Abstract
Sorting of membrane proteins in eukaryotic cells is a complex yet vital task that involves several 10,000 molecular players. Sorting takes place not only along the early secretory pathway, i.e., between the endoplasmic reticulum and the Golgi apparatus, but also between other organelles, including exchange with the cell's plasma membrane. Traditionally, specific binary interactions between proteins have been made responsible for most of the protein sorting. A more active role of lipids, however, became visible in recent years. Not only do lipids in complex membranes show domain formation that may support/suppress sorting events, but also collective, membrane-mediated interactions have emerged as a robust physico-chemical mechanism to drive protein sorting. Here, we will review recent insights into these aspects.
Introduction
Membranes are the defining envelopes of all living cells, and they control signal and mass exchange between the intra- and extracellular space. Membranes also play a vital role in the compartmentalization of eukaryotic cells (Alberts Citation2008). Being divided into organelles, eukaryotes need to ship material between separate reaction vessels to maintain vital functions, e.g., between the endoplasmic reticulum (ER) and the Golgi apparatus in the so-called early secretory pathway (see for a schematic representation). After translation at ER-attached ribosomes, virtually all nascent membrane proteins translocate into the ER through complex pores, the translocons (Rapoport Citation2007). The (driven) passage of a polypeptide chain through these translocons can have strongly anomalous characteristics (Kapahnke et al. Citation2010) and transport rates may be facilitated by ER-resident chaperones (Ambjornsson and Metzler Citation2004). In the ER, nascent proteins interact with members of the quality control machinery, e.g., with the calnexin/calreticulin cycle (Ellgaard and Helenius Citation2003), before properly folded proteins are allowed to exit the ER. Exiting the ER happens at so-called ER exit sites at which COPII coat proteins form small (i.e., 50 nm-sized) vesicles that ship cargo proteins to the Golgi apparatus (Mancias and Goldberg Citation2005). Within the Golgi apparatus, proteins are further sorted and are subject to anterograde and/or retrograde (COPI coat-mediated) transport into distinct cisternae (Glick and Nakano Citation2009). As a consequence of this complex sorting process, some proteins simply go through the ER and the Golgi apparatus and acquire necessary post-translational modifications whereas other proteins show non-uniform stationary distributions across the early secretory pathway that may be peaked, for example, in specific cisternae of the Golgi apparatus (Nilsson et al. Citation1993a, Rabouille et al. Citation1995, Rottger et al. Citation1998), cf. .
Figure 1. Schematic representation of the early secretory pathway. Nascent proteins are sorted into emerging COPII vesicles after clearing the quality control mechanisms in the endoplasmic reticulum (ER). COPII vesicles mediate anterograde transport via the ER-to-Golgi intermediate compartment (ERGIC) an eventually deliver their cargo at the cis-face of the Golgi apparatus. Here, proteins are either sorted to stay in the stack of membrane cisternae (called cis, medial and trans cisternae) and undergo further anterograde transport towards the trans-Golgi network (TGN), or they are marked to participate in retrograde transport via COPI vesicles. As shown by electron microscopy, many proteins feature non-uniform steady-state distributions along the secretory pathway, indicated here by overlapping color-coded distributions below the organelles. This Figure is reproduced in colour in the online version of Molecular Membrane Biology.
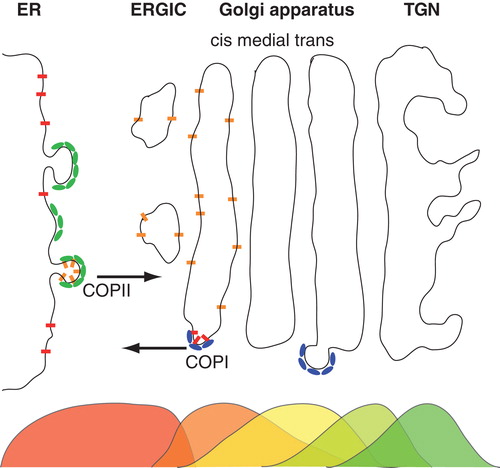
It is worth noting at this point, that not only transmembrane proteins are sorted and processed along the early secretory pathway. Also important morphogens like Wnt/Wg, which are initially soluble proteins, acquire one or more lipid anchors in the ER and travel subsequently along the secretory pathway (Bartscherer and Boutros Citation2008). Upon reaching the plasma membrane, they are secreted into the extracellular space.
Sorting processes on membranes, in particular within the early secretory pathway, have fascinated legions of researchers owing to the robustness and remarkable accuracy of the process in spite of a large number of involved proteins. At the advent of modern molecular biology, several important amino acid motifs have been uncovered that mediate specific interactions between cargo proteins and specific coat molecules which mediate long-range transport via the formation of vesicles. The most prominent of these sorting motifs are the cytoplasmic KDEL (Munro and Pelham Citation1987) and KKXX (Nilsson et al. Citation1989) motifs that promote retrograde transport (from the Golgi to the ER) via COPI-derived vesicles (Glick and Nakano Citation2009). Despite this major step forward, an understanding of the differential layers of sorting in terms of basic physico-chemical events prior to the interaction with coat molecules has remained controversial (see, e.g., Discussion in (Munro Citation1995)). A renewed interest in these events has emerged recently, driven by the observation that even protein-devoid lipid membranes can show rich phenomena like segregation events (Veatch and Keller Citation2005). Hence, the passive role of lipids in sorting events has been updated to a more active role. Here, we will review some recent advances in understanding protein sorting via lipid and membrane-mediated interactions and how these data fit into the larger context of protein trafficking in living cells.
Domains on (bio)membranes and diffusion-mediated protein partitioning
Classical textbook descriptions of biomembranes as two-dimensional fluids with few embedded proteins date back to the early 1970s when the ‘fluid mosaic model' was introduced (Singer and Nicolson Citation1972). By now, it has become clear that cellular membranes are more mosaic than fluid (Engelman Citation2005) due to a high density of proteins (about 50,000/μm2) and their manifold interactions with a plethora of different lipid species. Hence, also domain formation and segregation events are anticipated features of cellular membranes (Karnovsky et al. Citation1982). A prominent hypothesis along these lines is associated with the term ‘raft' (Simons and Ikonen Citation1997), i.e., the formation of functional protein-lipid complexes that have been assigned several important tasks in endo-/exocytosis and signalling. The original definition of raft domains as detergent-insoluble fractions, however, is vague and was a matter of heated discussions. More detailed biophysical analyses of putative rafts, e.g., by quantifying the nanoscale diffusion of lipids (Eggeling et al. Citation2009), have revealed that the size, mobility, composition, and stability of these domains most likely is very variable, hence challenging the original concept of stable rafts. Recently, a refined and more complex definition of the term ‘raft' has emerged (Pike Citation2006). Despite remaining uncertainties about rafts, the concept of inhomogeneous cellular membranes has been strongly supported by observations on binary and ternary lipid mixtures in vitro, e.g., in giant unilamellar vesicles (GUVs). Although lipid-lipid interactions alone are insufficient to explain the formation of functional cellular domains, these simplified artificial systems have served as valuable model systems that highlight the role of lipids during domain formation.
Phase separation in binary lipid mixtures is observed in the temperature interval between the chain melting transitions of the respective lipids, or in the gel phase in case the two species exhibit different packing properties. Typical mixtures involve two phosphatidylcholine (PC) species of different chain length, a PC-PE (PE: phosphatidylethanolamine) mixture or a PC mixture with a natural lipid such as brain ceramide. Evidence for a separation of domains was found via differential scanning calorimetry, X-ray scattering, and NMR experiments (Recktenwald and McConnell Citation1981, Koynova and Caffrey Citation2002). More recently, phases in binary mixtures also have been observed directly on giant unilamellar vesicles (GUVs) by fluorescence microscopy experiments (Fidorra et al. Citation2009). Direct visualization of domains via fluorescence microscopy experiments allows one to determine the shape of the domain, which reports on the interplay of line tension, lateral tension, bending stiffness, and curvature. For binary mixtures, gel domains are observed in a surrounding fluid phase and the domains can take on hexagonal, rhombic, six-cornered star, dumbbell, dendritic, and striped shapes (Bagatolli and Gratton Citation2000). In cells, however, domains are assumed to not assume a gel-like phase, i.e., many of the results on binary mixtures have been neglected as unphysiological.
Introducing a third lipid species alters the fluidity of existing domains without changing their shape (Bagatolli and Gratton Citation1999, Korlach et al. Citation1999). It may even induce a new set of phases (Veatch and Keller Citation2005). Commonly, ternary mixtures composed of a low melting lipid (e.g., an unsaturated or a short-chain saturated PC), a high melting lipid (e.g., sphingomyelin or ceramide), and cholesterol have been studied. In these systems, the phase behavior depends on the difference in melting transition temperatures. Frequently, a coexistence of two fluid phases, a liquid ordered and a liquid disordered phase, have been observed, e.g. via fluorescence microscopy on GUVs (Veatch and Keller Citation2005). These phases assume circular shapes to minimize the contact boundary and the related line tension. Right after preparation, a high number of very small, liquid-ordered domains emerge. These domains diffuse and coalesce upon contact, eventually forming a single domain, except in cases when small domains are stabilized by curvature effects (Baumgart et al. Citation2003, Semrau et al. Citation2009).
Liquid-ordered phases are strongly enriched in the high-melting lipid and slightly enriched in cholesterol, whereas liquid-disordered phases are enriched in low-melting lipids. Already few percent of the low-melting component are sufficient to induce liquid immiscibility. Similarly, small amounts of impurities in ternary mixtures, e.g., proteins or other lipids, can alter the miscibility transition temperature by several Kelvin. Impurities, e.g., proteins or labeled lipids, that partition equally into both liquid phases will lower the transition temperature while impurities that prefer one of the liquid phases will increase it. Even if ternary mixtures do not show a macroscopic phase separation still domains on the nanoscale may be present (Heberle et al. Citation2010).
Notably, gel phases but also liquid ordered and liquid disordered phases differ somewhat in thickness. This may be regarded as the simplest read-out of an inhomogeneous membrane for membrane-associated proteins. If the membrane-spanning domain of a transmembrane protein is longer/shorter than the hydrophobic thickness of the surrounding bilayer, the protein will experience a positive/negative hydrophobic mismatch. Via diffusion, the protein may explore the inhomogeneous membrane and it will partition with a higher probability into a domain with the least hydrophobic mismatch. This behaviour not only has been predicted by elastic theories like the mattress model (Mouritsen and Bloom Citation1984) but also has been observed in vitro (Morein et al. Citation2000) and in vivo (Ronchi et al. Citation2008). It is worth noting at this point that diffusion-driven partitioning is a stochastic process, i.e., the protein does not actively search for a more adequate environment, but it randomly probes other regions. Mean dwell times in regions with a strong or a vanishing hydrophobic mismatch, however, will be different since moving across the phase boundary into a less favourable region comes at a cost in the free energy. The latter enters into the Boltzmann factors that determine the stationary partitioning pattern.
Considering diffusion on membranes, one has to bear in mind two particularities. First, we note that the trajectory of Brownian motion provides a compact search algorithm in two dimensions which renders the assumption of a reaction constant invalid (Benichou et al. Citation2010). Rather, reaction coefficients become time-dependent and fractal kinetics as well as reactant segregation can emerge (Kopelman Citation1988, Hellmann et al. Citation2011). Second, calculating the translational diffusion constant is a formidable problem in two dimensions. While in three dimensions the well-known Einstein-Stokes equation holds (D = kBT/(6πηR) with kBT, R, η being thermal energy, particle radius, and fluid viscosity), deriving an analogue expression for diffusion on membranes fails due to the famous Stokes paradox (Lamb Citation1932). The problem can be solved, however, when considering that all membranes are embedded in three-dimensional space with adjacent fluids above and below. On this basis, Saffman and Delbruck (Citation1975) were able to derive an approximate expression for small protein radii, D ∼ ln(ηmh/ηcR), with ηm and ηc being the viscosities of the membrane and the adjacent fluid. For large radii, i.e., for R>>ηmh/ηc, this formula breaks down and a scaling D∼1/R in analogy to the edgewise motion of a thin disk emerges (Hughes et al. Citation1981). Still, the Saffman-Delbruck result is applicable to the motion of single proteins and small/intermediate oligomers. The weak logarithmic dependence on the object's radius R, however, may yield diffusion constants for protein oligomers that are almost as large as those for single proteins. Only if additional obstructions are present in the membrane, a more pronounced size-dependent anomalous diffusion may be observed (Malchus and Weiss Citation2010).
Interestingly, the Saffman-Delbruck relation not only is a good theoretical description for the diffusion of transmembrane proteins (Guigas and Weiss Citation2006) but also for peripheral/monotopic proteins that only dip into one leaflet of the membrane (Morozova et al. Citation2011). It should also be mentioned in this context that the viscosities ηm and ηc are effective parameters. The apparent membrane viscosity clearly depends on the penetration depth of the protein into the bilayer (Morozova et al. Citation2011). But also ηc is an effective parameter as it summarizes all drag forces acting on the protein from regions outside the bilayer. A straightforward contribution certainly is the pure viscous drag of the adjacent fluid. However, if proteins are sufficiently dense in a membrane so that they start interacting with their soluble residues, this can contribute an additional friction to ηc.
Structure formation of proteins due to membrane-mediated interactions
In the above discussion proteins were assumed to explore a membrane by diffusion, and they partitioned into regions that provide the least hydrophobic mismatch. Hence, proteins only read out a pre-existing lipid structure. However, embedding proteins into a bilayer perturbs the local lipid environment so that also lipids read out the presence/absence of a protein. Indeed, several simulation studies have examined local bilayer deformations due to embedding transmembrane proteins (Venturoli et al. Citation2005, de Meyer et al. Citation2008, Schmidt et al. Citation2008). The major findings were a local thickening (thinning) of the bilayer if a transmembrane protein had a positive (negative) hydrophobic mismatch (), and a tilting of the protein with respect to the bilayer normal if the positive mismatch was too strong. These theoretical results are corroborated by recent experimental and computational data on peptides in artificial bilayers containing cholesterol (Kaiser et al. Citation2011).
Figure 2. Mismatch-induced clustering of transmembrane proteins. (A) Embedding a transmembrane protein into a homogenous lipid bilayer leads to a local reduction or increase of the membrane thickness h(r); radial distances r from the protein are given in units of the size of a single lipid, r0. Insets show model proteins with a negative/positive hydrophobic mismatch (red and yellow indicate hydrophilic and hydrophobic residues, respectively). (B) A local minimum in the potential of mean force (PMF) between two proteins with a hydrophobic mismatch (given in units of thermal energy kBT) indicates a bound state for small distances. The minimum of the PMF becomes deeper for an increasing mismatch (black to red to blue). For large separations of the proteins no interaction is seen, i.e. the PMF is approximately constant. Inset: The mean first passage time τ to leave the bound state, i.e., to hop out of the minimum of the PMF, increases with the strength of the hydrophobic mismatch. Here, n indicates the length of the transmembrane domain with n <5 denoting a negative and n >5 denoting a positive mismatch. (C) Snapshot of a simulated membrane (grey) with mismatch-induced clusters of transmembrane proteins (red); water is not shown for better visibility. Figure adapted from (Schmidt et al. Citation2008). This Figure is reproduced in colour in the online version of Molecular Membrane Biology.
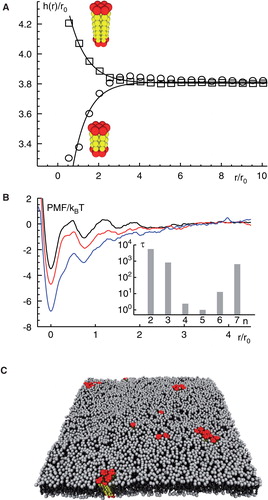
Changing the bilayer thickness was shown to come at a cost in the configuration of lipids near to the protein, i.e., an entropically unfavourable state emerged. Aiming at maximum entropy, the system will hence try to reduce the contact area between proteins and lipids which may result in the formation of clusters of transmembrane proteins (Schmidt et al. Citation2008). Therefore, local perturbations of the lipid bilayer can lead to a lipid-mediated attraction between transmembrane proteins if the mismatch is sufficiently strong. The net attraction between proteins can be quantified via the potential of mean force (PMF) (de Meyer et al. Citation2008, Schmidt et al. Citation2008, Morozova et al. Citation2011): The PMF is defined as the negative logarithm of the distance distribution (i.e., the pair correlation function) of two proteins, and it hence reports not only on attractive interactions due to specific binding events but also on collectively induced forces. The PMFs obtained for mismatched proteins shows a deep minimum at short inter-protein distances, indicating a bound state (). For large distances the potential becomes flat and featureless. Depth and width of the minimum of the PMF increase for enhanced positive and negative hydrophobic mismatches, so that binding energies of some 10 kBT can be reached (Schmidt et al. Citation2008). Calculating the mean first passage time from the PMF, i.e., solving the associated Kramer's problem for escaping the bound state, the lifetimes of the bound state can be estimated. These were seen to increase exponentially with increasing hydrophobic mismatching (Schmidt et al. Citation2008); cf. inset of .
Simulations of large ensembles of proteins further revealed that cluster formation correlated with the strength of the hydrophobic mismatch in that small mismatches only led to dimers and trimers whereas stronger mismatches yielded larger, long-lived assemblies (Schmidt and Weiss Citation2010); cf. . If proteins with different hydrophobic mismatch were mixed on a homogenous membrane, segregation into homo-oligomers was observed if the difference in mismatch was sufficiently strong (Schmidt and Weiss Citation2010). Proteins with similar mismatches formed hetero-oligomers. This indicates that ‘families' or ‘kins' of proteins with the same mismatch may group together to facilitate protein sorting.
Still, the segregation due to mismatching alone is a poor sorting strategy. Another layer of complexity can be achieved when considering that a number of transmembrane proteins are also acylated. While an additional lipid anchor seems to be irrelevant at first glance since the protein is membrane-bound already, one can show that an acylation alters the protein's tilting with respect to the bilayer normal, its partitioning behaviour, and eventually its ability to form mismatch-induced clusters (Morozova and Weiss Citation2010). These results compare favourably to experimental data on the transmembrane protein LRP6, for which an interplay between palmitoylation and length of the transmembrane domain was found to significantly affect the sorting and trafficking behaviour (Abrami et al. Citation2008). Hence, going beyond a mere hydrophobic mismatch, acylation may provide an efficient and reversible means to further exploit membrane perturbations for protein sorting.
Membrane-mediated interactions due to collective effects are not a mere speciality of transmembrane proteins. Also peripheral membrane proteins, sometimes also called monotopic proteins due to their association with only one leaflet of the bilayer, benefit from attractive interactions due to protein-induced changes in the local lipid environment (Morozova et al. Citation2011). Peripheral membrane proteins perturb their host leaflet in a similar way as transmembrane proteins altered the entire bilayer. There is a notable exception, though. Peripheral membranes can also perturb the opposing leaflet and the resulting changes in the leaflets can be quite different. Nevertheless, the consequence of lipid perturbation is again an entropy-driven, attractive net interaction that leads to an oligomerization. Binding energies can, depending on radius and penetration depth, be of 10 kBT or even more. Membrane-mediated cluster formation may happen within the same leaflet (in-plane oligomerization) but also cross-leaflet clusters can emerge ().
Figure 3. Structure formation of peripheral membrane proteins. (A) PMF for peripheral membrane proteins that reside in the same leaflet of a homogenous lipid bilayer. For large distances, proteins do not interact whereas for small distances a minimum in the PMF emerges that indicates a bound state. The binding strength increases for increasing penetration depths and radii of the proteins. (B) PMF for peripheral membrane proteins that reside in opposing leaflets of a homogenous lipid bilayer. In this case, proteins can form cross-leaflet dimers with a configuration and binding energy that depends again on the penetration depths and radii of the involved proteins. Figure adapted from (Morozova et al. Citation2011). This Figure is reproduced in colour in the online version of Molecular Membrane Biology.
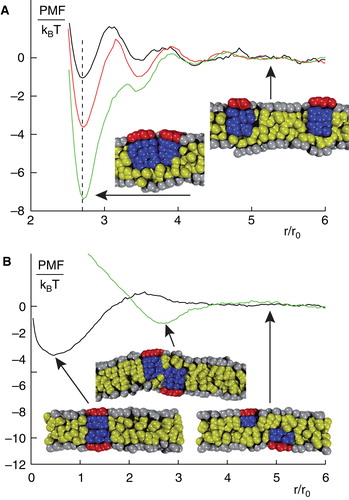
Cross-leaflet clusters are a very interesting phenomenon since they can mimic real transmembrane proteins. Even more striking, these effective transmembrane proteins can also form larger assemblies (Morozova et al. Citation2011). This observation challenges the common view that, for example, signal transduction across a membrane necessarily requires transmembrane proteins. Upon binding a ligand, a peripheral membrane protein may undergo a conformational change that makes it more prone to form a cross-leaflet dimer with an appropriate peripheral protein in the opposing leaflet. The resulting effective transmembrane protein assembly may even form higher-order clusters that act in much the same way as native transmembrane proteins. While some experimental evidence supports this view (Stuermer Citation2010), a clear-cut experimental test of this prediction is, as of yet, lacking.
Finally, we briefly would like to note that insertion/adsorption of proteins into/onto multi-component bilayers may induce lipid demixing. This scenario has been investigated theoretically (Sperotto and Mouritsen Citation1993) and in experiments with charged proteins that interact with bilayers composed of zwitterionic and charged lipids (Hartmann and Galla Citation1978, Gorbenko et al. Citation2009, Lai et al. Citation2011). In this case, proteins preferentially bind to charged lipids, therefore inducing a lateral segregation that creates a line tension (Mbamala et al. Citation2005). If the line tension is strong enough, proteins aggregate and induce macroscopic domains. A multi-pass membrane protein, lactose permease, reconstituted into POPG liposomes with different pyrene-labeled phospholipids analogs, has been shown to assemble lipids of similar hydrophobic length in its vicinity (Lehtonen and Kinnunen Citation1997). Thus, proteins not only follow a given template structure of lipids but also promote association of suitable lipids in their vicinity.
Protein traffic along the cell's secretory pathway
Having seen that membrane-mediated interactions can lead to a (transient) oligomerization of peripheral and transmembrane proteins, one may wonder if and how cells use these phenomena for sorting events, e.g., in the early secretory pathway. A long-standing hypothesis, which has been partially supported by experimental data (Mitra et al. Citation2004), is that organelle membranes along the secretory pathway show a slight increase in their thicknesses due to varying contents of cholesterol and sphingolipids. A gradient of membrane thickness therefore would mark directions for anterograde and retrograde transport along the secretory pathway. Based on the idea that ER membranes are thinner than Golgi membranes, and that these are thinner than the plasma membrane, an early hypothesis has predicted that the length of hydrophobic transmembrane domains determines the preferred localization of proteins (Bretscher and Munro Citation1993). While experimental data on transmembrane lengths were only few at that time, a more recent approach exploited the wealth of current protein data bases and strongly supported this claim (Sharpe et al. Citation2010). This result also gives indirect evidence for the validity of the idea of a membrane thickness gradient.
The claim of localizing to the best-matching membrane, however, requires not only a diffusive partitioning within the membrane of a single (sub)organelle, but it requires a means to explore and rate the properties of remote organelle membranes. Hence, entering transport intermediates, e.g., vesicles, and sensing bilayer properties need to be coupled to achieve a dynamic sorting scheme across different organelles. Indeed, the above described oligomerization of membrane proteins may be exploited for this very purpose ().
Figure 4. Model for sorting and trafficking facilitated by hydrophobic mismatching. If a membrane protein experiences a negative mismatch (lower left), e.g., in the ER, it will oligomerize and down-modulate the respective coat machinery, e.g., COPII. The emerging vesicle will transport the protein to a new compartment (middle right), e.g., the cis cisterna of the Golgi apparatus. Here, a slightly thicker membrane may extinguish cluster formation due to a lack of hydrophobic mismatching. As a consequence, proteins rarely leave this compartment (indicated by thinner arrows). If the protein experiences a positive mismatch (e.g., during a cisternal maturation process in the Golgi apparatus or after having left the best-matching bilayer by mistake), the protein will again start to form cluster that again will stabilize coat molecules at this locus (e.g., COPI at this time) which facilitates a retrieval to the better matching bilayer. This Figure is reproduced in colour in the online version of Molecular Membrane Biology.
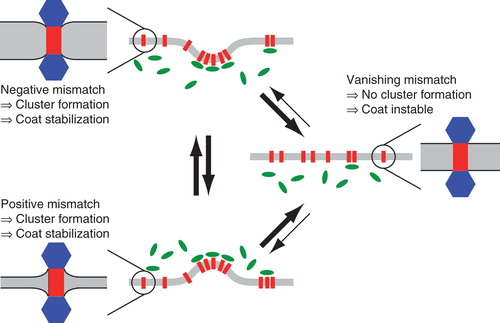
Let us assume that a nascent transmembrane protein, e.g., a Golgi-resident glycosylation enzyme, experiences a positive mismatch in ER membranes after having passed the ER quality control for proper folding. It will hence form mismatch-induced clusters that can down-modulate the local turnover rate of COPII proteins (Forster et al. Citation2006) to facilitate the formation of a COPII vesicle. The emerging vesicle eventually will carry the protein cluster to the so-called cis-face of the Golgi apparatus. Upon fusion of the vesicle with the cis-most cisterna of the Golgi apparatus, the protein cluster enters a new, slightly thicker membrane that may be just fine in terms of hydrophobic mismatching. When the cisterna grows thicker in time due to lipid metabolism (‘maturation'), the proteins will experience a negative hydrophobic mismatch. Again they will form mismatch-induced clusters and now will down-modulate the COPI vesicle machinery (Elsner et al. Citation2003) to stimulate a budding process that enables retrograde transport of the protein to another compartment with a more appropriate lipid composition. Much like the diffusive partitioning, also here the search process is dynamic and stochastic, yet it leads to an increased residence time in the compartment with the best-matching membrane due to modulation of the transport rates (cf. arrows of different thickness in ).
It is worth noting, that cisternal maturation is not a necessary ingredient for the outlined mechanism. If a bi-directional vesicular transport model is used instead of a maturation model, some proteins may accidentally escape their preferred compartment (e.g., the cis-most cisterna) and therefore end up in the less favourable medial or trans cisterna. Also in this case, mismatch-induced clusters will stimulate the making of well-filled COPI vesicles that retrieve the mislocalized proteins to the better matching membrane.
Indeed, the described trafficking scenario combines two earlier models: The first suggested that proteins ‘measure' the bilayer thickness and localize to the best-matching bilayer (Bretscher and Munro Citation1993) while the second suggested the formation of ‘kin oligomers' that form (transient) clusters to trigger a retrieval (Nilsson et al. Citation1993b). Since both suggestions have found experimental support, the synthesis via mismatch-induced clustering is an attractive synthesis of both models.
Perspective
As virtually all reviews on a topical and dynamically developing area of research, also the present manuscript surely is incomplete in referring all important aspects of mechanisms of protein sorting. Still, the outlined recent developments point a few directions along which further progress may develop within the next few years.
First, appropriate in vitro systems like GUVs and supported bilayers with reconstituted proteins and potentially asymmetric lipid bilayers will provide valuable systems with which some of the collective effects between lipids and proteins may be quantified within a relevant bio-mimetic setup. This approach will certainly benefit from recent developments in advanced light microscopy techniques that allow one to assess molecular features with a previously unknown spatiotemporal precision.
Second, simulation techniques and evolving computer power certainly will continue to deliver refined insights that will fuel and guide additional experiments. Not only have well-defined coarse-grained molecular dynamics approaches been developed in recent years, but also multi-scale approaches that bridge the gap between the nano- and the micron-world are currently being developed.
Third, concepts that have emerged from artificial membrane systems and simulations facilitate the choice of experiments to be done in vivo. Molecular biology, including silencing approaches, is by now well standardized so that fluorescently labeled and mutated proteins can be expressed in a variety of cells and organisms at will. Relying again on dynamic features that can be assessed by light microscopy techniques, these systems will be the ultimate test for any proposed physio-chemical mechanism of protein sorting.
Declaration of interest: Financial support by DFG grant WE4335/2-1 is gratefully acknowledged. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Abrami L, Kunz B, Iacovache I, Van Der Goot FG. 2008. Palmitoylation and ubiquitination regulate exit of the Wnt signaling protein LRP6 from the endoplasmic reticulum. Proc Natl Acad Sci USA 105:5384–5389.
- Alberts B. 2008. Molecular biology of the cell. New York: Garland Science.
- Ambjornsson T, Metzler R. 2004. Chaperone-assisted translocation. Phys Biol 1:77–88.
- Bagatolli LA, Gratton E. 1999. Two-photon fluorescence microscopy observation of shape changes at the phase transition in phospholipid giant unilamellar vesicles. Biophys J 77:2090–2101.
- Bagatolli LA, Gratton E. 2000. Two photon fluorescence microscopy of coexisting lipid domains in giant unilamellar vesicles of binary phospholipid mixtures. Biophys J 78:290–305.
- Bartscherer K, Boutros M. 2008. Regulation of Wnt protein secretion and its role in gradient formation. EMBO Rep 9:977–982.
- Baumgart T, Hess ST, Webb WW. 2003. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature 425:821–824.
- Benichou O, Chevalier C, Klafter J, Meyer B, Voituriez R. 2010. Geometry-controlled kinetics. Nat Chem 2:472–477.
- Bretscher MS, Munro S. 1993. Cholesterol and the Golgi-Apparatus. Science 261:1280–1281.
- De Meyer FJ, Venturoli M, Smit B. 2008. Molecular simulations of lipid-mediated protein-protein interactions. Biophys J 95:1851–1865.
- Eggeling C, Ringemann C, Medda R, Schwarzmann G, Sandhoff K, Polyakova S, 2009. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature 457:1159–1162.
- Ellgaard L, Helenius A. 2003. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 4:181–191.
- Elsner M, Hashimoto H, Simpson JC, Cassel D, Nilsson T, Weiss M. 2003. Spatiotemporal dynamics of the COPI vesicle machinery. EMBO Rep 4:1000–1004.
- Engelman DM. 2005. Membranes are more mosaic than fluid. Nature 438:578–580.
- Fidorra M, Garcia A, Ipsen JH, Hartel S, Bagatolli LA. 2009. Lipid domains in giant unilamellar vesicles and their correspondence with equilibrium thermodynamic phases: A quantitative fluorescence microscopy imaging approach. Biochim Biophys Acta 1788:2142–2149.
- Forster R, Weiss M, Zimmermann T, Reynaud EG, Verissimo F, Stephens DJ, 2006. Secretory cargo regulates the turnover of COPII subunits at single ER exit sites. Curr Biol 16:173–179.
- Glick BS, Nakano A. 2009. Membrane traffic within the Golgi apparatus. Annu Rev Cell Dev Biol 25:113–132.
- Gorbenko GP, Trusova VM, Molotkovsky JG, Kinnunen PK. 2009. Cytochrome c induces lipid demixing in weakly charged phosphatidylcholine/phosphatidylglycerol model membranes as evidenced by resonance energy transfer. Biochim Biophys Acta 1788:1358–1365.
- Guigas G, Weiss M. 2006. Size-dependent diffusion of membrane inclusions. Biophys J 91:2393–2398.
- Hartmann W, Galla HJ. 1978. Binding of polylysine to charged bilayer membranes: Molecular organization of a lipid-peptide complex. Biochim Biophys Acta 509:474–490.
- Heberle FA., Wu J, Goh SL, Petruzielo RS, Feigenson GW. 2010. Comparison of three ternary lipid bilayer mixtures: FRET and ESR reveal nanodomains. Biophys J 99:3309–3318.
- Hellmann M, Heermann DW, Weiss M. 2011. Anomalous reaction kinetics and domain formation on crowded membranes. Epl 94.
- Hughes BD, Pailthorpe BA, White LR. 1981. The translational and rotational drag on a cylinder moving in a membrane. J Fluid Mechan 110:349–372.
- Kaiser HJ, Orlowski A, Rog T, Nyholm TK, Chai W, Feizi T, 2011. Lateral sorting in model membranes by cholesterol-mediated hydrophobic matching. Proc Natl Acad Sci USA 108:16628–16633.
- Kapahnke F, Schmidt U, Heermann DW, Weiss M. 2010. Polymer translocation through a nanopore: The effect of solvent conditions. J Chem Phys 132:164904.
- Karnovsky MJ, Kleinfeld AM, Hoover RL, Klausner RD. 1982. The concept of lipid domains in membranes. J Cell Biol 94:1–6.
- Kopelman R. 1988. Fractal reaction-kinetics. Science 241:1620–1626.
- Korlach J, Schwille P, Webb WW, Feigenson GW. 1999. Characterization of lipid bilayer phases by confocal microscopy and fluorescence correlation spectroscopy. Proc Natl Acad Sci USA 96:8461–8466.
- Koynova R, Caffrey M. 2002. An index of lipid phase diagrams. Chem Phys Lipids 115:107–219.
- Lai AL, Tamm LK, Ellena JF, Cafiso DS. 2011. Synaptotagmin 1 modulates lipid acyl chain order in lipid bilayers by demixing phosphatidylserine. J Biol Chem 286:25291–25300.
- Lamb H. 1932. Hydrodynamics. Cambridge, UK: The University Press.
- Lehtonen JY, Kinnunen PK. 1997. Evidence for phospholipid microdomain formation in liquid crystalline liposomes reconstituted with Escherichia coli lactose permease. Biophys J 72:1247–1257.
- Malchus N, Weiss M. 2010. Anomalous diffusion reports on the interaction of misfolded proteins with the quality control machinery in the endoplasmic reticulum. Biophys J 99:1321–1328.
- Mancias JD, Goldberg J. 2005. Exiting the endoplasmic reticulum. Traffic 6:278–285.
- Mbamala EC, Ben-Shaul A, May S. 2005. Domain formation induced by the adsorption of charged proteins on mixed lipid membranes. Biophys J 88:1702–1714.
- Mitra K, Ubarretxena-Belandia I, Taguchi T, Warren G, Engelman DM. 2004. Modulation of the bilayer thickness of exocytic pathway membranes by membrane proteins rather than cholesterol. Proc Natl Acad Sci USA 101:4083–4088.
- Morein S, Koeppe IR, Lindblom G, De Kruijff B, Killian JA. 2000. The effect of peptide/lipid hydrophobic mismatch on the phase behavior of model membranes mimicking the lipid composition in Escherichia coli membranes. Biophys J 78:2475–2485.
- Morozova D, Guigas G, Weiss M. 2011. Dynamic structure formation of peripheral membrane proteins. Plos Computat Biol 7:e1002067. doi:10.1371/journal.pcbi.1002067
- Morozova D, Weiss M. 2010. On the role of acylation of transmembrane proteins. Biophys J 98:800–804.
- Mouritsen OG, Bloom M. 1984. Mattress model of lipid-protein interactions in membranes. Biophys J 46:141–153.
- Munro S. 1995. An investigation of the role of transmembrane domains in Golgi protein retention. Embo J 14:4695–4704.
- Munro S, Pelham HRB. 1987. A C-terminal signal prevents secretion of luminal Er proteins. Cell 48:899–907.
- Nilsson T, Jackson M, Peterson PA. 1989. Short cytoplasmic sequences serve as retention signals for transmembrane proteins in the endoplasmic-reticulum. Cell 58:707–718.
- Nilsson T, Pypaert M, Hoe MH, Slusarewicz P, Berger EG, Warren G. 1993a. Overlapping distribution of 2 Glycosyltransferases in the Golgi-apparatus of HeLa cells. J Cell Biol 120:5–13.
- Nilsson T, Slusarewicz P, Hoe MH, Warren G. 1993b. Kin recognition – a model for the retention of Golgi enzymes. Febs Lett 330:1–4.
- Pike LJ. 2006. Rafts defined: A report on the Keystone symposium on lipid rafts and cell function. J Lipid Res 47:1597–1598.
- Rabouille C, Hui N, Hunte F, Kieckbusch R, Berger EG, Warren G, 1995. Mapping the distribution of Golgi enzymes involved in the construction of complex oligosaccharides. J Cell Sci 108:1617–1627.
- Rapoport TA. 2007. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450:663–669.
- Recktenwald DJ, McConnell HM. 1981. Phase equilibria in binary mixtures of phosphatidylcholine and cholesterol. Biochemistry 20:4505–4510.
- Ronchi P, Colombo S, Francolini M, Borgese N. 2008. Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J Cell Biol 181:105–118.
- Rottger S, White J, Wandall HH, Olivo JC, Stark A, Bennett EP, 1998. Localization of three human polypeptide GalNAc-transferases in HeLa cells suggests initiation of O-linked glycosylation throughout the Golgi apparatus. J Cell Sci 111:45–60.
- Saffman PG, Delbruck M. 1975. Brownian-motion in biological-membranes. Proc Natl Acad Sci USA 72:3111–3113.
- Schmidt U, Guigas G, Weiss M. 2008. Cluster formation of transmembrane proteins due to hydrophobic mismatching. Phys Rev Lett 101:128104.
- Schmidt U, Weiss M. 2010. Hydrophobic mismatch-induced clustering as a primer for protein sorting in the secretory pathway. Biophys Chem 151:34–38.
- Semrau S, Idema T, Schmidt T, Storm C. 2009. Membrane-mediated interactions measured using membrane domains. Biophys J 96:4906–4915.
- Sharpe HJ, Stevens TJ, Munro S. 2010. A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 142:158–169.
- Simons K, Ikonen E. 1997. Functional rafts in cell membranes. Nature 387:569–572.
- Singer SJ, Nicolson GL. 1972. The fluid mosaic model of the structure of cell membranes. Science 175:720–731.
- Sperotto MM, Mouritsen OG. 1993. Lipid enrichment and selectivity of integral membrane proteins in two-component lipid bilayers. Eur Biophys J 22:323–328.
- Stuermer CA. 2010. The reggie/flotillin connection to growth. Trends Cell Biol 20:6–13.
- Veatch SL, Keller SL. 2005. Seeing spots: Complex phase behavior in simple membranes. Biochim Biophys Acta 1746:172–185.
- Venturoli M, Smit B, Sperotto MM. 2005. Simulation studies of protein-induced bilayer deformations, and lipid-induced protein tilting, on a mesoscopic model for lipid bilayers with embedded proteins. Biophys J 88:1778–1798.