Abstract
The transporter SbtA is a high affinity Na+-dependent HCO3 - uptake system present in a majority of cyanobacterial clades. It functions in conjunction with CO2 uptake systems and other HCO3 - uptake systems to allow cyanobacteria to accumulate high levels of HCO3 - used to support efficient photosynthetic CO2 fixation via the CO2 concentrating mechanism in these species. The phoA/lacZ fusion reporter method was used to determine the membrane topology of the cyanobacterial bicarbonate transporter, SbtA (predicted size of ∼ 39.7 kD), cloned from the freshwater strain, Synechocystis PCC6803. The structure conforms to a model featuring 10 transmembrane helices (TMHs), with a distinct 5 + 5 duplicated structure. Both the N- and C-terminus are outside the cell and the second half of the protein is inverted relative to the first. The first putative helix appears to lack sufficient topogenic signals for its correct orientation in the membrane and instead relies on the presence of later helices. The cytoplasmic loop between helices 5 and 6 is a likely location for regulatory mechanisms that could govern activation of the transporter, and the cytoplasmic loop between helices 9 and 10 also contains some conserved putative regulatory residues.
Introduction
In cyanobacteria, active HCO3 - transporters form essential components of the CO2 concentrating mechanism (CCM), which functions, in conjunction with active CO2 uptake systems, to allow active accumulation of HCO3 - inside the cell for photosynthesis. This HCO3 - pool is then used within carboxysome micro-compartments to support optimal rates of photosynthetic CO2 fixation via the primary carboxylase, ribulose bisphosphate carboxylase-oxygenase or Rubisco (Price et al. Citation2008). Notably, the uptake of CO2/HCO3 - represents the largest nutrient flux encountered by cyanobacteria. A functional CCM is an obligate growth requirement for all species of cyanobacteria so far examined. Thus, it is readily arguable that the cyanobacterial CCM transporters contribute to global primary productivity, especially in oceanic regions. In oceanic regions it is estimated that cyanobacteria contribute as much as 25% to annual net global primary productivity as giga-tonnes of carbon fixed per year (Liu et al. Citation1997, Field et al. Citation1998), with most of this primary CO2 fixation occurring in the open oceans.
The bicarbonate transporter, SbtA, is a high affinity, Na+-dependent transporter that is found in most, but not all, clades of cyanobacteria (Badger et al. Citation2006, Price et al. Citation2008). In all probability, SbtA acts as an electroneutral Na+/HCO3 - symporter, although proof of this function is not yet available. SbtA has been classified as Family TC.2.A.83 under the transporter classification database (www.tcdb.org), apparently falling within the wider sodium solute symporter (SSS) family. The available functional data on SbtA is presently restricted to a few β-cyanobacterial strains (those that possess Form-1B Rubisco and β-type carboxysomes) found in freshwater and estuarine/coastal habitats, and characteristically exposed to large fluctuations in CO2 availability and light intensity (Badger et al. Citation2006, Price et al. Citation2004, 2008). SbtA was first characterized in the freshwater model cyanobacterium, Synechocystis PCC6803 via gene knock-outs (Shibata et al. Citation2002). SbtA from the marine cyanobacterium, Synechococcus PCC7002, was also identified using gene knock-outs and was shown to possess a high photosynthetic affinity for HCO3 - (K0.5(Ci) of 2–5 μM) and able to support a relatively low flux rate (Price et al. Citation2004). Other SbtA orthologs in β-cyanobacteria have been identified by bioinformatics based on a high degree of sequence identity (Badger et al. Citation2006, Price et al. Citation2008), but it is the group of more distant SbtA homologs present in oceanic cyanobacteria (α-cyanobacteria; Form1A Rubisco and α-type carboxysomes) such as Prochlorococcus species that are likely to be the most ubiquitous SbtA homologues on a global scale (Badger et al. Citation2006, Price et al. Citation2008).
The SbtA transporter from Synechocystis PCC6803 can be considered the archetypal form and is predicted to be 374 amino acids in length (39.7 kD predicted) with topology predictions indicating it could have up to 10 transmembrane domains. Recent identification of the Sbt family arising through an intergenic duplication event also suggests a 10 helix structure comprising a duplication of an ancestral five helices (von Rozycki et al. Citation2004). It is probable that SbtA is a Na+/HCO3 - symporter, however the transport mechanism has not been definitively characterized. Proteomic data indicates that SbtA is present in the cytoplasmic membrane of Synechocystis PCC6803 at 160 kD on native gels (four times expected size) and therefore appears to form a tetramer (Zhang et al. Citation2004). The SbtA transporter, like all the active CO2 and HCO3 - uptake systems in cyanobacteria, is believed to be inactive in the dark, a process that potentially involves protein phosphorylation events (Price et al. Citation2008). Additionally we have identified SbtA as a possible transgene target for improving the efficiency of photosynthesis in the chloroplasts of crop plants; here knowledge of the post-translation events involved in regulation of SbtA is also needed information (Price et al. Citation2008). In attempting to understand the post-translational or allosteric activation of SbtA we have first determined a topology-map of SbtA so we can identify putative cytoplasmic loops that could be potentially involved in regulation or oligmerization.
The determination of the structure of membrane proteins is often hampered by notorious difficulties in obtaining protein crystals for hydrophobic membrane proteins. Instead, as a first approximation of topology, we have used of the phoA/lacZ reporter-fusion system (Alexeyev and Winkler Citation1999, van Geest and Lolkema Citation2000), combined with use of predictive models, an approach that we previously used to determine the structure of another class of cyanobacterial HCO3 - transporter, BicA (Shelden et al. Citation2010), as the most useful available method for determining the basic membrane topology of SbtA. The present report provides the first comprehensive topology information for the SbtA transporter from Synechocystis PCC6803, or any related member, a transporter likely to have an important role in global primary productivity.
Materials and methods
Bacterial strains and growth conditions
Escherichia coli JM109 was used as plasmid host in this study and strains were grown aerobically at 37°C in Luria Broth (LB). Low Na+, dual indicator plates (Alexeyev and Winkler Citation1999) contained 1% Bacto-tryptone, 0.5% yeast extract, 1 mM NaCl, 1.5% Bacto-agar, 80 mM K2HPO4 (pH 7.0), 0.1 mM IPTG, 80 μg/ml X-phos (5-bromo-4-chloro-3-indolyl phosphate, disodium salt, Research Organics), 100 μg ml-1 Red-Gal (6-chloro-3-indolyl-β-D-galactoside, Research Organics) and 30 μg ml-1 of chloramphenicol (Shelden et al. Citation2010). Dual indicator plates were incubated at room temperature since the lower temperature, plus lower IPTG and lower Na+ were effective in offsetting the general toxicity of expressing SbtA in E. coli (data not shown).
Cloning of SbtA into the dual reporter construct
Plasmid pNV1216 (Lehane et al. Citation2005) harbouring the pho-lac fusion was digested with Bam HI and Xba I to remove the gtrII gene and replaced with the 1.1 kb sbtA gene from Synechocystis PCC6803 that was PCR amplified from genomic DNA with forward primer SbtA (6803) BamH1(F), and reverse primer SbtA (6803) XbaI(R) – see . The phoA/lacZ reporter fusion was constructed according to Alexeyev and Winkler (Citation1999) as previously described (Shelden et al. Citation2010).
Table I. Primers used for generating SbtA (6803) phoA/lacZ constructs.
Construction of SbtA-phoA/lacZ fusions using Exonuclease-III
SbtA-6803/pNV1216 was linearized with the unique restriction sites, NsiI and XbaI. The Promega Erase-a-base kit was used to create a series of SbtA-phoA/lacZ fusions according to the manufacturer's recommendations. After ligation, plasmid DNA was transformed into JM109 cells and plated onto dual indicator plates. Plasmid DNA was purified from blue, red and purple colonies, digested with BamHI and SacI to confirm deletion size, and sequenced with the PhoSeq primer (cgctaagagaatcacgcaga). Big-dye Sequencing was carried out following the manufacturer's protocol (Applied Biosystems).
Construction of SbtA-phoA/lacZ fusions using PCR
PCR was used to generate a number of SbtA-phoA/lacZ fusions at desired locations to create SbtA-phoA/lacZ C-terminal fusions as shown in , with nomenclature referring to the last amino acid residue before the reporter. Primers were designed with an XbaI restriction site 3′ of the fusion site to allow in-frame directional cloning into pNV1216. SbtA (6803) was excised from pNV1216 using double digests with BamHI and XbaI and replaced with a designed PCR product. The specific 3′ PCR primer combinations used with the common 5′ forward primer are shown in . PCR products were ligated into BamHI/XbaI digested pNV1216 and transformed into JM109 cells, sequenced and then colonies were analysed on dual indicator plates, as above.
Colourmetric assays of alkaline phosphatase and β-galactosidase activities
E. coli JM109 cells containing SbtA-phoA/lacZ fusion constructs were grown overnight (37°C), diluted 1:10 in fresh low Na+-LB containing chloramphenicol and grown for a further 1 h until A600 ∼ 0.5, then induced for 1.5 h with 0.1 mM IPTG, then harvested by centrifugation and permeabilized as described previously (Domingues et al. Citation1997), but without the addition of chloroform. For the β-galactosidase (BG) assay, cells were harvested as above, except that prior to permeabilization, half the cells were resuspended in the BG buffer (25 mM Tris-HCl pH 7.5, 125 mM NaCl, 2 mM MgCl2 and 12 mM 2-mercaptoethanol) and half in alkaline phosphate (AP) buffer (25 mM Tris-HCl pH 8, 125 mM NaCl, 1 mM MgCl2 and 0.75 mM ZnCl2). Stock solutions (20 mg/ml) of X-gal (5-bromo-4-chloro-3-indolyl β-D-galacto-pyranoside) and X-Phos (2-Dicyclohexyl-phosphino-2′,4′,6′-triisopropylbiphenyl) were prepared in water and DMSO, respectively, and diluted in reaction buffer immediately prior to experiments, using a final concentrations of 2 mg/ml X-gal and 0.5 mg/ml X-Phos. BG and AP reactions in 96-well format were usually run for 1.5–2 h and 0.5–1 h, respectively, at (37°C) and timed assays were terminated with 100 μl DMSO. Assays were recorded in a 96-well plate reader with a 600 nm narrow-band filter. Cell densities (as controls without substrate) were measured just prior to assay termination. Assay reactions stopped with DMSO were measured with a 600 nm filter after 10 min and also after overnight colour development due to cell solubilization.
Topology prediction analysis
Several prediction models available on the Internet, were used to examine the SbtA protein sequence for predicted membrane topology. The TOPCONS programs (http://topcons.net/index.php) were used as before (Shelden et al. Citation2010) to fit experimentally-determined topology data to five different algorithms: SCAMPI (single sequence mode) (Bernsel et al. Citation2008), SCAMPI (multiple sequence mode) (Bernsel et al. Citation2008), PRODIV-TMHMM, PRO-TMHMM (Viklund and Elofsson Citation2004) and OCTOPUS (Alexeyev and Winkler Citation1999). A number of other programs were used including MEMSAT3 (Jones Citation2007) and TopPred 2 (Von Heijne Citation1992). The determination of helical wheel projections and calculation of hydrophobic moments (HM) were determined using tools at (http://rzlab.ucr.edu/scripts/wheel/wheel.cgi). Weblogo alignment representations were submitted to weblogo.threeplusone.com using aligned stacks of TMHs prepared using ClustalW2 at www.ebi.ac.uk/Tools. Searches for potential signal peptides using the SignalP prediction server (www.cbs.dtu.dk/services/SignalP) with gram negative bacteria selected as the organism group.
Results and discussion
PhoA-LacZ mapping of SbtA from Synechococcus PCC6803
The phoA/lacZ dual reporter construct was fused to successive C-terminal truncations of the SbtA transporter gene from Synechocystis PCC6803 (SbtA-6803) and the activity of both reporter enzymes determined from permeabilized cell lines. Alkaline phosphatase (AP) is active only in the periplasm while β-galactosidase (BG) is active only in the cytoplasm, so the relative activity of both reporters can be used to indicate the topological orientation of the construct in the E. coli membrane. An advantage of the dual reporter system is that the ratio of the two activities can be used to correct for any relative differences in the expression level of different fusion constructs. Thus, assay data were normalized and expressed as a ratio of AP activity to BG activity to allow for differences in the relative expression strength of each fusion construct (). Fusions showing a relatively equivalent activity ratio for both reporters were scored as being located in membrane spanning domains (TMHs); apparently TMH-located fusions have a fairly equivalent chance of being targeted to the periplast or cytoplast (van Geest and Lolkema Citation2000).
Table II. Enzyme activities of SbtA dual reporter fusions.
A total of 10 random in-frame fusions and 11 designed fusions, based on the predicted topology analysis () were initially identified on dual reporter plates and scored as red (putative cytoplasmic locations), blue (putative periplasmic locations) or purple colonies (putative membrane spanning domains; ). This collection of 21 fusion constructs were then quantitatively assessed for alkaline phosphatase (AP) and β-galactosidase (BG) activities (). Data obtained, primarily based on the AP/BG enzyme activity ratios of the C-terminal fusions, were interpreted to support a topology model of 10 transmembrane helices (). The eight C-terminal helices were clearly identified, but interpretation of the first five fusion constructs in the series (putative helices 1& 2) was problematic. While there are two clearly identifiable hydrophobic regions (von Rozycki et al. Citation2004) that are predicted to span the membrane, fusions in this region show equivalent levels of alkaline phosphatase and β-galactosidase activity.
Figure 1. Topology map of the Na+-dependent HCO3 - transporter, SbtA, from Synechocystis PCC6803. Experimentally determined phoA/lacZ mapping positions are shown as black (internal), mid-grey (TMH located) or light grey (external) based on the scored locations in (primarily based on AP/BG enzyme assay ratios); in the online version these are shown as blue, purple and red, respectively. The positions of positive charges on the inter-helical loops are also shown. This Figure is reproduced in colour in the online version of Molecular Membrane Biology. In addition, a Supplementary residue-based Figure (Supplementary Figure 1) is also provided online.
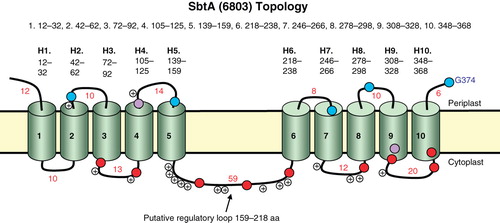
A difficulty in obtaining meaningful fusion data for the first two helices seems to arise from the lack of membrane positioning signals in the N terminus and first loop of the SbtA-6803 protein. Web-based prediction programs consistently predicted that the N- and C-termini are both located outside the cell. In particular, the TOPCONs SCAMPI-msa model (topcons.net; ) predicts 10 TMH, consistent with the existence of two N-terminal helices in addition to the eight identified from the data. This program compares the target sequence against a multiple alignment of sequences in generating the prediction (Bernsel et al. Citation2008). It was also found to give the best match to our previously determined topology of another cyanobacterial transporter, BicA (Shelden et al. Citation2010). No known targeting sequence exits in the first 12 amino acids of the N-terminus of SbtA-6803 using SignalP predictions, consistent with the finding that the S5 fusion remains cytoplasmically-located, forming purple colonies but with high BG activity () on indicator plates. Fusions I29, V37, A41 and T44, all of which occur in the first hydrophobic region, form purple colonies (a near unity ratio of normalized BG:AP activity). This could result from the TMH orientation in the membrane being largely random or could occur if the hydrophobic region is sometimes inserted N in and C out and sometimes completely fails to enter the membrane. In either case, the fusion inserts into the membrane in the opposite orientation to that predicted, at least some of the time. This could be because putative helix 1 is less hydrophobic at its N-terminal end. Shorter hydrophobic regions, and greater hydrophobicity at the C-terminus of a helix can result in preferential translocation of the C-terminal end (Dowhan and Bogdanov Citation2009). The helix may be inverted later in the biogenesis of the protein due to interaction with other helices and/or lipids (Dowhan and Bogdanov Citation2009).The charge asymmetry (positive inside rule) and hydrophobicity of the first two helices of SbtA appears to be sufficient to faithfully orientate the first two TMHs, and longer fusions, into the membrane (). A similar result was encountered when using phoA or lacZ fusions to map the topology of the E. coli Na+/proline transporter, PutP (Jung et al. Citation1998). Here the N-terminus was also determined to be periplasmic, and the correct targeting of the first 2–3 TMHs was determined to be dependent on charge asymmetry and partitioning of the hydrophobic TMHs into the membrane.
Further support for the 10 TMH topology of SbtA-6803 is that its sequence displays a distinct 5 + 5 duplicated structure ( and ), a feature that is also supported by hydropathy plots of 14 averaged SbtA members (von Rozycki et al. Citation2004). The duplicated nature of this structure can be seen by aligning helices 1–5 with helices 6–10 (); this results in a largely ungapped alignment with 23% identity and 53% similarity. This is interesting, since consideration of the simplistic diagram of a duplicated 5 + 5 structure indicted that the corresponding TMH pairs in the duplicated half must have opposite or inverted orientation in the membrane () requiring some evolutionary adaptation of inverted helices. Inverted repeats have been found in several other transporter families and it appears a common structural motif (Abramson and Wright Citation2009; Krishnamurthy et al. Citation2009). Without five helices in the N-terminal half of SbtA-6803, this symmetry would be lost.
Figure 2. The locations of the ten TMHs in the SbtA from Synechocystis PCC6803 based on experimentally determined phoA/lacZ mapping positions and the SCAMPI-msa prediction model. ‘i’ refers to cytoplasmic domains, while ‘o’ refers to periplasmic domains and grey blocks denote TMH domains.

Figure 3. Alignment of the two halves of the 5 + 5 repeated structure of SbtA-6803. The region encompassing helices 1–5 aligned to the region encompassing helices 6–10 of SbtA.
Figure 4. Simple cartoon showing the inversion of each helix in the repeat (H6–10) relative to each helix in the H1–5 region.
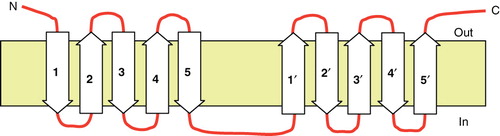
The concept of a duplicated, core 5 + 5 structure is common to a number of Na+-coupled transporters from prokaryotes and eukaryotes (Krishnamurthy et al. Citation2009), a situation that has become clear due to the recent availability of four crystal structures. These include the bacterial Na+/leucine symporter (LeuT), the bacterial Na+/galactose symporter (vSGLT), the nucleobase symporter (Mhp1) and the BetP glycine-betaine symporter (Abramson and Wright Citation2009, Krishnamurthy et al. Citation2009). A common mechanistic feature of these transporters is that they employ an alternating access mechanism, involving the two halves of the transporter alternately opening binding gates at the periplasmic and cytoplasmic faces. Until such time as a crystal structure for SbtA-6803 is available it will be hard to speculate on any structural similarities with these inverted repeat type Na+ symporters. One notable difference is that the symporters noted above transport substrates of significantly larger size than HCO3 -. Another difference is that the inverted structures that have been solved have the N-terminus of the first repeat, and the C-terminus of the second repeat, inside the cell, whereas in SbtA-6803 both termini appear to be periplasmic.
Phylogenetic diversity
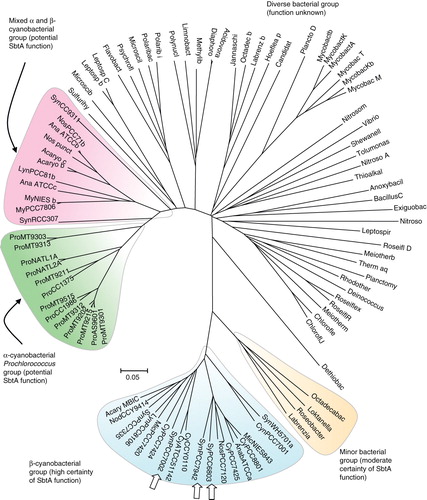
SbtA and related homologs are quite widespread within the cyanobacteria and a number of proteobacteria, but it appears that no easily identifiable homologs are present in the archaea. shows the relative relatedness of a 95-member family of SbtA protein homologs with alignment identity scores above 21%. Those 22 members with high probability of being functional SbtA HCO3 - transporters are present in two highlighted lobes at the bottom of the phylogenetic tree; these are comprised of 16 members from β-cyanobacteria (three of which are confirmed HCO3 - transporters from Synechocystis PCC6803, Synechococcus PCC7002 and Synechococcus PCC7942), two members from transition strains of α-cyanobacteria (Cyanobium PCC7001 and Synechococcus WH5701; freshwater /estuarine environments) and four members from some marine bacteria (Octadecabacter antarcticus 307, Loktanella vestfoldensis SKA53, Roseobacter sp. AzwK-3b, Labrenzia alexandrii DFL-11) that share 47–51% identity with SbtA-6803. Another lobe of interest is a more distant group of Prochlorococcus SbtA homologs (around 22–23% identity to SbtA-6803), although none of these have been confirmed to be functional HCO3 - transporters. Next to this lobe is a mixed group of homologs from the α- and β-cyanobacteria. The rest of the more distant homologs form a diverse group of members from a range of proteobacteria (). The 22 members in the set with a high probability of being functional SbtA transporter were subject to further analysis based on ClustalW alignments (Supplementary Figure 2, available online) and Weblogo representations (discussed below).
Figure 5. Phylogenetic tree showing the relationship of the extended SbtA family (95 prokaryotic members shown). Protein sequences were aligned using ClustalW and organized into a phylogenetic tree using nearest neighbour-joining routines in MEGA4 software (www.megasoftware.net). The scale marker represents 0.05 substitutions per residue. A list of defined abbreviations is available in Supplementary Table I, available online. This Figure is reproduced in colour in the online version of Molecular Membrane Biology.
Helical projections and buried charges
The determination of helical wheel projections and calculation of hydrophobic moments (HM), or determination of the most hydrophobic face of a helix, can provide a further test of how well the fine detail of the determined topology for SbtA-6803 fits expected tertiary structural principles for membrane transporter protein (Eisenberg et al. Citation1984, Brasseur Citation1988, Donnelly et al. Citation1993). In particular, there is an expectation that functional charge groups on each helix, potentially involved as substrate binding sites or charge-charge interlinks between TMHs, should be buried in the centre of the protein while the most hydrophobic faces should be lipid facing. shows the α-helical wheel projections for SbtA-6803. Seven of the helices, namely H2, H3, H5-H9, possess one or more charge groups, and significantly, all of these charges are on a face that is opposed to the most hydrophobic face, as determined by HM calculations (Eisenberg et al. Citation1984, Brasseur Citation1988, Donnelly et al. Citation1993) – see Methods. Furthermore, H3, H7 and H9 have the most hydrophobic helical faces in SbtA and are highly likely to be lipid facing. Many of the charge groups are also highly conserved ( and ) and may therefore play a functional role in transport.
Figure 6. Alpha-helical wheel projections of the ten TMH helices from SbtA-6803. The N-terminal residue in each helix is located at 12 o'clock position each following residue precedes 100° clockwise. A central arrowhead indicates the face with the highest hydrophobic moment (HM); charge residues are highlighted with a star symbol. Hydrophilic (circles), hydrophobic (diamonds), negatively charged (triangles) and positively charged (pentagons) residues are shown. This Figure is reproduced in colour in the online version of Molecular Membrane Biology.
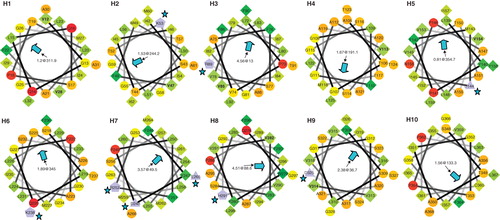
Figure 7. Weblogo representations of aligned helices 1–10 from the 22 member set of high probability SbtA homologs () and including the cytoplasmic loops H1–H2 and H9–H10 in the bottom panel. Conserved charge residues are highlighted with grey (yellow) arrowheads. Conserved S/T residues are indicated with asterisks (bottom panel). This Figure is reproduced in colour in the online version of Molecular Membrane Biology.

Putative regulatory domains
The topology of SbtA-6803 can also allow cytoplasmic domains involved in putative regulatory processes, such as phosphorylation, to be identified for further analysis. These regions are of particular importance in our goal of discovering how cyanobacterial HCO3 - transporters are activated on transition from darkness to illuminated conditions, and how such information can be used to adapt active HCO3 - transporters to the higher plant chloroplast. Of the five cytoplasmic TMH domains identified (H1-H2, H3-H4, H5-H6, H7-H8 and H9-H10) the 10-residue H1-H2 loop contains a conserved S/T34 residue in the high likelihood set of 22 SbtA/SbtA-like members (, bottom panel); this could potentially be a target for Ser/Thr protein kinase, and interestingly this S/T residue is predominantly conserved in the 95-member alignment (not shown). The 20-residue H9-H10 loop is potentially more interesting as it contains several conserved residues, T331, S338, S342 and Y244, that could be potential kinase targets (, bottom panel, marked with asterisks). S338 (red arrowhead) is particularly interesting in that this residue is present as a Glu in the four closely related bacteria. The possibility that S338 is a residue that is involved in dark inactivation is something we wish to pursue in future studies. The large H5–6 loop (59 residues in SbtA-6803) at the junction of the 5 + 5 structure was initially of interest as a potential regulatory domain, however this region is highly variable and is essentially missing in Prochlorococcus forms of SbtA, including the SbtA forms from two transition strains of α-cyanobacteria (Cyanobium PCC7001 and Synechococcus WH5701; Supplementary Figure 2, available online). This loop could still play a potential role in protein:protein interactions, although the loop is not highly conserved. Notwithstanding, at least two cytoplasmic loops harbour potential regulatory domains and these regions will be subjected to site-directed mutagenesis analysis in future studies.
Supplemental Material
Download PDF (293.4 KB)Acknowledgements
We thank Naresh Verma's lab for the gift of the pNV1216 plasmid.
Declaration of interest: This work was supported by an Australian Research Council Discovery grant to GDP, number DP0984773; we thank Loraine Tucker and Soumi Bala for technical support. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Abramson J, Wright EM. 2009. Structure and function of Na+-symporters with inverted repeats [Review]. Curr Opin Struct Biol 19:425–432.
- Alexeyev MF, Winkler HH. 1999. Membrane topology of the Rickettsia prowazekii ATP/ADP translocase revealed by novel dual pho-lac reporters. J Mol Biol 285:1503–1513.
- Badger MR, Price GD, Long BM, Woodger FJ. 2006. The environmental plasticity and ecological genomics of the cyanobacterial CO2 concentrating mechanism. J Exp Bot 57:249–265.
- Bernsel A, Viklund H, Falk J, Lindahl E, Von Heijne G, Elofsson A. 2008. Prediction of membrane-protein topology from first principles. Proc Nat Acad Sci USA 105:7177–7181.
- Brasseur R. 1988. Calculation of the three-dimensional structure of Saccharomyces cerevisiae cytochrome b inserted in a lipid matrix. J Biol Chem 263:12571–12575.
- Domingues L, Teixeira JA, Lima N. 1997. Rapid and sensitive detection of beta-galactosidase-producing yeasts by using microtiter plate assay. Biotechnol Techniques 11:399–402.
- Donnelly D, Overington JP, Ruffle SV, Nugent JH, Blundell TL. 1993. Modeling alpha-helical transmembrane domains: The calculation and use of substitution tables for lipid-facing residues. Protein Sci 2:55–70.
- Dowhan W, Bogdanov M. 2009. Lipid-dependent membrane protein topogenesis. Ann Rev Biochem 78:515–540.
- Eisenberg D, Weiss RM, Terwilliger TC. 1984. The hydrophobic moment detects periodicity in protein hydrophobicity. Proc Natl Acad Sci USA 81:140–144.
- Field CB, Behrenfeld MJ, Randerson JT, Falkowski P. 1998. Primary production of the biosphere – integrating terrestrial and oceanic components. Science 281:237–240.
- Jones DT. 2007. Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 23:538–544.
- Jung H, Rubenhagen R, Tebbe S, Leifker K, Tholema N, Quick M, Schmid R. 1998. Topology of the Na+/Proline transporter of Escherichia coli. J Biolog Chem 273:26400–26407.
- Krishnamurthy H, Piscitelli CL, Gouaux E. 2009. Unlocking the molecular secrets of sodium-coupled transporters [Review]. Nature 459:347–355.
- Lehane AM, Korres H, Verma NK, 2005. Bacteriophage-encoded glucosyltransferase GtrII of Shigella flexneri: membrane topology and identification of critical residues. Biochem J 389:137–143.
- Liu HB, Nolla HA, Campbell L. 1997. Prochlorococcus growth rate and contribution to primary production in the equatorial and subtropical North Pacific Ocean. Aquatic Microb Ecol 12:39–47.
- Price GD, Badger MR, Woodger FJ, Long BM. 2008. Advances in understanding the cyanobacterial CO2-concentrating-mechanism (CCM): functional components, Ci transporters, diversity, genetic regulation and prospects for engineering into plants. J Exp Bot 59:1441–1461.
- Price GD, Woodger FJ, Badger MR, Howitt SM, Tucker L. 2004. Identification of a SulP-type bicarbonate transporter in marine cyanobacteria. Proc Natl Acad Sci USA 101:18228–18233.
- Shelden MC, Howitt SM, Price GD, 2010. Membrane topology of the cyanobacterial bicarbonate transporter, BicA, a member of the SulP (SLC26A) family. Mol Membr Biol 27:12–23.
- Shibata M, Katoh H, Sonoda M, Ohkawa H, Shimoyama M, Fukuzawa H, Kaplan A, Ogawa T. 2002. Genes essential to sodium-dependent bicarbonate transport in cyanobacteria – function and phylogenetic analysis. J Biol Chem 277:18658–18664.
- Van Geest M, Lolkema JS. 2000. Membrane topology and insertion of membrane proteins: Search for topogenic signals [Review]. Micro Mol Biol Rev 64:13–33.
- Viklund H, Elofsson A. 2004. Best alpha-helical transmembrane protein topology predictions are achieved using hidden Markov models and evolutionary information. Protein Sci 13:1908–1917.
- Von Heijne G. 1992. Membrane-protein structure prediction – hydrophobicity analysis and the positive-inside rule. J Mol Biol 225:487–494.
- Von Rozycki T, Schultzel MA, Saier MH Jr. 2004. Sequence analyses of cyanobacterial bicarbonate transporters and their homologues. J Mol Microbiol Biotechnol 7:102–108.
- Zhang PP, Battchikova N, Jansen T, Appel J, Ogawa T, Aro EM. 2004. Expression and functional roles of the two distinct NDH-1 complexes and the carbon acquisition complex NdhD3/NdhF3/CupA/Sll1735 in Synechocystis sp PCC 6803. Plant Cell 16:3326–3340.