Abstract
Liposomes have found clinical application in cancer therapy in the delivery of cytostatic agents. As a result of the targeted delivery of these toxic molecules to the tumour cells coupled to avoidance of toxicity-sensitive tissues, the therapeutic window is widened. Over the past years the focus of cancer therapy has shifted towards the stromal cells that are present in the tumour. It appears that clinically relevant tumours have acquired the ability to modulate the microenvironment in such a way that a chronic pro-inflammatory and pro-angiogenic state is achieved that contributes to invasion and metastasis and continued proliferation. Over the past years, liposomal formulations have been designed that target key stromal cell types that contribute to tumour growth. At the same time, many promising cell types have not been targeted yet and most of the studies employ drugs that aim at depleting stromal cells rather than modulating their activity towards an anti-tumour phenotype. In this review these target cell types will be addressed. Complementing these targeted formulations with the appropriate drugs to optimally suppress tumour-promoting signals while preserving anti-tumour action will be the challenge for the future.
Introduction: From conventional to tumour cell-targeted therapies
Conventional cancer therapies, such as surgery, chemotherapy and radiotherapy, focus on eliminating tumour cells while minimizing the harm to the surrounding healthy tissue as much as possible. Surgery offers the greatest chance for cure for many types of cancer, especially those that have not spread to other parts of the body. It is usually followed by additional treatment to attack remaining micro-nodules and not all cancer types are amenable to surgical resection. Chemotherapy is designed to damage rapidly dividing cells and thus it affects not only tumour cells, causing a lot of discomfort for the patient during treatment. Radiotherapy can be directed to deliver the highest dose to the tumour mass and in addition cancer cells are most vulnerable because these cells are more sensitive to damage from radiation. Nevertheless, healthy tissue suffers from this treatment.
In 1975, 50% of the cancer patients in the U.S. survived five years after the diagnosis, which increased to 68% in 2006 [Surveillance Epidemiology and End Results [SEER] Citation2006). This increase in surviving patients is probably attributed to the combined action of improvements to conventional therapies, improved population awareness (introspection) and more advanced diagnostic tools. Although these conventional therapies have proven effective for many cancer patients, a large group of patients experience dose-limiting adverse effects or develop tumour therapy resistance. One of the key elements in improving cancer therapy lies in the development of drugs that are more selective. The selectivity can lie in attacking a molecular pathway that is preferentially active in tumour cells. The success of imatinib, a specific tyrosine kinase inhibitor, in the treatment of patients with chronic myelogenous leukemia (CML) shows the promise of such therapies (Deininger and Druker Citation2003). Imatinib inactivates the kinase activity of BCR-ABL, the fusion protein that causes CML. Compared to cytotoxic drugs, such targeted (kinase) inhibitors have more favourable levels of toxicity, yet their use is still associated with some level of side-effects (Stegmeier et al. Citation2010).
Another approach that may improve the selectivity for tumour cells is to increase the concentration of a therapeutic agent in the tumour while limiting systemic exposure. Site-specific drug delivery approaches often take advantage of the unique pathophysiology of tumour vasculature. Tumours contain abnormal, dilated, tortuous blood vessels that are poorly differentiated, with a higher vascular permeability than normal vessels (Hellberg et al. Citation2010). As a result local accumulation of macromolecules and nanoparticles occurs by virtue of the enhanced permeability and retention effect, which results from a combination of increased permeability of tumour blood vessels and a decreased rate of clearance caused by the lack of functional lymphatic vessels in the tumour (Hobbs et al. Citation1998, Gabizon et al. Citation2006, Maeda et al. Citation2009) Therefore drug carriers can passively accumulate over time in solid tumours after intravenous administration, provided their circulation time is extended. A formulation that is in the clinic, which exploits this phenomenon, is Doxil®. This pegylated, long-circulating, liposomal formulation of doxorubicin creates higher drug levels within the tumour and avoids uptake into the doxorubicin-sensitive cardiac tissue, thus creating a larger therapeutic window (Gabizon Citation2001).
Also molecular conjugates have been prepared that specifically bind to surface receptors that are overexpressed on the tumour cells (Hilgenbrink and Low Citation2005). One of these conjugates is EC145, a folic acid drug conjugate, which is the most advanced in its development, as it was the first folic acid drug conjugate to be evaluated in clinical trials (Li et al. Citation2009). This conjugate of folate and vinblastine targets the folate receptor alpha present on the tumour cell surface, strongly amplifying the therapeutic effect of the drug while limiting the side effects (Low and Kularatne Citation2009).
However, for these targeted therapies, the risk of development of resistance is looming. Irrespective of the nature of the targeting, be it attacking a cancer-cell specific pathway or delivering more drug molecules to the tumour cells, clonal selection of escape mutants of the genetically unstable tumour cells is a realistic threat. For example, many patients who were treated with imatinib have since become resistant, due to mutations in the target protein (Jabbour et al. Citation2007). One of the possible ways to support therapies directed towards tumour cells is by modulating the activity of the cells that constitute the ‘ecosystem’ of the tumour ().
Figure 1. Hallmarks of cancer. Seven processes define cancer and these processes are primarily associated with a variety of host cells that contribute to tumour growth and form the tumour's ecosystem.
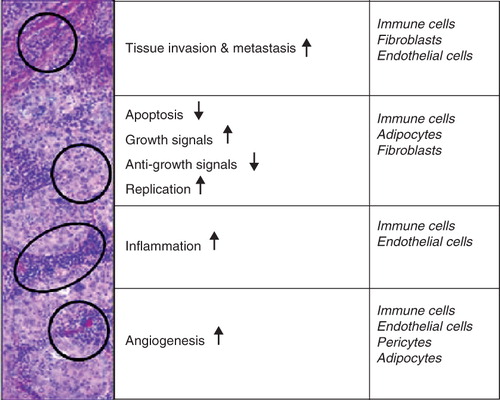
Research on tumour biology has shown that host cells in the tumour cells' environment are able to promote survival, progression and metastasis for the tumour and can be considered to be part of the tumour's ecosystem (Virchow Citation1863, Vasiliev Citation1964, Freitas and Baronzio Citation1994, Mor et al. Citation1998, Shain et al. Citation2000, Knezevic et al. Citation2001, Roskelley and Bissell Citation2002, Keibel et al. Citation2009). Although Virchow, already in 1863, introduced a possible role for inflammation and the immune system in cancer (Virchow Citation1863), it has become clear only recently that also other stromal cells, like adipocytes, fibroblasts, endothelial cells, innate and adaptive immune cells and pericytes are able to influence cancer growth (Freitas and Baronzio Citation1994, Baluk et al. Citation2003, Iyengar et al. Citation2003, Silzle et al. Citation2004). Here we review the various cell types within the tumour that are able to promote tumour growth and give an overview of liposomal formulations targeted to these cells.
Stromal cell types within the tumour
Immune cells
The immune system has been implicated to influence cancer for a long time. Virchow was the first to describe the presence of macrophages in tumour tissue (Virchow Citation1863). Initially it was thought that the immune system fights cancer. The immune system may indeed have a protective role in tumour development, known as cancer immunosurveillance (Dunn et al. Citation2002, Citation2004). This process seems especially effective in virus-induced cancers. A compromised immune system marks a sharp increase in cancer incidence (e.g., Kaposi sarcoma in HIV-patients) (Kinlen Citation1992). In general, however, immunosurveillance seems important in eliminating early-stage transformed cells, whereas clinically advanced tumours are able to avoid immunosurveillance by inducing immune tolerance (Zitvogel et al. Citation2006). Recently, the involvement of inflammation in the initiation, promotion and progression of cancer has received more attention (Balkwill and Mantovani Citation2010, Grivennikov et al. Citation2010, Moore et al. Citation2010).
Macrophages have a complex relationship with tumour cells. When appropriately activated they could be of value in anti-tumour strategies, however the vast majority of macrophages in the tumour is of the so called M2 phenotype. These alternatively activated macrophages contribute to tumour growth. They produce several molecules that help survival of the malignant cells, remodel extracellular membrane proteins, assist in the development of angiogenic blood vessels, down regulate adaptive immune responses by recruiting and stimulating T regulatory cells (Tregs) and recruiting Th2 lymphocytes, which inhibit cytotoxic T lymphocytes (CTL), and by inducing anergy of naïve T-cells [Balkwill et al. Citation2005).
Although mast cells are primarily known as the main effector cells in allergic, immunoglobulin (Ig) E-mediated immune reactions, mast cells can also be activated by many other immunological and non-immunological signals, such as IgG, immunoglobulin free light chains, complement factors, pathogens, inflammatory mediators, toll like receptors, and T-cells. Depending on the nature of the activating signals, activated mast cells can release a diverse array of pro-angiogenic and tumour growth stimulatory mediators (Detoraki et al. Citation2009). Increased numbers of mast cells are found in many tumours and it has been shown that the number of tumour infiltrating mast cells correlate with increased intratumoural microvessel density, enhanced tumour growth and tumour invasion, and poor clinical outcome (Crivellato et al. Citation2008, Cinel et al. Citation2009, Groot Kormelink et al. Citation2009, Strouch et al. Citation2010). Nevertheless, a couple of studies report a correlation with positive clinical prognosis, underlining that also the immune reaction orchestrated by mast cells can be beneficial as well as detrimental (Della Rovere et al. Citation2007, Hedstrom et al. Citation2007, Ali et al. Citation2009). Mast cells not only function as effector cells, they can also act as regulatory cells. Mast cell mediators can exert pro-inflammatory, anti-inflammatory and regulatory effects by influencing development, proliferation, migration or function of other immune cells of both adaptive and innate immune system. Recently, mast cells together with regulatory T-cells (Tregs) proved to be a strong prognostic factor for recurrence of hepatocellular carcinoma. Likely, these cells cooperate to shape the inflammatory microenvironment so that it promotes renewed outgrowth (Ju et al. Citation2009). The immunomodulatory functions of mast cells can be of importance to control tumour growth.
Polymorphonuclear neutrophils are the most abundant type of white blood cells. They are the first line of defence against microorganisms and can be rapidly recruited to sites of acute inflammation. In the tumour, however, the inflammatory response appears to be more chronic in nature. As a result, neutrophil infiltration in tumours is usually limited and their anti-tumour response is not very pronounced. Nevertheless, when an appropriate acute inflammatory signal can be delivered to the tumour, which should shift the chronic inflammatory state towards an acute inflammatory state, strong anti-tumour effects of neutrophils have been seen (Brú et al. Citation2010).
The cellular components of the adaptive immune system, i.e., B-cells, T-cells, and dendritic cells (DC), have primarily been targeted to elicit anti-tumour immune responses. Although these cell types are usually present inside the tumour, they are not effective because the tumour creates an environment with a state of immune paralysis (e.g., by M2 macrophages as described above). DC are normally in small quantities present in tissues and are powerful regulators of T- and B-cell immune responses as they are able to travel to the lymphoid tissues once they have been activated by antigens. There they present antigens to B- and T-cells to initiate the adaptive immune response (Chaput et al. Citation2008). Tumour cells seem to be able to suppress the functions of DC; DC from tumour tissue are less potent in stimulating T-cells as compared to DC from non-tumour tissue (Bluth et al. Citation2009). Several researchers have tried to modulate the DC in the tumour tissue towards the anti-tumour phenotype by using vaccines to recruit and activate DC (Lapteva et al. Citation2009a, Citation2009b), or by engineering DC to secrete antibodies for DC-based immunotherapy (Liu et al. Citation2009, Boczkowski et al. Citation2009). Targeting DC with immunomodulatory agents could be a promising route to turn on the anti-tumour immune response. Cancer antigen-specific CTL are the major effectors against cancer cells, but the immune suppressive networks limit their function. Also selection of antigenic variants that escape destruction may occur. It has been shown that sustained delivery of interleukin 12 (IL-12) and granulocyte macrophage colony-stimulating factor (GM-CSF) to tumours can break the paralysis and induce activation of CTL/memory cells within the tumour followed by CTL expansion, making this cell type an interesting target (Kilinc et al. Citation2006, Citation2009). In addition to CTL, regulatory T-cells (Tregs) seem important in preventing immune activation. There is increasing evidence that Tregs can migrate into tumours and suppress effective anti-tumour responses (Beyer and Schultze Citation2009). Modulating the activity of Tregs seems an interesting option to shift the balance in favour of immune activating signals.
Usually the targeting of adaptive immune cells involves activating these cells within the tumour microenvironment or at a remote location to elicit an immune response. But recent research indicates that even an activated adaptive immune system within the tumour microenvironment may contribute to tumour growth. In a study in a transgenic mouse model of inflammation-associated spontaneous epithelial carcinogenesis, without mature T- and B-lymphocytes, the neoplastic progression towards hyperplasia was limited. The authors suggested that in order to establish a chronic inflammatory state, which enables the promotion of spontaneous carcinogenesis, B-cells must be present (de Visser et al. Citation2005). This implicates that B-cells are required for tumour development in this model. One of the ways in which B-cells may contribute is their synthesis of immunoglobulins. Recently it has been shown that the locally produced antibodies can activate Fc gamma receptors on resident and recruited myeloid cells. The stromal production of auto-antibodies and activation of receptors can regulate recruitment, composition, and biological function of leukocytes in neoplastic tissue (Andreu et al. Citation2010). These studies also point to B-cells as possible targets for activity modulation.
At present, literature on the involvement of eosinophils and basophils is scarce and contradictory. For example, some have reported that eosinophils exert anti-tumoral activity (Caruso et al. Citation2002, Simson et al. Citation2007), while others also found protumoral activity (Wong et al. Citation1999).
Adipocytes
A cell type of which the involvement in cancer has not been studied extensively is the adipocyte (Iyengar et al. Citation2003). This cell type is present in most tissues and especially abundant in the breast, bone marrow and liver (Kim et al. Citation1987, Tillmann et al. Citation1999, Tokuda et al. Citation2003, Celis et al. Citation2005). A characteristic that is not commonly known for adipocytes is that they are able to secrete a variety of factors such as IL-6 and type VI collagen (Path et al. Citation2001, Iyengar et al. Citation2003). Iyengar has demonstrated that adipocyte conditioned medium (medium used to grow adipocytes in for a short period of time) can stimulate endothelial cells in vitro to form tube like structures, whereas fibroblast conditioned medium was far less effective in stimulating endothelial tube formation (Iyengar et al. Citation2003). This suggests that adipocytes are able to induce angiogenesis. Similar results were found for adipocytes co-cultured with human prostate cancer cells (Tokuda et al. Citation2003). Iyengar also performed an in vivo experiment in which they subcutaneously inoculated human breast cancer cells with and without adipocytes in nude mice. Amazingly, the tumours with adipocytes progressed rapidly to palpable tumours, whereas the tumours without adipocytes were barely able to grow (Iyengar et al. Citation2003), suggesting that adipocytes are also important for tumour progression and survival. In another study it was also shown that collagen VI expression of the adipocyte is related to these effects (Iyengar et al. Citation2005). Others have found matrix metalloproteinase 11 (MMP-11) expressed by adipocytes to be related to initial tumour cell survival (Andarawewa et al. Citation2005).
Endothelial cells and pericytes
The role of endothelial cells in tumour survival needs little explanation and has been evaluated by many authors with respect to neovascularisation (Pluda and Parkinson Citation1996, Tang and Conti Citation2004, Furuya et al. Citation2005, Ganss Citation2006, Balint et al. Citation2008, Furuya and Yonemitsu Citation2008, Legg et al. Citation2008). Indirectly, endothelial cells help the tumour cells survive by providing the vessel structure that delivers nutrients and oxygen, similarly as in processes where endothelial cells develop vessels for benign wound healing. Whether endothelial cells themselves are able to induce or influence tumour growth is difficult to determine. It has been determined that tumour angiogenic endothelium of various tumours expresses different receptors on their membrane surface compared to quiescent endothelium. Examples of such receptors are nucleolin, migration-stimulating factor (MSF), αvβ3 integrin, endosialin, Wilms' tumour suppressor and AKT-1 [Akalu et al. Citation2005, Landen et al. Citation2008, Danhier et al. Citation2009, Fogal et al. Citation2009, Hu et al. Citation2009, Sihto et al. Citation2009, Mitchell et al. Citation2010), which makes them promising targets for tumour-targeted delivery. Two studies have recently established that precancerous stem cells and breast tumour cells themselves are able to differentiate into the endothelial cell linage (Shen et al. Citation2008, Bussolati et al. Citation2009). It still needs to be investigated whether this phenomenon is also present in other tumour types, as this would implicate that the endothelial cell surface receptor profile would be tumour-specific rather than tissue specific or even tumour type-specific. If true, endothelial cell targeting would become a less attractive tumour supporting cell type for tumour drug targeting, not only because of differences in membrane receptors, but also because of the inherent genetic instability of the tumour cell-derived endothelial-like cells. The role of bone marrow-derived endothelial precursor cells (EPCs) in the development of tumour angiogenic neovasculature has been carefully reviewed by Le Bourhis et al. (Citation2010), but is expected to be of limited use as a target for drug delivery as EPCs only contribute 0.5–3% of the total neo-endothelial cells. Also, loss of proliferating endothelial cells by targeted drug delivery does not necessarily mean that the pericytes close to these endothelial cells and the basement membrane surrounding these cells are destructed, resulting in preservation of the conditions for rapid endothelial vessel re-growth [Baluk et al. Citation2005). Although pericytes have been found to be important in limiting metastasis (Xian et al. Citation2006), other studies have found a beneficial reduction in tumour growth, angiogenesis and metastasis when tumour cells, endothelial cells and pericytes were targeted simultaneously (Lang et al. Citation2008, Lu et al. Citation2010), showing that targeting more cell types simultaneously has good anti-tumour potential.
Fibroblasts
Another cell type abundantly present in tumour tissue is fibroblasts. These cells can be derived from bone-marrow or from epithelia or endothelial cells in the tumour tissue that turn into mesenchymal cells. Fibroblasts present in the tumour environment of various tumour types, such as prostate and colon cancer, have been found to actively support tumour growth (Silzle et al. Citation2004, Kaminski et al. Citation2006, Schmid et al. Citation2009, Xing et al. Citation2010). At the same time, fibroblasts are quite heterogeneous as different tumour-types have different gene expression and marker profiles. Fibroblasts can stimulate tumour growth via a variety of routes such as CXC motif ligand 12 (CXCL12)/stromal cell-derived factor-1 (SDF-1) mediated recruitment of bone marrow-derived endothelial precursor cells and stimulation of metastasis via chemokine ligand-5 (CCL5) (Ostman and Augsten Citation2009). Interfering with fibroblast's platelet-derived growth factor (PDGF)-, transforming growth factor (TGF)-beta- or hedgehog-signalling, could make them another promising target for specific targeting with liposomes (Ostman and Augsten Citation2009).
Liposomes targeting non-tumour cells in cancer
Passive targeting
Passive targeting of drug delivery systems, as illustrated in the first section for Doxil®, improves tumour accumulation and drastically reduces volume of distribution of doxorubicin (Gabizon et al. Citation2003). These passively targeted long-circulating systems are usually eventually taken up by tumour-associated macrophages, as demonstrated in . When these liposomes encapsulate clodronate, these phagocytic cells in the tumour tissue can be depleted. Zeisberger et al. showed that in murine models liposomal clodronate could almost completely inhibit tumour growth. Tumour inhibition was accompanied by a decreased blood vessel density in the tumour tissue, indicating that tumour-associated macrophages play an important role in tumour angiogenesis (Zeisberger et al. Citation2006). Many studies replicated these findings in a variety of tumour models with liposomal clodronate (Gazzaniga et al. Citation2007, Kimura et al. Citation2007, Banciu et al. Citation2008, Miselis et al. Citation2008, Meng et al. Citation2010). Also, when the liposomes were not designed to deplete the macrophages (as with clodronate) but to modulate macrophage activation status (with prednisolone), a marked reduction in pro-angiogenic/pro-inflammatory factors was observed. As a result, liposomal prednisolone appeared a very potent inhibitor of tumour growth by interfering with the pro-inflammatory signals from the macrophages, and shifting the balance towards an anti-inflammatory/anti-angiogenic state (Banciu et al. Citation2006).
Figure 2. (A) Illustration of tumour tissue with different kinds of stromal cells. In the middle a blood vessel is drawn to show that tumours tissue also contains blood vessels, which are more permeable than normal blood vessels. (B) Passive targeting: Liposomes passively targeted generally accumulate in tumour tissue because of the enhanced permeability and retention effect and are taken up by cells with phagocytic properties (macrophages). (C) Active targeting: Targeting of liposomes to specific cell types by using a cancer-associated stromal cell specific targeting ligand can influence the distribution pattern of the liposomes inside the tumour tissue. The distribution of the liposomes depends on the specificity of the targeting ligand to the target, here the possible distribution is shown when more than one cell type is targeted simultaneously.
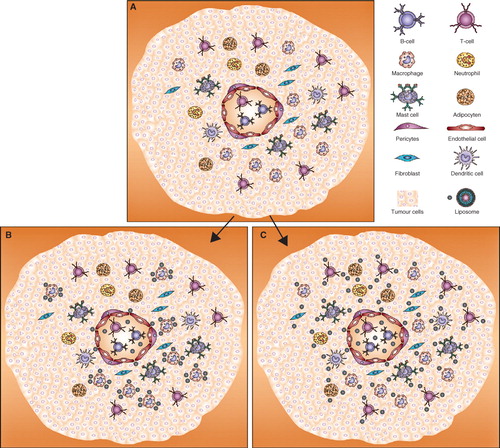
The passive targeting strategy offers little control in order to reach other cell types with which the drug delivery systems may interact. To improve this, targeting moieties can be coupled to the surface of particles to redirect them to the intended target cell type.
Targeting of stromal cells in general
A recent study by Prakash et al. (Citation2010) aimed at a broad targeting of the various tumour stromal cells that contribute to tumour growth. It was shown that many tumour stromal cell types abundantly express platelet-derived growth factor receptor-beta (PDGFR-beta) in human tumour biopsies. Also a number of cancer cells express this receptor. The carrier was composed of albumin decorated with a cyclic peptide (with the sequence CSRNLIDC) recognizing PDGFR-beta and specifically bound PDGFR-beta-expressing 3T3 fibroblasts, C26 and A2780 cancer cells in vitro. When, doxorubicin was conjugated to the carrier, the construct induced cell death in PDGFR positive cells in vitro, and in vivo, the conjugate rapidly accumulated into PDGFR-beta expressing cells in C26 tumours in mice and reduced tumour growth. With Prakash study it was shown that specific depletion of tumour-associated fibroblasts and pericytes is possible and this provides us with a tool to explore the potential roles of these cells in tumour progression (Park Citation2010).
Similarly, in a study on conditionally replicative oncolytic adenoviruses (CRAd), the inefficient infection of the tumour cells was addressed by additionally targeting tumour stromal cells (Lopez et al. Citation2009). It was hypothesized that, as the SPARC gene is expressed in both tumour and stromal cells, SPARC promoter-based CRAds could be more efficacious. Indeed, it appeared that tumour stromal cells can support viral replication and modulate the efficacy of a CRAd through the production of soluble factors.
These studies show beneficial effects of drug delivery systems with a limited target specificity where a broad variety of tumour stromal cells are the cell types that are aimed for (illustrated by ). Liposomes with such broad targeting capability have not been used, but could be of interest when loaded with cytostatics to deplete these cells. Modulating cell activity appears to be more challenging when targeting multiple cell types as each cell type requires its own signals to improve their anti-tumour action. The target cell type is therefore usually more defined as discussed below.
Targeting tumour vasculature
Hamzah et al. (Citation2009) developed liposomes engrafted with RGR, a vascular targeting peptide, selectively targeted to angiogenic endothelial cells, which were loaded with immunostimulatory oligodeoxynucleotides. The RGR-motif is thought to bind the PDGFR-beta receptor (Joyce et al. Citation2003) which was also the objective of Prakash et al. (Citation2010) with the modified albumin, as described above. The targeting specificity for the vasculature of the PDGFR-targeted liposomes should then come from the (much larger) liposome compared to the albumin, limiting extravasation and perfusion of the tumour interstitium, while promoting interaction with the endothelium. Apart from vascular accumulation, increased uptake by tumour resident macrophages was noted as well as increased tumour retention over time. The vascular targeting provoked an inflammatory response to the vessel wall with enhanced T-cell infiltration and activation of tumour specific cytotoxicity (Hamzah et al. Citation2009).
Another vascular targeting peptide known to preferentially bind to the integrin adhesion molecule αv is RGD. This simple motif provides strong targeting to activated endothelium and is one of the most popular targeting ligands (Temming et al. Citation2005). RGD-liposomes have been equipped with doxorubicin, the vascular disrupting agents ZD6126 and combretastatin A4, and paclitaxel, in most cases reporting enhanced effectiveness of the targeted therapy (Janssen et al. Citation2003, Schiffelers et al. Citation2003, Xiong et al. Citation2005, Fens et al. Citation2008, Zhao et al. Citation2009, Zhang et al. Citation2010). Somewhat surprisingly, for the endothelial cell-specific vascular disrupting agent ZD6126 there was no benefit of endothelial cell-specific delivery by RGD-liposomes compared to non-targeted liposomes. It would have been expected that a drug that is specifically active in angiogenic endothelium would profit more from endothelial delivery. Presumably, the non-targeted liposomes were able to deliver the drug in the vicinity of the endothelium, allowing ZD6126 to reach the target cell type by diffusion after liposomal degradation (Fens et al. Citation2008). Others have used immunoliposomes equipped with an antibody against vascular cell adhesion molecule 1 (VCAM-1), which is over-expressed on tumour blood vessels, and demonstrated by fluorescence microscopy that VCAM-1 targeted liposomes accumulated in tumour vessels while control liposomes accumulated in the tumour tissue by passive mechanisms (Gosk et al. Citation2008).
A single study explored the benefits of combined targeting of endothelial cells and pericytes. Tumour vasculature was targeted via aminopeptidase N (APN), whereas a peptide ligand to aminopeptidase A was used to target the pericytes. Injection of a combination of both types of liposomes encapsulating doxorubicin in murine models of neuroblastoma increased the level of apoptosis and led to a pronounced destruction of tumour vasculature with almost complete depletion of both endothelial cells and pericytes (Loi et al. Citation2010). This suggests that active targeting to more than one target cell types (i.e., pericytes and tumour-endothelial cells) is more beneficial than targeting solely one target cell type. Whether this suggestion holds true remains to be established in the future.
Targeting immune cells
Macrophages
Due to the fact that macrophages are professional phagocytes and readily take up liposomes that arrive in the tumour, active targeting has not been vigorously pursued for macrophages. Usually peripheral macrophages have been targeted with mannose and muramyltripeptide (Gano and Kleinerman Citation1995, Nardin et al. Citation2006, Lu et al. Citation2007, Ikehara and Kojima Citation2007, Kuramoto et al. Citation2008). One study used folate to target macrophages within the tumour (Turk et al. Citation2004). As the folate receptor is also overexpressed on many epithelial cancers this dual targeting approach may attack both cell types. It was shown that folate-conjugation of liposomes significantly enhanced their uptake into folate-receptor positive ovarian cancer cells and tumour-associated macrophages within ascites fluid. However, compared to ovarian cancer cells, macrophages accumulated tenfold more liposomes, half of which was receptor mediated and the other half presumably by non-specific phagocytosis (Turk et al. Citation2004). Although this study clearly demonstrated that folate-conjugated liposomes show increased accumulation in macrophages and tumour cells, the true value of this specific targeting still remains to be established; thus far folate-conjugated liposomes were not tested with a proper drug encapsulated within the liposome to show the full potential of this tumour-associated macrophage specific targeting.
To reach B-cells, Loomis et al. used the the CD22 antigen (Loomis et al. Citation2010). Although they employed this target for therapeutic intervention for B-cell lymphomas, CD22 could also be used to reach B-cells in general. When an anti-CD22 single chain Fv region was coupled to the surface of liposomes binding to B-cells was improved and liposomes were taken up. Loaded with doxorubicin, these liposomes enhanced accumulation of doxorubicin inside cells leading to enhanced cytotoxicity. This study was only performed in vitro, so also for this study more (in vivo) experiments are needed to demonstrate the full potential of B-cell specific targeting with anti-CD22.
Targeting fibroblasts
To target fibroblasts, Baum et al. (Citation2007) used fibroblast activation protein (FAP) as this surface antigen is selectively expressed by reactive cancer-associated fibroblasts (CAF) of various cancers. Anti-FAP single chain Fv region antibody fragments were coupled to liposomes and incubated with FAP-expressing cells. The coupling of the antibody fragment led to internalization of intact liposomes into the endosomal compartment indicating that they could be employed for fibroblast targeting. Although this seems very promising for a new CAF-specific delivery system, the true value of this ligand still needs to be evaluated in vivo.
The search for good CAF targeting ligands continues. Sadlonova et al. (Citation2009) have tried to identify molecules that are differentially expressed by breast carcinoma CAF (in comparison to normal breast fibroblasts) by generating gene expression profiles of both types of fibroblasts. They found that 420 genes were differentially expressed of which 240 genes were overexpressed by CAF. Although their focus was on finding genes that influence the behaviour of the breast epithelial cells, their data could potentially be of importance in finding CAF-specific surface markers that can be used to find CAF-specific targeting ligands.
Targeting adipocytes
Gouranton et al. (Citation2008) investigated the delivery of lycopene with different vehicles (liposomes, beadlets, BSA and LDL) to 3T3-L1 adipocytes in vitro. They demonstrated that passively targeted liposomes are internalized by adipocytes, but to a smaller extent than LDL, BSA or beadlets. They also were able to quantify the concentrations of lycopene in the different cell compartments and they showed that lycopene was delivered with a localization pattern similar with all vehicles; the largest quantity ending up in the lipid droplets, then the plasma membrane, the nuclear membrane and the smallest quantity in the cytoplasm (Gouranton et al. Citation2008). Translating these results to an in vivo situation is difficult, as the phagocytic cells in the tumour environment are probably more prone to internalize the liposomes than adipocytes. The results do show however that adipocytes are able to take up drug delivery vehicles and should not be ruled out for possible specific stromal-cell targeted drug therapy development. A similar strategy performed by Sadlonova et al, i.e., generating gene expression profiles or protein expression profiles of normal adipocytes and cancer-associated adipocytes, could be a possible route to find new tumour-adipocyte specific targeting receptors.
Discussion
The involvement of stromal cells in cancer has introduced the possibility of actively targeting this tumour-associated cell ‘ecosystem’ to improve anti-tumour drug delivery. The advantage of a strategy that targets non-tumour cells is that the treatment likely can be used for different types of cancer; the tumour cell type may differ, but the associated cells are more likely to be similar and unlike the tumour cells they are genetically stable, reducing the chance of occurrence of resistance (Schiffelers and Storm Citation2008).
The two cell types that have been targeted most extensively with liposomes are the tumour-associated macrophages and endothelial cells. The macrophages, because they are professional phagocytes, have a natural tendency to internalize particulate systems and can thus be targeted without employing specific targeting ligands. The endothelial cell is attractive because it is the first cell type liposomal systems encounter after intravenous administration and thus targeting these cells avoids passing the endothelial cell lining as an additional barrier. Most studies that address these two cell types use drug molecules that are aiming for depletion of these cells. Especially with macrophages depletion has been shown to result in strong tumour growth inhibition. Occasional reports have targeted other cell types, but it is clear that an important challenge lies in the identification of targeting ligands that allow us to efficiently reach these cells, that are at the same time able to efficiently pass the endothelial lining and are not recognized by macrophages in the tumour or in the mononuclear phagocyte system. Most of these reports either demonstrated the potential of their targeting ligand in vitro only or they did show the potential in vivo, but did not encapsulate/attach a drug to the liposome/targeting ligand. Hence, the full potential of these ‘new’ tumour-stromal cell targeting-ligands still remains to be established, hopefully showing promising results in the future. Another important issue that cannot be overlooked is that targeting ligands tested in mice or rat may not have similar effects in humans. By using targeting ligands of which the target receptors (or other proteins) are preserved across species, such discrepancies hopefully can be avoided.
Another challenge lies in the more subtle interference with tumour stromal cell signalling. At present the majority of studies use cytotoxic agents that kill the stromal cells, whereas these cells could also contribute to anti-tumour activity. For example, diverting M2 macrophages to an anti-tumour M1 phenotype may be more beneficial than depleting M2 macrophages altogether. Finding drug molecules that allow tumour stromal cells to break free from the pro-tumour signalling in the microenvironment is an important next step.
Acknowledgements
We would like to thank our illustrator F.G. Kuijer of the Communication and Design department of the faculty of Science (Utrecht University) for the illustration she created.
Declaration of interest: This work was financially supported by a VIDI-Innovational Research Incentives scheme grant from the Technology Foundation STW of the Netherlands Organization for Scientific Research (UFA 7947).
References
- Akalu A, Cretu A, Brooks PC. 2005. Targeting integrins for the control of tumour angiogenesis. Expert Opin Investig Drugs 14:1475–1486.
- Ali G, Boldrini L, Lucchi M, Mussi A, Corsi V, Fontanini G. 2009. Tryptase mast cells in malignant pleural mesothelioma as an independent favorable prognostic factor. J Thorac Oncol 4:348–354.
- Andarawewa KL, Motrescu ER, Chenard MP, Gansmuller A, Stoll I, Tomasetto C, Rio MC. 2005. Stromelysin-3 is a potent negative regulator of adipogenesis participating to cancer cell-adipocyte interaction/crosstalk at the tumor invasive front. Cancer Research 65:10862–10871.
- Andreu P, Johansson M, Affara NI, Pucci F, Tan T, Junankar S, Korets L, Lam J, Tawfik D, Denardo DG, Naldini L, De Visser KE, De Palma M, Coussens LM. 2010. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell 17:121–134.
- Balint K, Conejo-Garcia JR, Buckanovich R, Coukos G 2008. Role of vascular leukocytes in ovarian cancer neovascularization. Eicosanoids and Other Bioactive Lipids in Cancer, Inflammation and Radiation Injury 4:622, 273–280.
- Balkwill F, Charles KA, Mantovani A. 2005. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 7:211–217.
- Balkwill F, Mantovani A. 2010. Cancer and inflammation: Implications for pharmacology and therapeutics. Clin Pharmacol Ther 87:401–406.
- Baluk P, Hashizume H, McDonald DM. 2005. Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev 15:102–111.
- Baluk P, Morikawa S, Haskell A, Mancuso M, McDonald DM. 2003. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am J Pathol 163:1801–1815.
- Banciu M, Fens MH, Storm G, Schiffelers RM. 2008. Antitumor activity and tumor localization of liposomal glucocorticoids in B16 melanoma-bearing mice. J Control Release 127:131–136.
- Banciu M, Schiffelers RM, Fens MH, Metselaar JM, Storm G. 2006. Anti-angiogenic effects of liposomal prednisolone phosphate on B16 melanoma in mice. J Control Release 113:1–8.
- Baum P, Muller D, Ruger R, Kontermann RE. 2007. Single-chain Fv immunoliposomes for the targeting of fibroblast activation protein-expressing tumor stromal cells. J Drug Target 15:399–406.
- Beyer M, Schultze JL. 2009. Regulatory T cells: Major players in the tumor microenvironment. Curr Pharm Des 15:1879–1892.
- Bluth MJ, Zaba LC, Moussai D, Suarez-Farinas M, Kaporis H, Fan L, Pierson KC, White TR, Pitts-Kiefer A, Fuentes-Duculan J, Guttman-Yassky E, Krueger JG, Lowes MA, Carucci JA. 2009. Myeloid dendritic cells from human cutaneous squamous cell carcinoma are poor stimulators of T-cell proliferation. J Investigat Dermatol 129:2451–2462.
- Boczkowski D, Lee J, Pruitt S, Nair S. 2009. Dendritic cells engineered to secrete anti-GITR antibodies are effective adjuvants to dendritic cell-based immunotherapy. Cancer Gene Ther 16:900–911.
- Brú A, Souto JC, Alcolea S, Antón R, Remacha A, Camacho M, Soler M, Brú I, Porres A, Vila L. 2010. Tumour cell lines HT-29 and FaDu produce proinflammatory cytokines and activate neutrophils in vitro: possible applications for neutrophil-based antitumour treatment. Mediators Inflamm. 817498.
- Bussolati B, Grange C, Sapino A, Camussi G. 2009. Endothelial cell differentiation of human breast tumour stem/progenitor cells. J Cellular Molec Med 13:309–319.
- Caruso RA, Bersiga A, Rigoli L, Inferrera C. 2002. Eosinophil-tumor cell interaction in advanced gastric carcinoma: An electron microscopic approach. Anticancer Res 22:3833–3836.
- Celis JE, Moreira JM, Cabezon T, Gromov P, Friis E, Rank F, Gromova I. 2005. Identification of extracellular and intracellular signaling components of the mammary adipose tissue and its interstitial fluid in high risk breast cancer patients: Toward dissecting the molecular circuitry of epithelial-adipocyte stromal cell interactions. Molec Cellular Proteomics 4:492–522.
- Chaput N, Conforti R, Viaud S, Spatz A, Zitvogel L. 2008. The Janus face of dendritic cells in cancer. Oncogene 27:5920–5931.
- Cinel L, Aban M, Basturk M, Ertunc D, Arpaci R, Dilek S, Camdeviren H. 2009. The association of mast cell density with myometrial invasion in endometrial carcinoma: A preliminary report. Pathol Res Pract 205:255–258.
- Crivellato E, Nico B, Ribatti D. 2008. Mast cells and tumour angiogenesis: New insight from experimental carcinogenesis. Cancer Lett 269:1–6.
- Danhier F, Vroman B, Lecouturier N, Crokart N, Pourcelle V, Freichels H, Jerome C, Marchand-Brynaert J, Feron O, Preat V. 2009. Targeting of tumor endothelium by RGD-grafted PLGA-nanoparticles loaded with Paclitaxel. J Control Release 140:166–173.
- de Visser KE, Korets LV, Coussens LM. 2005. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell 7:411–423.
- Deininger MW, Druker BJ. 2003. Specific targeted therapy of chronic myelogenous leukemia with imatinib. Pharmacol Rev 55:401–423.
- Della Rovere F, Granata A, Familiari D, D'arrigo G, Mondello B, Basile G. 2007. Mast cells in invasive ductal breast cancer: Different behavior in high and minimum hormone-receptive cancers. Anticancer Res 27:2465–2471.
- Detoraki A, Staiano RI, Granata F, Giannattasio G, Prevete N, De Paulis A, Ribatti D, Genovese A, Triggiani M, Marone G. 2009. Vascular endothelial growth factors synthesized by human lung mast cells exert angiogenic effects. J Allergy Clin Immunol 123:1142–1149, 1149 e1–5.
- Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. 2002. Cancer immunoediting: From immunosurveillance to tumor escape. Nat Immunol 3:991–998.
- Dunn GP, Old LJ, Schreiber RD. 2004. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 21:137–148.
- Fens MH, Hill KJ, Issa J, Ashton SE, Westwood FR, Blakey DC, Storm G, Ryan AJ, Schiffelers RM. 2008. Liposomal encapsulation enhances the antitumour efficacy of the vascular disrupting agent ZD6126 in murine B16.F10 melanoma. Br J Cancer 99:1256–1264.
- Fogal V, Sugahara KN, Ruoslahti E, Christian S. 2009. Cell surface nucleolin antagonist causes endothelial cell apoptosis and normalization of tumor vasculature. Angiogenesis 12:91–100.
- Freitas I, Baronzio GF. 1994. Neglected factors in cancer treatment: Cellular interactions and dynamic microenvironment in solid tumors. Anticancer Res 14:1097–1101.
- Furuya M, Nishiyama M, Kasuya Y, Kimura S, Ishikura H. 2005. Pathophysiology of tumor neovascularization. Vasc Health Risk Manag 1:277–290.
- Furuya M, Yonemitsu Y. 2008. Cancer neovascularization and proinflammatory microenvironments. Curr Cancer Drug Targets 8:253–265.
- Gabizon A, Shmeeda H, Barenholz Y. 2003. Pharmacokinetics of pegylated liposomal Doxorubicin: Review of animal and human studies. Clin Pharmacokinet 42:419–436.
- Gabizon AA. 2001. Pegylated liposomal doxorubicin: Metamorphosis of an old drug into a new form of chemotherapy. Cancer Invest 19:424–436.
- Gabizon AA, Shmeeda H, Zalipsky S. 2006. Pros and cons of the liposome platform in cancer drug targeting. J Liposome Res 16:175–183.
- Gano JB, Kleinerman ES. 1995. Liposomal MTP-PE: A promising new biologic response modifier. Oncol Nurs Forum 22:809–816.
- Ganss R. 2006. Tumor stroma fosters neovascularization by recruitment of progenitor cells into the tumor bed. J Cellular Molec Med 10:857–865.
- Gazzaniga S, Bravo AI, Guglielmotti A, Van Rooijen N, Maschi F, Vecchi A, Mantovani A, Mordoh J, Wainstok R. 2007. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J Invest Dermatol 127:2031–2041.
- Gosk S, Moos T, Gottstein C, Bendas G. 2008. VCAM-1 directed immunoliposomes selectively target tumor vasculature in vivo. Biochim Biophys Acta 1778:854–863.
- Gouranton E, El Yazidi C, Cardinault N, Amiot MJ, Borel P, Landrier JF. 2008. Purified low-density lipoprotein and bovine serum albumin efficiency to internalise lycopene into adipocytes. Food and Chemical Toxicology 46:3832–3836.
- Grivennikov SI, Greten FR, Karin M. 2010. Immunity, inflammation, and cancer. Cell 140:883–899.
- Groot Kormelink T, Abudukelimu A, Redegeld FA. 2009. Mast cells as target in cancer therapy. Curr Pharm Des 15:1868–1878.
- Hamzah J, Altin JG, Herringson T, Parish CR, Hammerling GJ, O'Donoghue H, Ganss R. 2009. Targeted liposomal delivery of TLR9 ligands activates spontaneous antitumor immunity in an autochthonous cancer model. J Immunol 183:1091–1098.
- Hedstrom G, Berglund M, Molin D, Fischer M, Nilsson G, Thunberg U, Book M, Sundstrom C, Rosenquist R, Roos G, Erlanson M, Amini RM, Enblad G. 2007. Mast cell infiltration is a favourable prognostic factor in diffuse large B-cell lymphoma. Br J Haematol 138:68–71.
- Hellberg C, Ostman A, Heldin CH. 2010. PDGF and vessel maturation. Recent Results Cancer Res 180:103–114.
- Hilgenbrink AR, Low PS. 2005. Folate receptor-mediated drug targeting: From therapeutics to diagnostics. J Pharm Sci 94:2135–2146.
- Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, Jain RK. 1998. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proc Natl Acad Sci USA 95:4607–4612.
- Hu H, Ran YL, Zhang YS, Zhou Z, Harris SJ, Yu L, Sun LX, Pan J, Liu J, Lou JN, Yang ZH. 2009. Antibody library-based tumor endothelial cells surface proteomic functional screen reveals migration-stimulating factor as an anti-angiogenic target. Molec Cellular Proteomics 8:816–826.
- Ikehara Y, Kojima N. 2007. Development of a novel oligomannose-coated liposome-based anticancer drug-delivery system for intraperitoneal cancer. Curr Opin Mol Ther 9:53–61.
- Iyengar P, Combs TP, Shah SJ, Gouon-Evans V, Pollard JW, Albanese C, Flanagan L, Tenniswood MP, Guha C, Lisanti MP, Pestell RG, Scherer PE. 2003. Adipocyte-secreted factors synergistically promote mammary tumorigenesis through induction of anti-apoptotic transcriptional programs and proto-oncogene stabilization. Oncogene 22:6408–6423.
- Iyengar P, Espina V, Williams TW, Lin Y, Berry D, Jelicks LA, Lee H, Temple K, Graves R, Pollard J, Chopra N, Russell RG, Sasisekharan R, Trock BJ, Lippman M, Calvert VS, Petricoin EF 3rd, Liotta L, Dadachova E, Pestell RG, Lisanti MP, Bonaldo P, Scherer PE. 2005. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J Clin Investigat 115:1163–1176.
- Jabbour E, Cortes J, O'Brien S, Giles F, Kantarjian H. 2007. New targeted therapies for chronic myelogenous leukemia: Opportunities to overcome imatinib resistance. Semin Hematol 44:S25–31.
- Janssen AP, Schiffelers RM, Ten Hagen TL, Koning GA, Schraa AJ, Kok RJ, Storm G, Molema G. 2003. Peptide-targeted PEG-liposomes in anti-angiogenic therapy. Int J Pharm 254:55–58.
- Joyce JA, Laakkonen P, Bernasconi M, Bergers G, Ruoslahti E, Hanahan D. 2003. Stage-specific vascular markers revealed by phage display in a mouse model of pancreatic islet tumorigenesis. Cancer Cell 4:393–403.
- Ju MJ, Qiu SJ, Gao Q, Fan J, Cai MY, Li YW, Tang ZY. 2009. Combination of peritumoral mast cells and T-regulatory cells predicts prognosis of hepatocellular carcinoma. Cancer Sci 100:1267–1274.
- Kaminski A, Hahne JC, Haddouti EM, Florin A, Wellmann A, Wernert N. 2006. Tumour-stroma interactions between metastatic prostate cancer cells and fibroblasts. Int J Molec Med 18:941–950.
- Keibel A, Singh V, Sharma MC. 2009. Inflammation, microenvironment, and the immune system in cancer progression. Curr Pharm Des 15:1949–1955.
- Kilinc MO, Aulakh KS, Nair RE, Jones SA, Alard P, Kosiewicz MM, Egilmez NK. 2006. Reversing tumor immune suppression with intratumoral IL-12: Activation of tumor-associated T effector/memory cells, induction of T suppressor apoptosis, and infiltration of CD8(+) T effectors. J Immunol 177:6962–6973.
- Kilinc MO, Gu T, Harden JL, Virtuoso LP, Egilmez NK. 2009. Central role of tumor-associated CD8+ T effector/memory cells in restoring systemic antitumor immunity. J Immunol 182:4217–4225.
- Kim YI, Yu ES, Lee KW, Park EU, Song HG. 1987. Dedifferentiated liposarcoma of the liver. Cancer 60:2785–2790.
- Kimura YN, Watari K, Fotovati A, Hosoi F, Yasumoto K, Izumi H, Kohno K, Umezawa K, Iguchi H, Shirouzu K, Takamori S, Kuwano M, Ono M. 2007. Inflammatory stimuli from macrophages and cancer cells synergistically promote tumor growth and angiogenesis. Cancer Sci 98:2009–2018.
- Kinlen LJ. 1992. Malignancy in autoimmune diseases. J Autoimmun 5(Suppl. A):363–371.
- Knezevic V, Leethanakul C, Bichsel VE, Worth JM, Prabhu VV, Gutkind JS, Liotta LA, Munson PJ, Petricoin EF 3rd, Krizman DB. 2001. Proteomic profiling of the cancer microenvironment by antibody arrays. Proteomics 1:1271–1278.
- Kuramoto Y, Kawakami S, Zhou S, Fukuda K, Yamashita F, Hashida M. 2008. Use of mannosylated cationic liposomes/immunostimulatory CpG DNA complex for effective inhibition of peritoneal dissemination in mice. J Gene Med 10:392–399.
- Landen CN, Kim TJ, Lin YG, Merritt WM, Kamat AA, Han LY, Spannuth WA, Nick AM, Jennnings NB, Kinch MS, Tice D, Sood AK. 2008. Tumor-selective response to antibody-mediated targeting of alphavbeta3 integrin in ovarian cancer. Neoplasia 10:1259–1267.
- Lang SA, Schachtschneider P, Moser C, Mori A, Hackl C, Gaumann A, Batt D, Schlitt HJ, Geissler EK, Stoeltzing O. 2008. Dual targeting of Raf and VEGF receptor 2 reduces growth and metastasis of pancreatic cancer through direct effects on tumor cells, endothelial cells, and pericytes. Molec Cancer Therapeut 7:3509–3518.
- Lapteva N, Aldrich M, Rollins L, Ren W, Goltsova T, Chen SY, Huang XF. 2009a. Attraction and activation of dendritic cells at the site of tumor elicits potent antitumor immunity. Molec Ther 17:1626–1636.
- Lapteva N, Aldrich M, Weksberg D, Rollins L, Goltsova T, Chen SY, Huang XF. 2009b. Targeting the intratumoral dendritic cells by the oncolytic adenoviral vaccine expressing RANTES elicits potent antitumor immunity. J Immunother 32:145–156.
- Le Bourhis X, Romon R, Hondermarck H. 2010. Role of endothelial progenitor cells in breast cancer angiogenesis: From fundamental research to clinical ramifications. Breast Cancer Res Treat 120:17–24.
- Legg JA, Herbert JMJ, Clissold P, Bicknell R. 2008. Slits and roundabouts in cancer, tumour angiogenesis and endothelial cell migration. Angiogenesis 11:13–21.
- Li J, Sausville EA, Klein PJ, Morgenstern D, Leamon CP, Messmann RA, Lorusso P. 2009. Clinical pharmacokinetics and exposure-toxicity relationship of a folate-Vinca alkaloid conjugate EC145 in cancer patients. J Clin Pharmacol 49:1467–1476.
- Liu Q, Zhai B, Yang W, Yu LX, Dong W, He YQ, Chen L, Tang L, Lin Y, Huang DD, Wu HP, Wu MC, Yan HX, Wang HY. 2009. Abrogation of local cancer recurrence after radiofrequency ablation by dendritic cell-based hyperthermic tumor vaccine. Molec Ther 17:2049–2057.
- Loi M, Marchio S, Becherini P, Di Paolo D, Soster M, Curnis F, Brignole C, Pagnan G, Perri P, Caffa I, Longhi R, Nico B, Bussolino F, Gambini C, Ribatti D, Cilli M, Arap W, Pasqualini R, Allen TM, Corti A, Ponzoni M, Pastorino F. 2010. Combined targeting of perivascular and endothelial tumor cells enhances anti-tumor efficacy of liposomal chemotherapy in neuroblastoma. J Control Release 145:66–73.
- Loomis K, Smith B, Feng Y, Garg H, Yavlovich A, Campbell-Massa R, Dimitrov DS, Blumenthal R, Xiao X, Puri A. 2010. Specific targeting to B cells by lipid-based nanoparticles conjugated with a novel CD22-ScFv. Exp Mol Pathol 88:238–249.
- Lopez MV, Viale DL, Cafferata EG, Bravo AI, Carbone C, Gould D, Chernajovsky Y, Podhajcer OL. 2009. Tumor associated stromal cells play a critical role on the outcome of the oncolytic efficacy of conditionally replicative adenoviruses. PLoS One 4:e5119.
- Low PS, Kularatne SA. 2009. Folate-targeted therapeutic and imaging agents for cancer. Curr Opin Chem Biol 13:256–262.
- Lu C, Shahzad MM, Moreno-Smith M, Lin YG, Jennings NB, Allen JK, Landen CN, Mangala LS, Armaiz-Pena GN, Schmandt R, Nick AM, Stone RL, Jaffe RB, Coleman RL, Sood AK. 2010. Targeting pericytes with a PDGF-B aptamer in human ovarian carcinoma models. Cancer Biol Ther 9:176–182.
- Lu Y, Kawakami S, Yamashita F, Hashida M. 2007. Development of an antigen-presenting cell-targeted DNA vaccine against melanoma by mannosylated liposomes. Biomaterials 28:3255–3262.
- Maeda H, Bharate GY, Daruwalla J. 2009. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharmaceut Biopharmaceut 71:409–419.
- Meng Y, Beckett MA, Liang H, Mauceri HJ, Van Rooijen N, Cohen KS, Weichselbaum RR. 2010. Blockade of tumor necrosis factor alpha signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res 70:1534–1543.
- Miselis NR, Wu ZJ, Van Rooijen N, Kane AB. 2008. Targeting tumor-associated macrophages in an orthotopic murine model of diffuse malignant mesothelioma. Mol Cancer Ther 7:788–799.
- Mitchell D, Pobre EG, Mulivor AW, Grinberg AV, Castonguay R, Monnell TE, Solban N, Ucran JA, Pearsall RS, Underwood KW, Seehra J, Kumar R. 2010. ALK1-Fc inhibits multiple mediators of angiogenesis and suppresses tumor growth. Mol Cancer Ther 9:379–388.
- Moore MM, Chua W, Charles KA, Clarke SJ. 2010. Inflammation and cancer: Causes and consequences. Clin Pharmacol Ther 87:504–508.
- Mor G, Yue W, Santen RJ, Gutierrez L, Eliza M, Berstein LM, Harada N, Wang J, Lysiak J, Diano S, Naftolin F. 1998. Macrophages, estrogen and the microenvironment of breast cancer. J Steroid Biochem Mol Biol 67:403–411.
- Nardin A, Lefebvre ML, Labroquere K, Faure O, Abastado JP. 2006. Liposomal muramyl tripeptide phosphatidylethanolamine: Targeting and activating macrophages for adjuvant treatment of osteosarcoma. Curr Cancer Drug Targets 6:123–133.
- Ostman A, Augsten M. 2009. Cancer-associated fibroblasts and tumor growth – bystanders turning into key players. Curr Opin Genet Dev 19:67–73.
- Park K. 2010. A new ligand for targeted drug delivery to tumor stromal cells. J Control Release 145:75.
- Path G, Bornstein SR, Gurniak M, Chrousos GP, Scherbaum WA, Hauner H. 2001. Human breast adipocytes express interleukin-6 (IL-6) and its receptor system: Increased IL-6 production by beta-adrenergic activation and effects of IL-6 on adipocyte function. J Clin Endocrinol Metab 86:2281–2288.
- Pluda JM, Parkinson DR. 1996. Clinical implications of tumor-associated neovascularization and current antiangiogenic strategies for the treatment of malignancies of pancreas. Cancer 78:680–687.
- Prakash J, De Jong E, Post E, Gouw AS, Beljaars L, Poelstra K. 2010. A novel approach to deliver anticancer drugs to key cell types in tumors using a PDGF receptor-binding cyclic peptide containing carrier. J Control Release 145:91–101.
- Roskelley CD, Bissell MJ. 2002. The dominance of the microenvironment in breast and ovarian cancer. Seminars Cancer Biol 12:97–104.
- Sadlonova A, Bowe DB, Novak Z, Mukherjee S, Duncan VE, Page GP, Frost AR. 2009. Identification of molecular distinctions between normal breast-associated fibroblasts and breast cancer-associated fibroblasts. Cancer Microenviron 2:9–21.
- Schiffelers RM, Koning GA, Ten Hagen TL, Fens MH, Schraa AJ, Janssen AP, Kok RJ, Molema G, Storm G. 2003. Anti-tumor efficacy of tumor vasculature-targeted liposomal doxorubicin. J Control Release 91:115–122.
- Schiffelers RM, Storm G. 2008. Liposomal nanomedicines as anticancer therapeutics: Beyond targeting tumor cells. Int J Pharmaceut 364:258–264.
- Schmid SA, Dietrich A, Schulte S, Gaumann A, Kunz-Schughart LA. 2009. Fibroblastic reaction and vascular maturation in human colon cancers. Int J Radiation Biol 85:1013–1025.
- Surveillance Epidemiology and End Results (SEER). 2006. SEER Cancer Statistics Review 1975–2006. Surveillance Research Program, NCI.
- Shain KH, Landowski TH, Dalton WS. 2000. The tumor microenvironment as a determinant of cancer cell survival: A possible mechanism for de novo drug resistance. Curr Opin Oncol 12:557–563.
- Shen R, Ye Y, Chen L, Yan Q, Barsky SH, Gao JX. 2008. Precancerous stem cells can serve as tumor vasculogenic progenitors. Plos One 3:e1652.
- Sihto H, Tynninen O, Halonen M, Puputti M, Karjalainen-Lindsberg ML, Kukko H, Joensuu H. 2009. Tumour microvessel endothelial cell KIT and stem cell factor expression in human solid tumours. Histopathology 55:544–553.
- Silzle T, Randolph GJ, Kreutz M, Kunz-Schughart LA. 2004. The fibroblast: Sentinel cell and local immune modulator in tumor tissue. Int J Cancer 108:173–180.
- Simson L, Ellyard JI, Dent LA, Matthaei KI, Rothenberg ME, Foster PS, Smyth MJ, Parish CR. 2007. Regulation of carcinogenesis by IL-5 and CCL11: A potential role for eosinophils in tumor immune surveillance. J Immunol 178:4222–4229.
- Stegmeier F, Warmuth M, Sellers WR, Dorsch M. 2010. Targeted cancer therapies in the twenty-first century: Lessons from imatinib. Clin Pharmacol Ther 87:543–552.
- Strouch MJ, Cheon EC, Salabat MR, Krantz SB, Gounaris E, Melstrom LG, Dangi-Garimella S, Wang E, Munshi HG, Khazaie K, Bentrem DJ. 2010. Crosstalk between mast cells and pancreatic cancer cells contributes to pancreatic tumor progression. Clin Cancer Res 16:2257–2265.
- Tang DG, Conti CJ. 2004. Endothelial cell development, vasculogenesis, angiogenesis, and tumor neovascularization: An update. Semin Thromb Hemost 30:109–117.
- Temming K, Schiffelers RM, Molema G, Kok RJ. 2005. RGD-based strategies for selective delivery of therapeutics and imaging agents to the tumour vasculature. Drug Resist Updat 8:381–402.
- Tillmann T, Kamino K, Dasenbrock C, Germann PG, Kohler M, Morawietz G, Campo E, Cardesa A, Tomatis L, Mohr U. 1999. Ito cell tumor: Immunohistochemical investigations of a rare lesion in the liver of mice. Toxicol Pathol 27:364–369.
- Tokuda Y, Satoh Y, Fujiyama C, Toda S, Sugihara H, Masaki Z. 2003. Prostate cancer cell growth is modulated by adipocyte-cancer cell interaction. Bju Int 91:716–720.
- Turk MJ, Waters DJ, Low PS. 2004. Folate-conjugated liposomes preferentially target macrophages associated with ovarian carcinoma. Cancer Lett 213:165–172.
- Vasiliev JM. 1964. The role of the changes on the microenvironment of epithelial and connective-tissue cells in the carcinogenesis. Acta Unio Int Contra Cancrum 20:1431–1432.
- Virchow R. 1863. Die Cellularpathologie in ihrer Begrundung auf physiologische und pathologische Gewebelehre. Berlin: August Hirschwald Verlag.
- Wong DTW, Bowen SM, Elovic A, Gallagher GT, Weller PF. 1999. Eosinophil ablation and tumor development. Oral Oncology 35:496–501.
- Xian XJ, Hakansson J, Stahlberg A, Lindblom P, Betsholtz C, Gerhardt H, Semb H. 2006. Pericytes limit tumor cell metastasis. J Clin Investigat 116:642–651.
- Xing F, Saidou J, Watabe K. 2010. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Frontiers Biosci 15:166–179.
- Xiong XB, Huang Y, Lu WL, Zhang X, Zhang H, Nagai T, Zhang Q. 2005. Enhanced intracellular delivery and improved antitumor efficacy of doxorubicin by sterically stabilized liposomes modified with a synthetic RGD mimetic. J Control Release 107:262–275.
- Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjallman AH, Ballmer-Hofer K, Schwendener RA. 2006. Clodronate-liposome-mediated depletion of tumour-associated macrophages: A new and highly effective antiangiogenic therapy approach. Br J Cancer 95:272–281.
- Zhang YF, Wang JC, Bian DY, Zhang X, Zhang Q. 2010. Targeted delivery of RGD-modified liposomes encapsulating both combretastatin A-4 and doxorubicin for tumor therapy: In vitro and in vivo studies. Eur J Pharm Biopharm 74:467–473.
- Zhao H, Wang JC, Sun QS, Luo CL, Zhang Q. 2009. RGD-based strategies for improving antitumor activity of paclitaxel-loaded liposomes in nude mice xenografted with human ovarian cancer. J Drug Target 17:10–18.
- Zitvogel L, Tesniere A, Kroemer G. 2006. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat Rev Immunol 6:715–727.