA 101:18228–18233.
- Badger MR, Price GD, Long BM, Woodger FJ. 2006. The environmental plasticity and ecological genomics of the cyanobacterial CO2 concentrating mechanism. J Exp Bot 57:249–265.
- Price GD, Badger MR, Woodger FJ, Long BM. 2008. Advances in understanding the cyanobacterial CO2-concentrating-mechanism (CCM): Functional components, Ci transporters, diversity, genetic regulation and prospects for engineering into plants. J Exp Bot 59:1441–1461.
- Field CB, Behrenfeld MJ, Randerson JT, Falkowski P. 1998. Primary production of the biosphere – integrating terrestrial and oceanic components. Science 281:237–240.
- Liu HB, Nolla HA, Campbell L. 1997. Prochlorococcus growth rate and contribution to primary production in the equatorial and subtropical North Pacific Ocean. Aquatic Microb Ecol 12:39–47.
- Hawkesford MJ, De Kok LJ. 2006. Managing sulphur metabolism in plants [Review]. Plant Cell Environ 29:382–395.
- Mount DB, Romero MF. 2004. The SLC26 gene family of multifunctional anion exchangers [Review]. Eur J Physiol 447:710–721.
- Chernova MN, Jiang LW, Shmukler BE, Schweinfest CW, Blanco P, Freedman SD, Stewart AK, Alper SL. 2003. Acute regulation of the SLC26A3 congenital chloride diarrhoea anion exchanger (DRA) expressed in Xenopus oocytes. J Physiol (Lond) 549:3–19.
- Ko SBH, Zeng WZ, Dorwart MR, Luo X, Kim KH, Millen L, Goto H, Naruse S, Soyombo A, Thomas PJ, Muallem S. 2004. Gating of CFTR by the STAS domain of SLC26 transporters. Nature Cell Biol 6:343–350.
- Rouached H, Berthomieu P, El Kassis E, Cathala N, Catherinot V, Labesse G, Davidian JC, Fourcroy P. 2005. Structural and functional analysis of the C-terminal STAS (sulfate transporter and anti-sigma antagonist) domain of the Arabidopsis thaliana sulfate transporter SULTR1.2. J Biol Chem 280:15976–15983.
- Shibagaki N, Grossman AR. 2006. The role of the STAS domain in the function and biogenesis of a sulfate transporter as probed by random mutagenesis. J Biol Chem 281:22964–22973.
- Lohi H, Lamprecht G, Markovich D, Heil A, Kujala M, Seidler U, Kere J. 2003. Isoforms of SLC26A6 mediate anion transport and have functional PDZ interaction domains. Am J Physiol – Cell Physiol. 284:C769–779.
- Alexeyev MF, Winkler HH. 1999. Membrane topology of the Rickettsia prowazekii ATP/ADP translocase revealed by novel dual pho-lac reporters. J Mol Biol 285:1503–1513.
- van Geest M, Lolkema JS. 2000. Membrane topology and insertion of membrane proteins: Search for topogenic signals [Review]. Micro Mol Biol Rev 64:13–33.
- Cassel M, Seppala S, von Heijne G. 2008. Confronting fusion protein-based membrane protein topology mapping with reality: The Escherichia coli ClcA H+/Cl- exchange transporter. J Mol Biol 381:860–866.
- Lehane AM, Korres H, Verma NK. 2005. Bacteriophage-encoded glucosyl-transferase GtrII of Shigella flexneri: Membrane topology and identification of critical residues. Biochem J 389:137–143.
- Manoil C. 1991. Analysis of membrane-protein topology using alkaline-phosphatase and gamma-galactosidase gene fusions. Methods Cell Biol 34:61–75.
- Bernsel A, Viklund H, Falk J, Lindahl E, von Heijne G, Elofsson A. 2008. Prediction of membrane-protein topology from first principles. Proc Nat Acad Sci USA 105:7177–7181.
- Viklund H, Elofsson A. 2004. Best alpha-helical transmembrane protein topology predictions are achieved using hidden Markov models and evolutionary information. Protein Sci 13:1908–1917.
- Jones DT. 2007. Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 23:538–544.
- Von Heijne G. 1992. Membrane-protein structure prediction – hydrophobicity analysis and the positive-inside rule. J Mol Biol 225:487–494.
- von Heijne G, Gavel Y. 1988. Topogenic signals in integral membrane proteins. Eur J Biochem 174:671–678.
- Saier MH, Eng BH, Fard S, Garg J, Haggerty DA, Hutchinson WJ, Jack DL, Lai EC, Liu HJ, Nusinew DP, Omar AM, Pao SS, Paulsen IT, Quan JA, Sliwinski M, Tseng TT, Wachi S, Young GB. 1999. Phylogenetic characterization of novel transport protein families revealed by genome analyses. Biochim Biophys Acta – Rev Biomemb 1422:1–56.
- Leves FP, Tierney ML, Howitt SM. 2008. Polar residues in a conserved motif spanning helices 1 and 2 are functionally important in the SulP transporter family. Int J Biochem Cell Biol 40:2596–2605.
- Sültemeyer D, Klughammer B, Badger MR, Price GD. 1998. Fast induction of high-affinity HCO3- transport in cyanobacteria. Plant Physiol 116:183–192.
- Screpanti E, Hunte C. 2007. Discontinuous membrane helices in transport proteins and their correlation with function. J Struct Biol 159:261–267.
- Zheng J, Long KB, Shen WX, Madison LD, Dallos P. 2001. Prestin topology: Localization of protein epitopes in relation to the plasma membrane. Neuroreport 12:1929–1935.
- Byeon MK, Westerman MA, Maroulakou IG, Henderson KW, Suster S, Zhang XK, Papas TS, Vesely J, Willingham MC, Green JE, Schweinfest CW. 1996. The down-regulated in adenoma (Dra) gene encodes an intestine-specific membrane glycoprotein. Oncogene 12:387–396.
- Matsuda K, Zheng J, Du GG, Klocker N, Madison LD, Dallos P. 2004. N-linked glycosylation sites of the motor protein prestin: Effects on membrane targeting and electrophysiological function. J Neurochem 89:928–938.
- Navaratnam D, Bai JP, Samaranayake H, Santos-Sacchi J. 2005. N-terminal-mediated homomultimerization of prestin, the outer hair cell motor protein. Biophys J 89:3345–3352.
- Coyle B, Reardon W, Herbrick JA, Tsui LC, Gausden E, Lee J, Coffey R, Grueters A, Grossman A, Phelps PD, Luxon L, Kendalltaylor P, Scherer SW, Trembath RC. 1998. Molecular analysis of the Pds gene in Pendred-syndrome (Sensorineural Hearing Loss and Goitre). Human Mol Genet 7:1105–1112.
- Pera A, Dossena S, Rodighiero S, Gandia M, Botta G, Meyer G, Moreno F, Nofziger C, Hernandez-Chico C, Paulmichl M. 2008. Functional assessment of allelic variants in the SLC26A4 gene involved in Pendred syndrome and nonsyndromic EVA. Proc Nat Acad Sci USA 105:18608–18613.
- Karniski LP. 2001. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene: Correlation between sulfate transport activity and chondrodysplasia phenotype. Human Mol Genet 10:1485–1490.
- Khurana OK, Coupland LA, Shelden MC, Howitt SM. 2000. Homologous mutations in two diverse sulphate transporters have similar effects. FEBS Lett 477:118–122.
- Aravind L, Koonin EV. 2000. The STAS domain – a link between anion transporters and antisigma-factor antagonists. Curr Biol 10:R53–55.
- Loque D, Lalonde S, Looger LL, von Wiren N, Frommer WB. 2007. A cytosolic trans-activation domain essential for ammonium uptake. Nature 446:195–198.
- Neuhaus HE. 2007. Transport of primary metabolites across the plant vacuolar membrane. Febs Lett 581:2223–2226.