Abstract
Autotransporters produced by Gram-negative bacteria consist of an N-terminal signal sequence, a C-terminal translocator domain (TD), and a passenger domain in between. The TD facilitates the secretion of the passenger across the outer membrane. It generally consists of a channel-forming β-barrel that can be plugged by an α-helix that is formed by a polypeptide fragment immediately N-terminal to the barrel domain in the sequence. In this work, we characterized the TD of the hemoglobin protease Hbp of Escherichia coli by comparing its properties with the TDs of NalP of Neisseria meningitidis and IgA protease of Neisseria gonorrhoeae. All TDs were produced in inclusion bodies and folded in vitro. In the case of the TD of Hbp, this procedure resulted in autocatalytic intramolecular processing, which mimicked the in vivo processing. Liposome-swelling assays and planar lipid bilayer experiments revealed that the pore of the Hbp TD was largely obstructed. In contrast, an Hbp TD variant that lacked only one amino-acid residue from the N terminus showed the opening and closing of a channel comparable to what was reported for the TD of NalP. Additionally, the naturally processed helix contributed to the stability of the TD, as shown by chemical denaturation monitored by tryptophan fluorescence. Overall these results show that Hbp is processed by an autocatalytic intramolecular mechanism resulting in the stable docking of the α-helix in the barrel. In addition, we could show that the α-helix contributes to the stability of TDs.
Introduction
Proteins secreted by Gram-negative bacteria have to cross two membranes, the inner membrane and the outer membrane (OM), as well as the peptidoglycan-containing periplasm in between. The autotransporter pathway, on first sight, seems the simplest protein secretion mechanism to overcome this cell-envelope barrier, since all the required elements for translocation across the OM appear present within the protein sequence itself. Autotransporters consist of an N-terminal signal sequence, a C-terminal translocator domain (TD) and, in between, a secreted passenger domain (Dautin and Bernstein Citation2007). The signal sequence is required for transport across the inner membrane via the Sec system, whereas the TD mediates passenger transport across the OM (Pohlner et al. Citation1987). However, the exact mechanism of OM translocation is largely unknown and is currently under intense debate.
The autotransporter Hbp (Hemoglobin protease) is produced by an Escherichia coli strain that causes peritonitis in humans. The protein interacts specifically with human hemoglobin, degrades it, and subsequently binds the released heme (Otto et al. Citation1998). This specific heme-scavenging protein most likely makes heme accessible for bacterial growth under the iron-restricted conditions encountered in the human body, not only for E. coli, but also for Bacteroides fragilis during the mixed infections found in peritonitis patients (Otto et al. Citation2002). Apart from being a virulence factor, Hbp is also a model protein for the secretion of classical monomeric autotransporters. The structure of the passenger of Hbp has been solved (Otto et al. Citation2005) and shows a β-helical backbone that shares extensive similarity with that of another autotransporter, pertactin (Emsley et al. Citation1996), although it is less regular and slightly kinked in the center. Three separate non-helical domains protrude from the backbone of which domain 1, located at the N terminus of the passenger, is the largest one. This domain shows homology to trypsin and includes six β-strands rolled into a barrel-like structure, with several long β-hairpins over its surface and the serine-protease active site. The very open conformation of that active site presumably helps Hbp to attack its substrate, the globular hemoglobin. The passenger structure has been a useful tool to perform mutagenesis in order to gain insight into the secretion mechanism of Hbp (Jong et al. Citation2007, Sauri et al. Citation2009). However, until now, nothing is known about the structure and conformation of the TD of Hbp.
To date crystal structures have been solved of two TDs of classical autotransporters, i.e., those of NalP from Neisseria meningitidis (Oomen et al. Citation2004) and EspP from E. coli (Barnard et al. Citation2007). The latter is a member of the SPATE family (Serine Protease AuTotransporters of Enterobacteriaceae) of autotransporters to which also Hbp belongs. The crystal structure of the TD of NalP was obtained from in vitro folded protein. Electrophysiological experiments with this TD reconstituted in planar lipid bilayers revealed the opening and closing of channels of two distinct sizes. The structure revealed a 12-stranded β-barrel with an α-helix blocking the hydrophilic channel within the barrel (Oomen et al. Citation2004). Likewise, the structure of the TD of EspP, which was obtained from protein purified from the OM, displayed a 12-stranded β-barrel (Barnard et al. Citation2007). However, the channel within the barrel contained only a small part of the α-helix that had remained after an autocatalytic intramolecular cleavage event (Dautin et al. Citation2007). This proteolytic cleavage event involved residues that are part of the β-barrel wall and stick out into the channel lumen, rendering passenger release completely independent of the protease activity of the serine-protease domain in the passenger (Dautin et al. Citation2007). By contrast, the release of the neisserial autotransporters NalP and IgA protease from the cell surface is mediated by the protease domain found in the passenger (van Ulsen et al. Citation2003).
The goal of the present study was to characterize the TD of the E. coli autotransporter Hbp. To this end, we produced this TD in inclusion bodies and folded it in vitro into its native conformation. Its biophysical properties were then compared with those of the autotransporters NalP of N. meningitidis, of which the structure is known, and IgA protease of Neisseria gonorrhoeae, the first-ever studied autotransporter (Pohlner et al. Citation1987).
Materials and methods
Bacterial strains and growth conditions
E. coli BL21(DE3) (Invitrogen) was grown at 37°C in Luria-Bertani (LB) medium. For plasmid maintenance, ampicillin was added to the culture at a final concentration of 100 μg/ml. When appropriate, isopropyl-β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM to induce gene expression from the lac promoter. N. meningitidis strain H44/76 and N. gonorrhoeae strain MS11 were from our laboratory collection. The neisserial strains were grown on GC agar (Oxoid) supplemented with Vitox (Oxoid) at 37°C in candle jars.
Plasmid constructions
The T7 expression system was used for IPTG-induced synthesis of the different TD variants (Studier et al. Citation1990). Plasmid pPU320 encoding the TD of NalP was described previously (Oomen et al. Citation2004). For the Hbp TD constructs, DNA fragments were obtained by PCR using plasmid pACYC-Hbp (Otto et al. Citation1998) as the DNA template. The primers used were BarNdeI (5′-CATTGCATATGAACAAACGCATGGGCGATTTG-3′) and BarBamHI (5′-TATAACCTAGGTCTCTACACAAGTCCTCAATC-3′) for construct Hbpβ-1 and Bar13NdeI (5′-CTTCCCATATGAACGACGGCCAGGGTAAGGTG-3′) and BarBamHI for construct Hbpβ13. The DNA fragment encoding the NalP TD without the α-helix (NalP TDΔα was amplified by PCR using chromosomal DNA of N. meningitidis strain H44/76 as the template and primers Nmb0478end (5′-CAAGATCTCAGAACCGGTAGCCTACGCCGA-3′) and NalPB3_1 (5′-GCAATTCCATATGTTGGACCACAACGGCACGGGTCT-3′). The DNA fragment coding for the TD of the IgA protease of N. gonorrhoeae was obtained by PCR using primers IgATD MS11NdeI (5′-CATATGCCGCCAGTATTTTCATGG-3′) and IgAend (5′-TTAGAAACGGAATCTGTATTTTAATTTGTCCGGA-3′) and chromosomal DNA of strain MS11 as the template. In all cases, the resulting PCR products were first cloned into pCRII-TOPO (Invitrogen). From there they were excised using the NdeI and BamHI or BglII restriction sites that were included in the primers (underlined) and ligated into plasmid pET11a (Novagen) cut with NdeI and BamHI, placing the ORFs downstream of the IPTG-inducible T7 promoter.
Isolation and solubilization of inclusion bodies
Transformants of strain BL21(DE3) containing the different plasmids were grown to an optical density at 600 nm (OD600) of 0.7, after which IPTG was added to induce expression of the recombinant gene. After another 2-h incubation, cells were harvested, washed with 10 mM Tris-HCl (pH 8.0), and disrupted by sonication (3 × 5 min at level 8, 40% output, Branson sonifier 450; Branson Ultrasonics Corporation, Danbury, CT, USA). Inclusion bodies were collected by centrifugation (10 min, 2000 g, 4°C) and solubilized in 20 mM Tris-HCl (pH 8.0), 8 M urea, 100 mM glycine. Residual insoluble material and membranes were removed by ultracentrifugation (200,000 g, 1 h, 4°C).
In vitro folding and purification
Refolding of the recombinant proteins was initiated by rapid 10-fold dilution of the protein from the 8 M urea-containing stock solution into a buffer containing 0.5% (w/v) N-dodecyl-N,N-dimethyl-1-ammonio-3-propanesulfonate (SB-12), 20 mM Tris-HCl and 1 M NaCl with pH 8.0 or 7.4 for the NalP and Hbp variants, respectively. Refolding was carried out for 72 h at 37°C and evaluated by semi-native SDS-PAGE (described below). Refolded proteins were dialyzed against buffers A1 [0.5% (w/v) SB-12, 20 mM Tris-HCl (pH 8.0)] for NalP variants or A2 [0.5% (w/v) SB-12, 20 mM Tris-HCl (pH 7.4)] for Hbp variants. Proteins were purified by anion-exchange chromatography on a resource-Q column (GE Healthcare) and eluted with a linear gradient of 0–1 M NaCl in buffer A1 or A2. PorA and LpxR were obtained as described in the literature (Jansen et al. Citation2000, Rutten et al. Citation2009).
Semi-native SDS-PAGE
To assess their folding, proteins were analyzed by semi-native SDS-PAGE with reduced amounts of SDS. Samples were dissolved in SDS-PAGE-loading buffer [0.062 M Tris-HCl, pH 6.8, 10% (v/v) glycerol, 0.01% (w/v) bromophenol blue, 20 mM dithiothreitol containing either 2% (w/v) SDS for denatured samples or 0.2% (w/v) SDS for native samples] and incubated for 10 min at either 100°C for denatured samples or room temperature for native samples. For electrophoresis, a protean III minigel system (Biorad) and 10% (w/v) polyacrylamide gels without SDS in the stacking and running gels were used. Electrophoresis was done at 14 mA in a temperature-controlled room at 4°C to prevent denaturation of proteins by heating of the gels. Proteins in the gels were stained with Coomassie brilliant blue G250.
N-terminal sequencing
Refolded Hbpβ13 was dissolved in SDS-PAGE loading buffer and incubated for 10 min at 100°C. Samples were separated on a 10% (w/v) SDS-PAGE gel and blotted onto a 0.45-μm PVDF Immobilon membrane (Millipore, Bedford, USA). The proteins on the membrane were stained with Coomassie R250 in 50% methanol. Bands of interest were cut from the membrane and send to Alphalyse (Odense, Denmark) for N-terminal sequencing by Edman degradation for six cycles.
Trypsin digestion
Refolded proteins were incubated with 20 μg/ml of trypsin for 20 min on ice. Trypsin digestion was stopped by adding phenylmethylsulfonyl fluoride to a final concentration of 1 mM, followed by 10 min incubation on ice. In the fluorescence spectroscopy experiments, the NalP TDΔα was in 1200-fold excess compared to trypsin.
Liposome-swelling assay
Liposome-swelling assays were performed as described (Van Gelder et al. Citation2000). In short, liposomes were prepared from 4 μmol 1,2-dioleoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids, Alabaster, AL, USA) and 1 μmol egg phosphatidyl-DL-glycerol (Avanti). After addition of the different proteins, proteoliposomes were generated in 5 mM Tris-HCl (pH 7.6) containing 17% dextran T40 (Amersham Biosciences). The isotonic concentration was determined by diluting the proteoliposomes into different concentrations of raffinose in 5 mM Tris-HCl (pH 7.6). The test solutes used for diffusion into the proteoliposomes were arabinose, glucose, maltose, and saccharose (all from Sigma), solubilized in 5 mM Tris-HCl (pH 7.6). Swelling of the proteoliposomes was monitored at 550 nm and at least three independent measurements were performed.
Planar lipid bilayer measurements
Planar lipid bilayers were produced as described previously (Van Gelder et al. Citation2000), except that the analogous output voltage signal was converted by a Powerlab/4SP (ADInstruments) and recorded with a 2000 datapoints/s resolution on a computer using the Chart4 program (ADInstruments). In brief, lipid membranes were formed from 1% L-α-lecithin (Sigma) in hexane/chloroform (9:1 v/v) at room temperature across an orifice (100–500 μm diameter) in a Teflon membrane pretreated with a solution of hexadecane/hexane (1:50), separating two electrolyte (1 M KCl, 5 mM CaCl2, 10 mM Tris-HCl, pH 7.4)-containing chambers of 2.5 ml each. Proteins were added to the aqueous subphase, and insertions were monitored after applying a transmembrane potential of 100 mV. The channel conductance of the pores was determined from the stepwise current increments. The pore size was calculated by applying: G = κ[πa 2/(d + πa/2)] where G is the conductance, κ the bulk conductance (11.3 S/m), a the pore radius and d the membrane thickness (4 nm) (Van Gelder et al. Citation2000). For voltage-ramp experiments, an increasing potential from 0 to +200 or −200 mV was applied over a time span of 200 s.
Fluorescence spectroscopy
A Perkin Elmer LS55 luminescence spectrometer was used for the tryptophan fluorescence measurements. Samples with a protein concentration of 1 μM in 20 mM Tris-HCl, pH 7.4, 0.5% SB-12 were incubated for 1 h at 25°C with different concentrations of guanidine hydrochloride (GdnHCl). Emission spectra from 300–450 nm were collected at 25°C using an excitation wavelength of 280 nm and a slit width of 5.0 nm in a 10-mm cuvette. Denaturation curves were plotted at the emission wavelength where the fluorescence was maximal for the native protein, i.e., at 337.5 nm for Hbpβ-1; 339.5 nm for Hbpβ13; 330 nm for NalP TD; and 328.5 for NalP TDΔα. Fit of the graphs was realized using the following equation (Pace and Scholtz Citation1997):
where y = fluorescent signal; [D] = concentration of the denaturant; a = yfolded; b = mfolded; c = yunfolded; d = munfolded; m = slope of the transition curve; h = [D]1/2; t = temperature in Kelvin.
ΔGs were calculated using the methods described by Pace and Scholtz (Citation1997). In short, ΔGs of several points of the transition zone were calculated by using the following formula: ΔG = −RTlnK = −RTln[(yF-y)/(y-yU)] where R is the gas constant (1.987 cal mol-1 K-1) and T the absolute temperature in Kelvin. Fluorescence values (arbitrary units) of the folded (yF) and unfolded (yU) proteins were determined by extrapolation from the pre- and post-transition regions. Then, the ΔGs obtained were plotted against the urea concentration and the ΔG in absence of denaturant was determined by linear extrapolation.
Circular dichroism (CD) spectroscopy
CD spectra were recorded at 25°C on a Jasco J-715 spectropolarimeter (Jasco, Tokyo, Japan). Samples containing 10 μM of the different proteins in 20 mM phosphate buffer, pH 8, 0.5% SB-12, 200 mM NaCl were analyzed using quartz cells with a path-length of 1 mm. The bandwidth was 1 nm and measurements were carried out each 0.1 nm with an averaging time of 4 s. Five spectra were averaged and the contribution of the buffer was subtracted. The percentages of secondary structure, as deduced from the CD spectra, were predicted by using the online software tool K2D2 as described (Perez-Iratxeta and Andrade-Navarro Citation2008).
Results
Expression and purification of the TD variants
High-level expression of OM proteins, such as the TDs of autotransporters, without their signal sequence usually results in their accumulation in cytoplasmic inclusion bodies, which can be easily isolated. To this end, DNA fragments encoding the TDs of three different autotransporters were cloned into the pET11a expression vector under control of an IPTG-inducible T7 promoter. In total, plasmids for five different TD variants were constructed (). Since Hbp belongs to the SPATE family, the protein is expected to be autocatalytically cleaved inside the β-barrel between residues 1100 and 1101 of the predicted α-helix. Two constructs for the Hbp TD were tested. One encodes Hbpβ-1, which starts at amino-acid residue 1103 after the initiation methionine (which then substitutes Leu1102) and therefore lacks the predicted natural cleavage site of Hbp. The other encodes Hbpβ13, which starts at amino-acid residue 1088 being 13 residues upstream of the natural cleavage site. Two constructs for the TD of NalP were analyzed, one encoding the NalP TD consisting of the β-barrel and the α-helix that plugs the barrel as previously described (Oomen et al. Citation2004), and the other encoding a polypeptide, designated NalP TDΔα, that lacks the N-terminal 42 amino-acid residues, which includes the α-helix. Finally, a construct encoding the TD of IgA protease (IgA TD) from N. gonorrhoeae strain MS11 was included. Of note, after autocatalytic processing, the IgA TD of MS11 consists of a surface-exposed domain of ∼12 kDa, which is sensitive to externally added proteases, and a membrane-embedded domain of ∼33 kDa, which is protease resistant and constitutes the β-barrel core with the α-helix as found in other autotransporter TDs (Klauser et al. Citation1993) (). Expression of all constructs in E. coli strain BL21(DE3) resulted in the formation of inclusion bodies, which were isolated and solubilized in 8 M urea.
Figure 1. Schematic representation of the proteins used in this study. The translocator domains are shown with β representing the β-barrel domain (black box) and α representing the α-helix (hatched box); in the case of IgA protease, these assignments were based upon PSIpred secondary structure predictions [Bryson et al. Citation2005]. The surface-exposed N-terminal extension of the IgA protease TD is also indicated (vertically hatched box). The arrows indicate the natural processing sites. In the case of NalP, the exact position of the processing is not known and the approximate position is indicated. Numbers refer to the positions in the amino-acid sequences of the precursors of the wild-type proteins.
![Figure 1. Schematic representation of the proteins used in this study. The translocator domains are shown with β representing the β-barrel domain (black box) and α representing the α-helix (hatched box); in the case of IgA protease, these assignments were based upon PSIpred secondary structure predictions [Bryson et al. Citation2005]. The surface-exposed N-terminal extension of the IgA protease TD is also indicated (vertically hatched box). The arrows indicate the natural processing sites. In the case of NalP, the exact position of the processing is not known and the approximate position is indicated. Numbers refer to the positions in the amino-acid sequences of the precursors of the wild-type proteins.](/cms/asset/98b0ead7-9477-4ae0-809f-b2e960adf42e/imbc_a_550328_f0001_b.jpg)
In vitro folding of TDs
Non-denatured β-barrel OM proteins display an electrophoretic mobility that is different to that of their heat-denatured form when analyzed by semi-native SDS-PAGE (Nakamura and Mizushima Citation1976), a feature that is called heat modifiability. It is well established that the TDs of autotransporters show the same property (Oomen et al. Citation2004). We used heat modifiability to monitor and optimize the folding of the different TD variants after their dilution from urea-solubilized inclusion bodies into various buffers containing detergents. Hbpβ-1, Hbpβ13 and NalP TD folded readily in the presence of SB-12 micelles, and the resulting proteins showed heat-modifiability (). After in vitro folding, the heat-denatured sample of Hbpβ13 showed an extra band that migrated slightly faster in the gel than did the protein directly from inclusion bodies, i.e., it migrated at a position similar to that of Hbpβ-1 (, left panel). This band was not present in the inclusion bodies and, therefore, resulted from the folding procedure. This result suggested that folding of at least a portion of the Hbpβ13 molecules resulted in natural processing between the asparagine residues at positions 1100 and 1101 in wild-type Hbp resulting in the release of the N-terminal fragment. N-terminal sequencing of the extra band confirmed that Hbpβ13 is, indeed, processed at the expected site. Thus, in vitro folding resulted in an, apparently, correct conformation of the TD that allowed for proper processing. Further purification of the in vitro folded Hbpβ13 resulted in a batch of protein composed of only one species, being the form remaining after cleavage (, right panel). Apparently, the intramolecular processing proceeded during the purification process. Consequently, the Hbpβ13 used in our further experiments is only one amino-acid residue longer than the Hbpβ-1 variant and includes the natural N-terminal asparagine. Of note, the Hbpβ-1 variant not only lacks that asparagine, but also has a methionine substituted for the leucine residue at the +2 position.
Figure 2. In vitro folding of the TD variants of Hbp and NalP. (A) Heat-modifiability of refolded Hbp TD variants before (left panel) and after purification (right panel) was analyzed by semi-native SDS-PAGE. Inclusion bodies (IB) were incubated at 100°C and refolded samples either at 100°C (d) or at room temperature (n) prior to electrophoresis. F, folded form; U, unfolded form. The arrow indicates the processed form of Hbpβ13 after refolding, which is visible in the denatured sample. (B) Heat-modifiability of refolded NalP TD and NalP TDΔα. Samples were incubated at 100°C (d) or at room temperature (n) prior to electrophoresis. F, folded form; U, unfolded form. (C) Heat-modifiability of the in vitro folded NalP TDΔα digested by trypsin. Where indicated, the sample was incubated with 20 μg/ml of trypsin before SDS-PAGE analysis. (D) CD spectra of the in vitro folded Hbp (top) and NalP (bottom) TD variants. Numbers at the left of panels A-C refer to the positions of molecular mass standard proteins (in kDa).
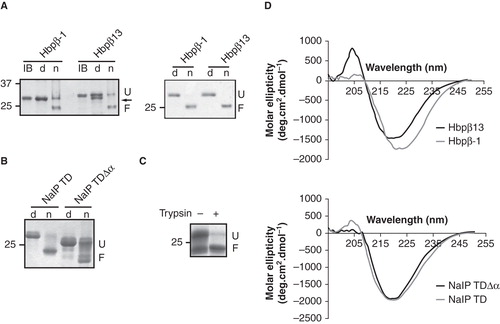
The in vitro folding of NalP TDΔα was not as efficient as that of NalP TD with the α-helix as judged from the smeary protein pattern of the native sample on SDS-PAGE (). Such a pattern could result from inefficient folding in the absence of the helix or from intrinsic instability of the barrel without the helix resulting in denaturation during the semi-native SDS-PAGE analysis. To discriminate between these possibilities, we treated in vitro-folded NalP TDΔα with trypsin prior to electrophoresis. After this treatment, only one distinct band was visible in the non-heated sample, which migrated at the position of the folded protein (). The band at the position of unfolded protein was not detected anymore. Therefore, this band was not generated by denaturation of the folded form during SDS-PAGE analysis, but was already present as a trypsin-sensitive, unfolded form in the protein preparation, suggesting that the absence of the α-helix negatively influences the folding efficiency of the barrel; however once folded, this barrel is stable, at least to an extent that it withstands the mild denaturing conditions of semi-native SDS-PAGE as well as protease treatment.
CD spectroscopy indicated that all in vitro folded proteins had a high content in β-sheets, in accordance with the expected TD conformations (). However, the two variants of the Hbp TD showed somewhat different minima, being 222 nm and 220 nm for Hbpβ-1 and Hbpβ13, respectively. The CD spectra were used to predict the secondary structure content, which resulted for both constructs in 36% of β-sheets and 25% of α-helix, the rest being random coiled. This result indicates that, even though the minima were slightly different, the secondary-structure content appeared identical for both Hbp variants. Remarkably, the CD spectra of the two NalP TD variants were almost indistinguishable in spite of the presence of a long α-helix in NalP TD and the presence of a considerable amount of unfolded protein in NalP TDΔα. Apparently, this unfolded protein had attained some β-sheet structure in spite of not being folded in a stable β-barrel conformation.
Unfortunately, as judged from the electrophoretic mobility in semi-native SDS-PAGE, the in vitro folding of the TD of IgA protease did not yield sufficient amounts of folded protein to be used directly in the following experiments (results not shown).
In vitro folded proteins form pores in liposomes
Previously, the liposome-swelling assay had been applied to demonstrate that the TD of IgA protease of N. gonorrhoeae forms pores (Veiga et al. Citation2002). We used this assay to study the pore activities of Hbpβ-1 and Hbpβ13 and to compare them with those of NalP TD and NalP TDΔα. The in vitro folded proteins were incorporated into proteoliposomes that were subsequently suspended in a solution containing an iso-osmotic concentration of arabinose. As a positive control, we measured the pore-forming activity of the porin PorA of N. meningitidis. Liposomes containing LpxR of Salmonella typhimurium, a lipopolysaccharide deacylase that does not form pores (Rutten et al. Citation2009), served as a negative control. Incubation of proteoliposomes containing PorA with arabinose resulted in a rate of swelling that was proportional to the amount of PorA at low protein concentrations and reached saturation at higher concentrations, whereas, as expected, the liposomes that contained LpxR had a swelling rate close to zero, consistent with the absence of a pore (). For NalP TDΔα, the assay failed because we were unable to establish isotonic conditions for these proteoliposomes; this observation, however, does suggest the formation of pores. The three remaining TDs showed pore activity in this assay (). The swelling rate of the Hbpβ13-containing liposomes was less than that of the Hbpβ-1-containing liposomes (), indicating either a smaller pore size of the Hbpβ13 pores or a lower probability of these pores to be in an open conformation. The liposomes containing the NalP TD showed a higher permeability than those of Hbp, but the swelling rate was only about half of that of PorA-containing proteoliposomes.
Figure 3. In vitro folded proteins form pores in proteoliposomes. (A) Swelling rates of proteoliposomes reconstituted with the indicated amounts of the tested proteins in an iso-osmotic solution of L-arabinose. (B) Relative swelling rates of proteoliposomes containing PorA, NalP TD, Hbpβ-1 or Hbpβ13 in solutions of sugars with different MW. The sugars used were arabinose (150 Da), glucose (180 Da), saccharose (342 Da), and maltose (360 Da). The data are shown relative to the swelling in arabinose and are the averages of at least three independent experiments. The swelling rate corresponding to 10% of that in arabinose was used to calculate the size of the channel.
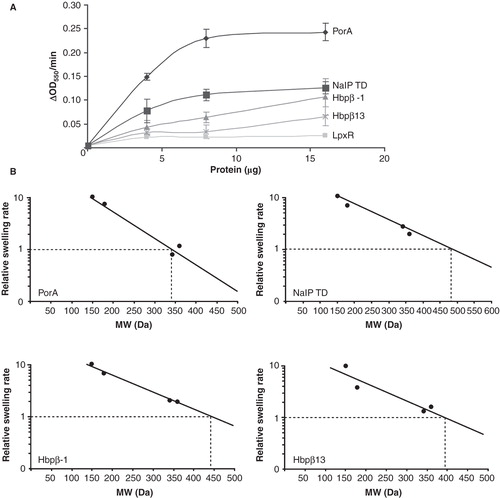
To estimate the sizes of the pores, we used the method described by Nikaido et al (Citation1991). First, proteoliposomes were incubated with sugars of different molecular weights (MWs) and the relative swelling rates were plotted against the MWs of the sugars used (). Then, from the plots, the MW of a solute that would diffuse at 10% of the rate of arabinose was determined. This MW can then be used to calculate a theoretical pore size (Nikaido et al. Citation1991). Our measurements and subsequent calculations yielded pore-sizes for the three TDs () that were quite similar to that reported for the TD of IgA protease (Veiga et al. Citation2002), which was ∼2 nm. However, this value is not consistent with the pore size of ∼10 × 12 Å observed in the crystal structure of the TD of NalP (Oomen et al. Citation2004). Furthermore, while the swelling rate of the Hbpβ13-containing proteoliposomes was clearly lower than that of the Hbpβ-1-containing proteoliposomes (), the calculated pore sizes appeared very similar (). Apparently, Hbpβ13 had a lower probability than Hbpβ-1 of being in the open conformation, which suggests a better obstruction of the pore, either because of the increased length of the α-helix by one residue, or because the proteolytic cleavage facilitated a better docking of the short α-helix within the barrel, or a combination of both.
Table I. Pore sizes of the TDs measured.
Pore activity of the translocator domains in black lipid films
The pore properties of the various TDs were also determined in planar lipid bilayer experiments. After insertion of the TDs in planar lipid bilayers, a transmembrane potential was applied and changes in conductance, representing openings and closings of pores, were measured. Similar to our earlier experiments (Oomen et al. Citation2004), the NalP TD showed openings and closings of pores with a conductance of 1.3 nS as well as smaller conductance steps of 0.15 nS (). Based on the correspondence between the pore sizes calculated from these conductances and the channel dimensions observed in the crystal structure, we have previously postulated that the 1.3-nS conductance steps of NalP correspond to open β-barrel channels without the α-helix inserted, whereas the small conductance steps result from pores with the α-helix positioned within the barrel (Oomen et al. Citation2004), although actual proof for this hypothesis is lacking. Hbpβ-1 clearly showed large ∼1.2-nS conductance steps (), whereas Hbpβ13 only showed conductance steps of 0.2 nS (). The similarity of these conductance steps to those measured for the NalP TD suggested that the auto-cleavage of the Hbpβ13 variant resulted in a blocked pore, presumably because the short α-helix is stably positioned inside the β-barrel, while the large conductances of the Hbpβ-1 variant may indicate that the α-helix in that variant is not stably docked within the barrel. The experiments with NalP TDΔα revealed irregular openings and closings of pores with conductances mainly between 1 and 1.5 nS (). These irregular openings and closings of channels suggest that NalP TDΔα is able to form active channels, but of a variable nature due to a flexible β-barrel structure in the absence of the α-helix.
Figure 4. Pore-forming activity of TDs in planar lipid bilayers. Pore activity was measured at an applied potential of 100 mV. In all panels, the zero-conductance level is indicated with a closed arrowhead (0 nS). (A) Recording for NalP TD. Conductance steps of 1.3 nS and of 0.15 nS are indicated by filled and open arrowheads, respectively, above the recording; the 1.45-nS level represents the simultaneous presence of an open 1.3-nS channel and an open 0.15-nS channel. (B-E) Recordings of Hbpβ-1 (B), Hbpβ13 (C), NalP TDΔα (D), and IgA TD (E). (F) The influence of polarity switching on the conductivity of the IgA TD channels. The moment of switching is indicated by the open arrowheads. The left panel shows a recording with an inversion of the polarity from positive to negative; immediately after the polarity switch a second 1.3-nS channel has opened that closes within a second, while one channel remains open. The right panel shows a recording with a reversion of the polarity from negative to positive; the channel, which had a conductance of 1.3 nS before the polarity switch, shows a conductance of 0.2 nS after the switch. (G) Small channels recorded for IgA TD after the polarity had been reversed from negative to positive.
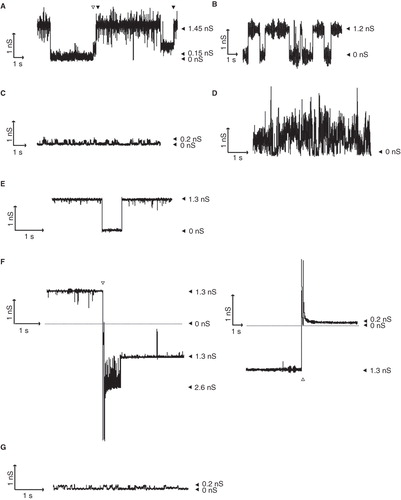
We did not succeed to fold the IgA TD efficiently in vitro. Nevertheless, we tried to obtain recordings of channels formed by this TD by incubating the planar lipid bilayers with urea-solubilized inclusion bodies of IgA TD. Extensive incubation times (4–5 h) occasionally yielded recordings of channels that, apparently, resulted from proteins that spontaneously folded and inserted into the lipid bilayer. The observed conductances of the channels formed, i.e., ∼1.3 nS (), were in line with those observed for Hbpβ-1 and the larger channels of NalP TD, suggesting that the IgA TD attained a similar conformation.
Stepwise increase of the applied potential from 0 to −200 or to +200 mV did not result in closure of the pores for any of the TDs studied suggesting that the channels are not voltage-gated (data not shown). The symmetry of the channels was tested by switching the polarity of the applied current, which did not result in a different conductivity for any of the proteins tested (data not shown and left panel of for IgA TD). However, when we then switched the polarity of the potential back to the original set-up, we measured in the case of the IgA TD only small conductance steps of 0.2 nS (right panel of and ) that did not open to the ∼1.3 nS channels anymore. The small conductances observed were again very similar to the smaller channels found for NalP TD, which could indicate that the IgA protease channels were stably blocked as a result of the polarity switching. This induced stability may be explained by the influence of the extension in IgA TD (), which may prevent the predicted α-helix, once inserted, from leaving the barrel.
The pore dimensions of the TDs can be calculated from the conductance steps observed in black lipid bilayer experiments, if one assumes that the proteins form perfect cylinders (). Although such calculations have to be interpreted with care (Nikaido Citation2003), the calculated pore sizes of the larger ∼1.2/1.3-nS channels were all very similar and corresponded to the dimensions of the pore in the crystal structure of the NalP TD, i.e., 10 × 12 Å (Oomen et al. Citation2004, Barnard et al. Citation2007). Overall, the planar lipid bilayer measurements indicated that the different TDs formed comparable channels that showed similar conductances. Furthermore, they indicated that the α-helix in the processed Hbpβ13 construct is stably fixed within the interior of the barrel, thereby blocking the channel. In the other constructs, both open and blocked channels or only open channels were observed, except in the case of IgA TD, where the channels were blocked after polarity switching.
Contribution of the α-helix to the stability of the β-barrel
The results presented above indicate that the α-helices of the TDs assist the folding of the β-barrels and/or plug the channels after secretion of the passenger domain. In addition, the helix may stabilize the β-barrel structure. To assess the latter possibility further, we measured the stability of the Hbp and NalP TD variants using chemical denaturation monitored by tryptophan fluorescence. Chemical denaturation was performed by incubating the proteins at 25°C with various concentrations of GdnHCl. Tryptophan residues were excited at 280 nm. The resulting fluorescence was measured at the wavelength where the fluorescence was maximal for the native sample, and the values obtained were plotted (). Hbpβ13 was more resistant to chemical denaturation than Hbpβ-1, since higher concentrations of GdnHCl were required to denature the protein (). The denaturation was reversible, which enabled us to determine the ΔG. The ΔG of Hbpβ13 unfolding was 1.8 times higher than that of Hbpβ-1 (), underlining the increased stability of the Hbp TD that resulted from intramolecular processing. The fluorescence spectrum of NalP TDΔα was difficult to interpret due to presence of two shoulders in the fluorescence peak (results not shown), probably due to the presence of unfolded proteins. To remove the unfolded protein, the NalP TDΔα sample was treated with trypsin before incubation with GdnHCl. For an accurate comparison, NalP TD was also treated with trypsin. Denaturation curves for trypsin-treated NalP variants showed that the protein lacking the α-helix is less stable than that with α-helix ( and ). In conclusion, the presence of the α-helix appears to contribute to the stability of the proteins.
Figure 5. Stability of the TD β-barrels with and without α-helices. Chemical denaturation by GdnHCl was monitored by tryptophan fluorescence. AU, arbitrary units.
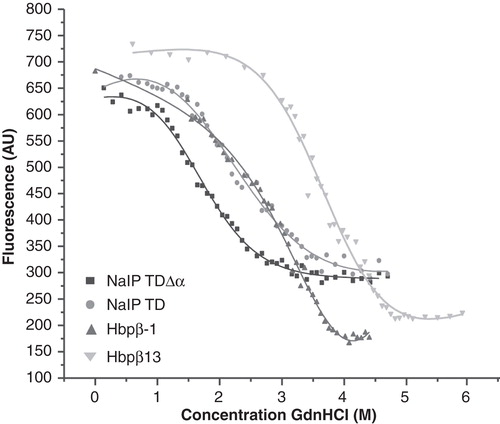
Table II. Calculated ΔG for chemical denaturation.
Discussion
Hbp of E. coli is used as a model to elucidate the mechanism of autotransporter secretion, but its TD had not been characterized so far. In this study, two variants of the Hbp TD were produced in inclusion bodies and successfully folded in vitro. The longer variant was cleaved during folding within the predicted α-helix that was presumed to occupy the channel in the lumen of the β-barrel. The cleavage site was mapped between two asparagine residues (Asn1100 and Asn1101 in full-length Hbp) that are part of a conserved sequence (EVNNLNKRMGDLRD) found in all SPATEs and that were previously reported to be targeted for intramolecular processing in the case of the autotransporter EspP of E. coli (Dautin et al. Citation2007). Apparently, the in vitro folding of Hbpβ13 resulted in a native conformation that allows for correct processing. In the case of EspP, cleavage between the conserved asparagines (Asn1023 and Asn1024 in EspP) requires an aspartic acid residue (Asp1120) located in the barrel wall (Barnard et al. Citation2007, Dautin et al. Citation2007). After translocation of the passenger, Asn1023 is hydrogen bonded to Asp1120, after which the amide group of Asn1023 performs a nucleophilic attack on the Asn1023–Asn1024 peptide bond. This reaction releases the passenger domain and induces a conformational change in the remaining five residues long N-terminal α-helix that runs perpendicular to the barrel axis and efficiently blocks the channel lumen in the final structure (Dautin et al. Citation2007). The catalytic aspartic acid is conserved in Hbp (Asp1197).
After in vitro folding, the pore activities of the TDs were determined. Both the liposome-swelling assays and the planar lipid bilayer measurements clearly indicated that the channel in the processed Hbpβ13 construct was much better blocked than in Hbpβ-1, which is just a single amino-acid residue shorter. The structure of the TD of EspP (Barnard et al. Citation2007) showed that the short α-helix that remains after cleavage is predominantly positively charged due to the Lys1027 and Arg1028 residues. Arg1028 interacts with Asp1120 in an acidic cluster (Asp1120, Glu1154, Glu1172) in the barrel wall to confer stability. The acidic cluster and the positively charged residues in the helix are all conserved in Hbp, suggesting that in the naturally processed Hbp the α-helix is stably anchored to the barrel wall, like in EspP. Although these residues are also present in Hbpβ-1, it is entirely possible that the five amino-acid residues long α-helix is not formed in the absence of the N-terminal asparagine and that, therefore, the positively charged residues are not appropriately positioned to interact with the acidic cluster. This could explain why the Hbpβ-1 channels, in contract to Hbpβ13, are not effectively blocked in the bilayer experiments and it could also explain the slight differences in the CD spectra of the two constructs (). In one of the two β-barrels in the PDB file of the EspP TD structure, the N-terminal Asn1024 and Leu1025 form a network of ionic and hydrogen bonds with Glu1154, Glu1172, and Arg1205, all again conserved in the Hbp TD. The in vitro folded Hbpβ-1 lacks the corresponding Asn1101, while Leu1102 is replaced by Met. Based upon the analogy to the EspP TD structure, the expected interactions of Asn1101 to Glu1249 and Arg1282 would be lacking in the Hbpβ-1. Thus, even if a short N-terminal α-helix is formed in Hbpβ-1, the reduced number of interactions with the barrel wall might explain the different pore properties of Hbpβ13 and Hbpβ-1.
The α-helix in the TD of NalP does not seem to be as stably docked to the barrel wall as that of Hbp, since both the liposome-swelling assays and the planar lipid bilayer experiments indicated that the pore can be open. However, the construct used is not identical to the naturally processed TD (Oomen et al. Citation2004), and it is conceivable that after natural processing also this TD is blocked as a result of additional contacts between the helix and the barrel that are not present in the in vitro folded protein. The unfolded TD of IgA protease occasionally inserted into the lipid bilayers and formed channels with conductances comparable to the large open channels of the other TDs, suggesting that the IgA TD adopted a similar open conformation. The TD of IgA protease of N. gonorrhoeae strain MS11 consists of a core domain, which encompasses the predicted α-helix and β-barrel, and a 12-kDa extracellularly exposed domain (). Changing the polarity of the potential in the planar lipid bilayer experiments appeared to fix the IgA TD in a conformation with the α-helix inserted, since the small 0.2-nS conductances observed were consistent with a closed pore. Consequently, this would then imply that the entire 12-kDa domain could travel through the pore formed by the IgA TD β-barrel. The source of energy necessary for the translocation of the passenger domain across the OM is still unknown and it is often suggested that it is provided by the folding of the passenger domain at the cell surface. However, if the conformation attained by IgA TD indeed resembles that of the NalP TD, the observed conductances also suggest that translocation of at least a small part of the passenger domain through the channel can be induced by applying an electric field. In vivo such a field could perhaps be provided by the Donnan potential present across the OM (Stock et al. Citation1977), which is variable but can be as high as 100 mV (Sen et al. Citation1988).
All TDs showed pore activity in the liposome-swelling assays and planar lipid bilayer experiments, but the calculated pore diameters differed dependent on the method used (). However, regardless the method used, the measured values were similar when the translocator domains of different autotransporters were compared. By contrast, the C-terminal domain of BrkA from Bordetella pertussis was reported to form pores with a conductance of 3 nS (Shannon and Fernandez Citation1999), which is much larger than the maximal conductance of 1.3 nS found in our studies. The basis of this discrepancy is unclear, but the methods used vary considerably. Firstly, in the BrkA planar lipid bilayer measurements, re-folding of the protein was not monitored, and also the different composition of the membranes (for BrkA oxidized cholesterol was used) might have some influence. In addition, some recordings showed a weak resolution of the steps, so that multiple insertions could have been confused for a single insertion.
Our results support the notion that plugging of the β-barrel is a major function for the α-helix of the TD of Hbp; it will block the diffusion of solutes across the OM after release of the passenger. Additionally, the α-helix could improve the folding efficiency of the TD. For Hbp, we did not observe such a function, since both Hbp constructs folded with similar efficiencies, despite the difference in length of the predicted α-helices. However, in the case of the NalP TD constructs the folding efficiency was decreased in the absence of the α-helix (, ). Also in vivo, the insertion of NalP TD constructs into the OM was considerably reduced when the α-helix was lacking (Oomen et al. Citation2004). Apparently, the α-helix acts in this case as a folding scaffold. A similar contribution of the α-helix to the correct assembly of the TD of EspP into the OM has been reported (Ieva et al. Citation2008). In that case, the α-helix was hypothesized to facilitate the folding of the TD by allowing the β-barrel to fold around it. Perhaps, in the case of Hbp, the small α-helix that is still present in Hbp β-1 is sufficient to mediate this function.
An additional function of the α-helix could be to stabilize the β-barrel. Indeed, Hbpβ13 was more resistant to chemical denaturation than Hbp-1. The extensive docking of the α-helix of Hbpβ13 to the barrel wall after processing, inferred from the EspP TD structure, is in agreement with such a stabilizing effect. Similarly, NalP TD was also more stable than NalP TDΔα. The NalP data corroborated predictions of molecular dynamics simulations (Khalid and Sansom Citation2006), which suggested that removing the α-helix would lead to a minor relaxation and a slightly increased conformational flexibility of the barrel. Such increased flexibility might also explain the irregular openings and closings of the NalP TDΔα channel observed in the planar lipid bilayer experiments. Similarly, noisy signals were reported for the TD of SphB1of B. pertussis in the absence of the α-helix (Dé et al. Citation2008). However, it is interesting to note that the planar lipid bilayer recordings indicated that the open channels of NalP TD and Hbpβ-1 were stable suggesting that the helices do not completely leave the channel or that they establish new stabilizing interactions when out of the channel.
In conclusion, we could refold the TD of the autotransporter Hbp in vitro resulting in its processing via a mechanism similar to what has been described for EspP in vivo. Overall, the biophysical properties of the Hbp TD were very similar to those of the TDs of other autotransporters, such as NalP. However, in the processed TD, the remaining α-helix stably blocked the pore in the β-barrel and stabilized the protein. While this paper was under review the crystal structure of a mutant form of the Hbp TD was published (Tajima et al. Citation2010). This mutant protein, encompassing residues 1075–1377, is not processed due to the substitution of the catalytic Asn1100 by Asp, resulting in a pore that is occupied by an N-terminal α-helix that would be released after processing and an already tilted short α-helix that could block the pore after processing. The overall structure and its channel dimensions corroborate fully the results reported here. Furthermore, analysis of the tilted α-helix and its position in the pore show that the α-helix is not yet stably docked in this mutant protein. Apparently, the stable docking requires the autocatalytic processing. The availability of the structure will help in designing appropriate mutants to study the secretion mechanism and the role of the TD therein.
Acknowledgements
We would like to thank Fabrice Homblé for his helpful suggestions for the planar bilayer experiment using unfolded proteins.
Declaration of interests: This work was supported by the Council for Chemical Sciences of the Netherlands Organization for Scientific Research (NWO-CW) (grant 700.52.309) and The Netherlands Organization for Health Research and Development (Zon-Mw). Virginie Roussel-Jazédé was also supported by a FEMS short-term fellowship. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Barnard TJ, Dautin N, Lukacic P, Bernstein HD, Buchanan SK. 2007. Autotransporter structure reveals intra-barrel cleavage followed by conformational changes. Nat Struct Mol Biol 14:1214–1220.
- Bryson K, McGuffin LJ, Marsden RL, Ward JJ, Sodhi JS, Jones DT. 2005. Protein structure prediction servers at University College London. Nucl Acids Res 33(Web Server issue): W36–38. Available from: http://bioinf.cs.ucl.ac.uk/psipred/.
- Dautin N, Bernstein HD. 2007. Protein secretion in Gram-negative bacteria via the autotransporter pathway. Annu Rev Microbiol 61:89–112.
- Dautin N, Barnard TJ, Anderson DE, Bernstein HD. 2007. Cleavage of a bacterial autotransporter by an evolutionarily convergent autocatalytic mechanism. EMBO J 26:1942–1952.
- Dé E, Saint N, Glinel K, Meli AC, Levy D, Jacob-Dubuisson F. 2008. Influence of the passenger domain of a model autotransporter on the properties of its translocator domain. Mol Membr Biol, 25, 192–202.
- Emsley P, Charles IG, Fairweather NF, Isaacs NW. 1996. Structure of Bordetella pertussis virulence factor P.69 pertactin. Nature 381:90–92.
- Ieva R, Skillman KM, Bernstein HD. 2008. Incorporation of a polypeptide segment into the β-domain pore during the assembly of a bacterial autotransporter. Mol Microbiol 67:188–201.
- Jansen C, Wiese A, Reubsaet L, Dekker N, de Cock H, Seydel U, Tommassen J. 2000. Biochemical and biophysical characterization of in vitro folded outer membrane porin PorA of Neisseria meningitidis. Biochim Biophys Acta 1464:284–298.
- Jong WSP, ten Hagen-Jongman CM, den Blaauwen T, Slotboom DJ, Tame JRH, Wickström D, de Gier JW, Otto BR, Luirink J. 2007. Limited tolerance towards folded elements during secretion of the autotransporter Hbp. Mol Microbiol 63:1524–1536.
- Khalid S, Sansom MS. 2006. Molecular dynamics simulations of a bacterial autotransporter: NalP from Neisseria meningitidis. Mol Membr Biol 23:499–508.
- Klauser T, Kramer J, Otzelberger K, Pohlner J, Meyer TF. 1993. Characterization of the Neisseria Iga β-core. The essential unit for outer membrane targeting and extracellular protein secretion. J Mol Biol 234:579–593.
- Nakamura K, Mizushima S. 1976. Effects of heating in dodecyl sulfate solution on the conformation and electrophoretic mobility of isolated major outer membrane proteins from Escherichia coli K-12. J Biochem 80:1411–1422.
- Nikaido H, Nikaido K, Harayama S. 1991. Identification and characterization of porins in Pseudomonas aeruginosa. J Biol Chem 266:770–779.
- Nikaido H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67:593-656.
- Oomen CJ, van Ulsen P, Van Gelder P, Feijen M, Tommassen J, Gros P. 2004. Structure of the translocator domain of a bacterial autotransporter. EMBO J 23:1257–1266.
- Otto BR, van Dooren SSJ, Nuijens JH, Luirink J, Oudega B. 1998. Characterization of a hemoglobin protease secreted by the pathogenic Escherichia coli strain EB1. J Exp Med 188:1091–1103.
- Otto BR, van Dooren SJ, Dozois CM, Luirink J, Oudega B. 2002. Escherichia coli hemoglobin protease autotransporter contributes to synergistic abscess formation and heme-dependent growth of Bacteroides fragilis. Infect Immun 70:5–10.
- Otto BR, Sijbrandi R, Luirink J, Oudega B, Heddle JG, Mizutani K, Park SY, Tame JRH. 2005. Crystal structure of hemoglobin protease, a heme binding autotransporter protein from Escherichia coli. J Biol Chem 280:17339–17345.
- Pace CN, Scholtz JM. 1997. Measuring the conformational stability of a protein. In: Creighton TE, editor. Protein structure: A practical approach. 2nd ed. New York: Oxford University Press. pp 302–324.
- Perez-Iratxeta C, Andrade-Navarro MA. 2008. K2D2: Estimation of protein secondary structure from circular dichroism spectra. BMC Struct Biol 8:25. Available from http://www.embl.de/∼andrade/k2d/.
- Pohlner J, Halter R, Beyreuther K, Meyer TF. 1987. Gene structure and extracellular secretion of Neisseria gonorrhoeae IgA protease. Nature 325:458–462.
- Rutten L, Mannie JP, Stead CM, Raetz CRH, Reynolds CM, Bonvin AM, Tommassen JP, Egmond MR, Trent MS, Gros P. 2009. Active-site architecture and catalytic mechanism of the lipid A deacylase LpxR of Salmonella typhimurium. Proc Natl Acad Sci USA 106:1960–1964.
- Sauri A, Soprova Z, Wickström D, de Gier JW, Van der Schors RC, Smit AB, Jong WSP, Luirink J. 2009. The Bam (Omp85) complex is involved in secretion of the autotransporter haemoglobin protease. Microbiology 155:3982–3991.
- Sen K, Hellman J, Nikaido H. 1988. Porin channels in intact cells of Escherichia coli are not affected by Donnan potentials across the outer membrane. J Biol Chem 263:1182–1187.
- Shannon JL, Fernandez RC. 1999. The C-terminal domain of the Bordetella pertussis autotransporter BrkA forms a pore in lipid bilayer membranes. J Bacteriol 181:5838–5842.
- Stock JB, Rauch B, Roseman S. 1977. Periplasmic space in Salmonella typhimurium and Escherichia coli. J Biol Chem 252:7850–7861.
- Studier FW, Rosenberg AH, Dunn JJ, Dubbendorff JW. 1990. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol 185:60–89.
- Tajima N, Kawai F, Park SY, Tame JRH. 2010. A novel intein-like autoproteolytic mechanism in autotransporter proteins. J Mol Biol 402:645–656.
- Van Gelder P, Dumas F, Rosenbusch JP, Winterhalter M. 2000. Oriented channels reveal asymmetric energy barriers for sugar translocation through maltoporin of Escherichia coli. Eur J Biochem 267:79–84.
- van Ulsen P, van Alphen L, ten Hove J, Fransen F, van der Ley P, Tommassen J. 2003. A Neisserial autotransporter NalP modulating the processing of other autotransporters. Mol Microbiol 50:1017–1030.
- Veiga E, Sugawara E, Nikaido H, de Lorenzo V, Fernadez LA. 2002. Export of autotransported proteins proceeds through an oligomeric ring shaped by C-terminal domains. EMBO J 21:2122–2131.