Abstract
A number of recent studies revealed that epigenetic modifications play a central role in the regulation of lipid and of other metabolic pathways such as cholesterol homeostasis, bile acid synthesis, glucose and energy metabolism. Epigenetics refers to aspects of genome functions regulated in a DNA sequence-independent fashion. Chromatin structure is controlled by epigenetic mechanisms through DNA methylation and histone modifications. The main modifications are histone acetylation and deacetylation on specific lysine residues operated by two different classes of enzymes: Histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively. The interaction between these enzymes and histones can activate or repress gene transcription: Histone acetylation opens and activates chromatin, while deacetylation of histones and DNA methylation compact chromatin making it transcriptionally silent. The new evidences on the importance of HDACs in the regulation of lipid and other metabolic pathways will open new perspectives in the comprehension of the pathophysiology of metabolic disorders.
Introduction
In recent years the scientific community has witnessed a growing interest in epigenetics, as demonstrated by the increasing number of publications on this topic. Epigenetic modifications play a role in many DNA-related processes including transcription, recombination, DNA repair and replication. Epigenetic modifications also regulate several important physiological and pathological processes such as embryonic development, aging and cancer. However nowadays, the importance of epigenetics has been recognized in many other fields relevant to human health, such as inflammation, obesity, insulin resistance, type 2 diabetes mellitus, cardiovascular, neurodegenerative and immune diseases.
As many of these diseases are associated to lipid disorders, the purpose of this review is to outline the role of histone deacetylases (HDACs) in the regulation of lipid metabolism and of tightly related metabolic pathways. We will focus on the importance of HDACs as key regulators of the association of transcription factors and cofactors to target promoters of genes involved in the regulation of lipid and other metabolic pathways. To this end, we will briefly review the epigenetic mechanisms controlling bile acid synthesis, cholesterol metabolism, glucose homeostasis and energy metabolism.
We will discuss how bile acids, which are synthesized from cholesterol, act as signaling molecules, by promoting the association of several chromatin-remodeling factors and consequently modulating gene transcription. We will also analyze the recent results on the role of class II HDACs in glucose metabolism. Finally, we will present recent experimental evidences reporting that modifications of chromatin structure induced by class I HDACs affect energy metabolism and insulin sensitivity suggesting the epigenetic approach as a novel frontier in the treatment of disorders linked to altered lipid metabolic pathways.
Histone deacetylases as epigenetic regulators
Epigenetics can be defined as a set of processes and mechanisms that regulate gene activity, by modifying chromatin structure without changes in DNA sequence. Nucleosomes, the typical structure formed by histone in which the genomic DNA is folded in eukaryotic cells, are building blocks of the dynamic structure called chromatin. During activation of gene transcription, chromatin adopts a locally accessible and transcriptionally active form referred to as euchromatin, while highly condensed and transcriptionally less active genetic material is known as heterochromatin () (Richards and Elgin Citation2002).
Figure 1. Conformational changes of chromatin induced by histone modifications. On the left, euchromatin represents acetylated and transcriptionally active state. On the right, DNA methylation and histone deacetylation induce chromatin packaging (heterochromatin) making it less accessible to transcriptional factors.
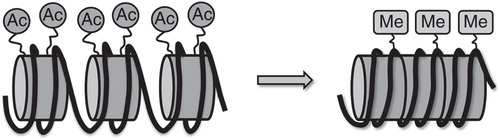
The main epigenetic modifications include DNA methylation and histone modifications (Vaillant and Paszkowski Citation2007). Amino acid residues on the histone tails are modified by post-translational acetylation, methylation, ADP-ribosylation and phosphorylation leading to significant changes in the chemical structure of the histone protein tails: Acetylation of lysine residues of histone tails carried out by HAT increases the distance between DNA and histones and the accessibility of transcription factors to gene promoter regions, while deacetylation by HDACs induces chromatin packaging and reduces the accessibility of transcription factors to local chromatin regions (López-Rodas et al. Citation1993). Mammalian HDACs are listed into four classes based on their homology with yeast HDACs () (Haberland et al. Citation2009).
Figure 2. Superfamily of mammalian histone deacetylases.
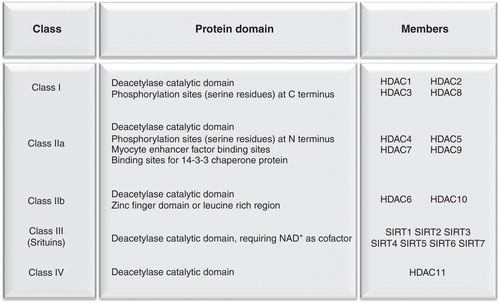
Class I HDACs are ubiquitously expressed and are mainly localized in the nuclear compartment where they exert the most relevant histone deacetylase activity. Class II HDACs localize in the cytoplasm, shuttling into the nucleus in response to specific cellular signals. Class IIa HDACs are characterized by post-translational regulation such as phosphorylation that determines their cytosolic localization, whereas dephosphorylation of Class IIa HDACs enables their translocation to the nucleus (Lu J et al. Citation2000, McKinsey et al. Citation2000, Passier et al. Citation2000, Vega et al. Citation2004). Class IIa HDACs feature only minimal histone deacetylase activity due to a swap of a key tyrosine residue in the catalytic domain with a histidine (Lahm et al. Citation2004); nonetheless, they act as scaffold molecules to recruit class I HDACs. Class IIb HDACs are localized in the cytoplasm and act mainly on non-histone substrates such as cytoskeletal and transmembrane proteins (Haberland et al. Citation2009). Class III HDACs, also known as sirtuins, are sensitive to changes in the intracellular NAD+/NADH ratio and they rely upon NAD+ hydrolysis for their deacetylase activity. In mammals, sirtuins regulate several functions, ranging from the control of cellular stress to energy metabolism (Finkel et al. Citation2009). Rodgers et al. (Citation2005) investigated the molecular mechanisms responsible for the adaptive metabolic response to fasting and found that SIRT1 is involved in the metabolic control in mammals. These authors showed that in the fasted state, SIRT1 is induced in the liver and deacetylates PPAR gamma coactivator 1α (PGC-1α) at specific lysine residues in a NAD+-dependent manner: Deacetylated PGC-1α is more active and transactivates the transcription of genes encoding enzymes of fatty acid β-oxidation and gluconeogenesis. In fact, knock-down of SIRT1 in the liver leads to reduced glucose production and fatty acid oxidation in the liver, under fasting conditions (Rodgers and Puigserver Citation2007). These studies demonstrate that SIRT1 regulates metabolic adaptation and that PGC-1α mediates most of SIRT1 effects. Finally, HDAC11 is the only member of class IV HDACs but its function is still poorly understood.
Epigenetics and its role in the regulation of lipid homeostasis and other metabolic pathways
Recently it has been reported that chromatin-remodeling events regulate metabolic homeostasis. In recent years, HDACs have been shown to regulate genes involved in bile acid biosynthesis. Bile acids are amphipathic metabolites of cholesterol that promote solubilization, transport and absorption of lipids. Bile acid biosynthesis represents the chief metabolic pathway to maintain cholesterol balance in mammals and it occurs in the liver through two different pathways: The classical or ‘neutral' pathway, exclusively hepatic, and the alternative or ‘acidic' pathway starting from precursors in endothelial cells, fibroblasts and macrophages. Under physiological conditions the classical pathway is predominant and leads to the formation of cholic and chenodeoxycholic acids through complex and highly regulated steps. Cholesterol 7α-hydroxylase (CYP7A1) is the major check-point and the rate limiting enzyme in this pathway catalyzing the hydroxylation of cholesterol at the 7α position and it has a deep impact on cholesterol homeostasis. Pullinger et al. (Citation2002) showed that patients bearing homozygous deletion mutation in CYP7A1, resulting in loss of the active site and enzyme function, develop hypercholesterolemia with high levels of LDL cholesterol.
This study underscores the critical role of CYP7A1 and bile acid synthesis in the maintenance of cholesterol homeostasis. The transcription of CYP7A1 gene is feedback regulated by bile acids. In liver, in fact, bile acids interact with their nuclear receptor farnesoid X receptor (FXR, NR1H4) inducing the expression of the orphan nuclear receptor small heterodimer partner (SHP, NR0B2), a transcriptional repressor that inhibits CYP7A1 transcription (Goodwin et al. Citation2000, Lu TT et al. Citation2000). Although the FXR pathway is responsible for most of the biological effects exerted by bile acids on gene transcription, loss of SHP impairs but does not eliminate completely the bile acid negative feedback on their own synthesis and particularly on CYP7A1 gene, as shown by analyzing SHP-deficient mice, suggesting also the involvement of additional mechanisms (Kerr et al. Citation2002, Wang et al. Citation2002). To corroborate these observations, we showed that bile acids repress the transactivation potential of hepatocyte nuclear factor 4α (HNF-4α, NR2A1) (De Fabiani et al. Citation2001), a potent CYP7A1 transcription activator, in a FXR/SHP-independent manner consequently inhibiting CYP7A1 expression and bile acid biosynthesis. In another study we also found that bile acids interfere with the recruitment of PGC-1α and CBP, two transcriptional coactivators, to CYP7A1 promoter (De Fabiani et al. Citation2003). These findings lead us to investigate chromatin dynamics in bile acid-sensitive regulatory cascades in the liver. By analyzing CYP7A1 promoter it has been shown that bile acid treatment remodels chromatin, making DNA less accessible to endonucleases in euchromatin (Boulias and Talianidis Citation2004, Kemper et al. Citation2004, Fang et al. Citation2007).
Subsequently we showed that the SHP-dependent mechanism on the regulation of CYP7A1 transcription does not occur as first response to bile acid treatment, because the kinetics of bile acid-induced SHP mRNA is delayed as compared to the reduction of CYP7A1 mRNA (Mitro et al. Citation2007). By analyzing chromatin remodeling elicited by bile acids, we observed dissociation of PCG-1α and cAMP response element binding protein-binding protein (CBP) from CYP7A1 promoter and recruitment of several corepressors after bile acid treatment in a liver cell line. More interestingly, we also found that bile acids affect chromatin acetylation state. Following bile acid treatment, sequential recruitment of HDAC 7, 3, and 1, and of the corepressors SMRTα and NCoR1 into the nucleus builds a repressive complex on CYP7A1 promoter that leads to HNF-4α export out of the nucleus and, ultimately, to the repression of CYP7A1 gene transcription ().
Figure 3. Model of feedback regulation of CYP7A1 by bile acids. Bile acids (BA) induce HDAC7 translocation from cytoplasm to nucleus and the sequential recruitment of HDAC7, 3, 1, SMRT and NCoR1 on CYP7A1 gene promoter. Histone deacetylation mediated by this repressor complex prevents the recruitment of transcriptional factors leading to CYP7A1 inhibition.
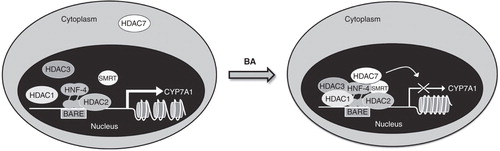
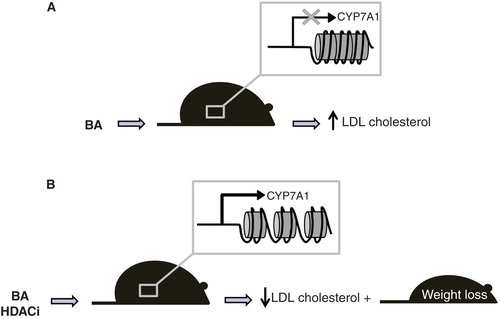
To better investigate the contribution of specific HDACs in this regulatory pathway, we used RNAi to silence the HDACs recruited in the repressor complex on the promoter of CYP7A1 and they showed that knock-down of HDAC7 prevents bile acid-mediated CYP7A1 transcription repression. Furthermore, we observed that bile acids promote HDAC7 translocation to the nucleus and that calyculin, a phosphatase inhibitor, causes HDAC7 retention in the cytoplasm that prevents the feedback inhibition of bile acids on CYP7A1 gene transcription. We also showed that valproic acid and tricostatin A, two non-selective HDAC inhibitors (HDACi), prevent the repressive effect exerted by bile acids on CYP7A1 mRNA levels in a liver cell line, increasing the expression of CYP7A1 also in vivo. Indeed, the administration of HDACi to LDL receptor deficient (Ldlr-/- ) mice, a genetic model of hypercholesterolemia, induces CYP7A1 expression along with increased bile acid biosynthesis and decreases serum cholesterol in the LDL-fraction () (Mitro et al. Citation2007). The effects of HDACi on bile acid synthesis and cholesterol homeostasis were accompanied by a significant loss of body weight (Mitro et al., unpublished work) (). These observations highlight the importance of epigenetics and of HDACs in the regulation of specific lipid metabolic pathways.
Figure 4. HDACi in Ldlr-/- mice induce CYP7A1 expression and decrease serum LDL-cholesterol and body weight. (A) Bile acids repress CYP7A1 expression leading to LDL cholesterol increase. (B) Treatment with HDACi derepresses CYP7A1 and increase conversion of cholesterol to bile acids, ultimately reducing LDL cholesterol and body weight.
Role of HDACs in hepatic lipid and glucose metabolism
In the liver the genes encoding the rate-limiting gluconeogenic enzymes glucose-6-phophatase (G6pc) and phosphoenolpyruvate carboxykinase (Pck1) are regulated by transcription factors such as cAMP response element binding protein (CREB), forkhead box protein O1 (FOXO1), hepatocyte nuclear factor 4 alpha (HNF4α), the glucocorticoid receptor (GR, NR3C1), CCATT/enhancer binding proteins (C/EBPs) and the coactivatorPGC-1α (Viollet et al. Citation2009). These proteins can be phosphorylated and consequently inactivated via two different pathways: Insulin signaling pathway (Matsumoto et al. Citation2007, Haeusler et al. Citation2010) and the LKB1/AMPK pathway (Shaw et al. Citation2005, Viollet et al. Citation2009, Cantó and Auwerx Citation2010).
Hepatic gluconeogenesis is also regulated by the acetylation of histones and transcription factors in response to fasting and feeding (Guarente Citation2006); in this context class IIa HDACs (HDAC4, 5, 7, and 9) play a peculiar role. These HDACs can be phosphorylated on specific conserved residues (Ser259 and Ser498 in human HDAC5) and consequently bound to 14-3-3 protein. This binding leads to the cytoplasmic sequestration of class IIa HDACs (reviewed in Haberland et al. Citation2009).
Recently, Mihaylova et al. (Citation2011) demonstrated that in the liver class IIa HDACs phosphorylation is modulated by LKB1-dependent kinases whereas they can be dephosphorylated in response to glucagon and consequently translocate into the nucleus. Also, class IIa HDACs associate with the G6pc and Pck1 promoters and recruit HDAC3 to gluconeogenic loci. Furthermore, they showed that in hepatocytes and liver depletion of class IIa HDACs via RNAi results in FOXO hyperacetylation. This hyperacetylation leads to decreased expression of FOXO target genes that results in the reduction of hyperglycemia in several mouse models of type 2 diabetes, highlighting the key role of HDACs in mammalian glucose homeostasis.
In another study, Knutson et al. (Citation2008) demonstrated that HDAC3 deletion disrupts normal lipid homeostasis. Liver specific Hdac3 knock outmice, in fact, show hepatomegaly as a result of hepatocyte hypertrophy, together with altered carbohydrate and lipid metabolism. The authors also observed a strong increase of hepatic and circulating triglycerides and cholesterol. The de-repression of several genes regulating lipid biosynthesis is due to increased expression of peroxisome proliferator-activated receptor γ2 (Pparγ2, NR1C3), which is consistent with previous reports showing that hepatic induction of Pparγ2 in a mouse model of obesity induces lipogenesis and liver steatosis (Yu et al. Citation2003, Zhang et al. Citation2006).
Recently Feng et al. (Citation2011) demonstrated that genomic recruitment of HDAC3 in the liver shows a circadian rhythm and is related to the expression pattern of the circadian nuclear receptor Rev-erbα. Rev-erbα recruits HDAC3 along with NCoR1 (Zamir et al. Citation1996, Yin and Lazar Citation2005), and they colocalize near genes involved in lipid metabolism. Deletion of HDAC3 or Rev-erbα in mouse liver causes hepatic steatosis, suggesting that Rev-erbα-dependent recruitment of HDAC3 regulates hepatic lipid homeostasis.
HDACs regulate energy metabolism
In recent years it has become clear that epigenetic changes, consequent to disruption of HAT or HDAC activity, are associated to several pathological conditions, such as cancer (Miremadi et al. Citation2007), insulin resistance and diabetes. One of the first evidences of the involvement of HDACs in the pathogenesis of diabetes is a genome wide analysis showing significant linkage of HDAC2 located in the region 6q21 with both type 1 and type 2 diabetes loci (Gray and Ekstrom Citation2001, Nerup and Pociot Citation2001, Xiang et al. Citation2004). No other mutations of HDACs have been so far reported, also for other diseases like cancer (Dawson and Kouzarides Citation2012) in which epigenetic regulation has been extensively studied in the last decade. Furthermore, several studies revealed that HDACs, in particular HDAC4 and HDAC5, regulate the expression of metabolic genes in skeletal muscle (Czubryt et al. Citation2003, Potthoff et al. Citation2007, McGee et al. Citation2008). Interestingly, a recent study by Gao et al. (Citation2009) emphasized the involvement of HDACs in energy expenditure. They reported that sodium butyrate, a short chain fatty acid often found in the diet that inhibits class I and class II HDAC activity, improves metabolic dysfunction in diet-induced obese mice. These authors observed that supplementation with sodium butyrate increases energy expenditure and fatty acid oxidation, and protects mice from high fat diet-induced insulin resistance. Sodium butyrate also enhances thermogenic function and reduces fat accumulation and size of brown adipocytes. In skeletal muscles sodium butyrate increases the number of oxidative fibers and enhances mitochondrial function. Indeed, in vivo butyrate is an activator of PGC-1α, the master regulator of mitochondrial biogenesis. Based on these results, the hypothesis is that butyrate, by inhibiting histone deacetylases, opens chromatin in the PGC-1α gene promoter thus inducing the transcription of this gene. However, butyrate is known to affect energy metabolism via other mechanisms (Donohoe et al. Citation2011), as it has been shown to affect adipogenesis via HDAC-independent mechanisms (Haberland et al. Citation2009). Therefore it will be necessary to perform ad hoc studies with more specific inhibitors to better address the role of HDACs in energy metabolism in the context of obesity and type 2 diabetes.
Another strong evidence demonstrating that epigenetic modifications regulate energy metabolism arises from a recent study by Yamamoto et al. (Citation2011), in which the authors showed that nuclear receptor corepressor 1 (NCoR1) is a regulator of muscle mass and its oxidative capacity. NCoR1 is a transcriptional corepressor that participates in the formation of repressive complexes, together with several HDACs, in particular HDAC3, HDAC4, 5, 7 and 9 (reviewed in Perissi et al. Citation2010). These authors demonstrated that muscle-specific loss of NCoR1 in mice (NCoR1skm-/- ) increase muscle mass, locomotor activity (i.e., mice run for a significantly longer time and distance) and oxygen consumption, with a reduction in the respiratory exchange ratio (RER), suggesting that they preferentially use fat as energy source. In line with these observations, they detected increased mitochondrial content and activity in the gastrocnemius of NCoR1skm-/- , with concomitant reduction of the number of glycolytic MyHC2b-positive fibers and increased number of oxidative MyHC2x- and 2a- positive fibers. Interestingly, these authors showed that the expression of MEF2 family members increases in skeletal muscle of NCoR1skm-/- mice and negatively correlates with NCoR1 levels. It should be mentioned that MEF2 could be acetylated and activated by the acetyltransferases p300/CBP, whereas HDAC3 (Gregoire 2007), HDAC4, 5, 7 and 9 (McKinsey Citation2001, Haberland et al. Citation2009) and SIRT1 (Zhao Citation2005) interact with MEF2 and prevent its activation. Thus the absence of NCoR1 induces acetylation and activation of MEF2, leading to induction of MEF2 targets and to increased muscle mass. Moreover, NCoR1skm-/- mice show higher expression of genes related to mitochondrial respiration and fatty acid catabolism, whose expression in muscle is controlled by PPAR and estrogen-related receptor (ERR) families (Alaynick Citation2008). It should be noted that NCoR1 is recruited to PPAR responsive elements (PPREs) in the Ucp3 promoter and to the extended nuclear receptor half-sites (NR1/2) that binds members of ERR family (Zhang et al. Citation2006) in the pyruvate dehydrogenase kinase 4 (Pdk4) promoter ().
Figure 5. NCoR1 modulates transcription of oxidative genes in skeletal muscle. (A) NCoR1 is recruited on the responsive element in the promoter of PPAR and ERR target genes forming a repressive complex and inhibiting the expression of these genes. (B) The ablation of NCoR1 leads to HDACs dissociation and to the recruitment of the acetyltransferases p300/CBP with the consequent derepression of target gene transcription.
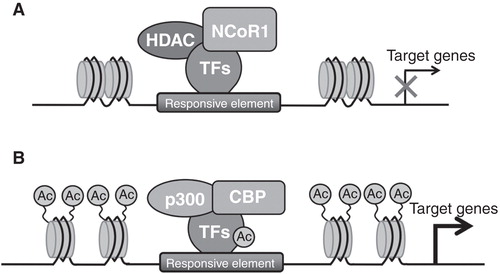
In addition, histone 4 is hyperacetylated on these promoters in the absence of NCoR1, suggesting that under this condition the coactivator PGC-1α activates the transcription of these genes (Handschin and Spiegelman Citation2006). For this reason, loss of NCoR1 phenocopies PGC-1α overexpression, in that both models show induction of mitochondrial fatty acid β-oxidation and improvement of exercise capacity (Lin et al. Citation2002).
A recent study by Li P. et al. (2011) demonstrated that NCoR1 also plays a key role in adipose tissue. PPARγ is highly expressed in this tissue and is a determinant of adipocyte differentiation, as well as a regulator of insulin sensitivity and adipokine secretion (Evans et al. Citation2004, Imai et al. Citation2004, Rangwala and Lazar Citation2004, Tontonoz and Spiegelman Citation2008). However, the transcriptional regulation exerted by PPARγ depends on the multi-protein coregulatory complexes recruited on target gene promoters and NCoR1 is one of the most important corepressors also in this tissue. In the same study, they found that adipocyte-specific NCoR1 knockout (NCoR1 AKO) mice become more obese than control mice when fed with high fat diet, due to increased subcutaneous and visceral adipose tissue mass. In epididymal white adipose tissue of NCoR1 AKO mice they detected increased expression of typical adipogenic genes such as Fas, Acc, Srebp1c, Scd1 and Scd2. Nonetheless, inflammation of adipose tissue is reduced, indicating improved functionality of this tissue and enhanced systemic insulin sensitivity. In adipose tissue, insulin promotes storage of free fatty acids into triglycerides and, consistently, the authors observed reduced circulating levels of free fatty acids in NCoR1 AKO mice, suggesting a reduction of lipolysis in adipose tissue. Since it has been demonstrated that down-regulation of NCoR1 expression in 3T3-L1 cells enhances adipocyte differentiation in part by increasing PPARγ transcriptional activity (Yu et al. Citation2005), Li P. et al. (2011) and collaborators focused on the repressive effect of NCoR1 on PPARγ. In this regard it should be mentioned that Choi et al. (Citation2010) showed that high fat diet induces phosphorylation of PPARγ at serine 273, which dysregulates the expression of many genes of the adipogenic program. On the contrary, PPARγ agonists prevent phosphorylation at this site and allow the functional transcriptional cascade in adipose tissue. Consistently, in adipose tissue from NCoR1 AKO mice serine 273 phosphorylation decreases, leading to increased expression of typical markers of adipocyte functionality.
A recent paper by Weems et al. (Citation2012) demonstrated that class II HDACs regulate gene expression in adipocytes as a result of adrenergic activation. The authors observed that HDAC5 plays a central role in repression of the insulin-dependent glucose transporter GLUT4 transcription in preadipocytes. Since it is known that repression of GLUT4 expression is correlated with insulin resistance in adipose tissue, this paper underlines the importance of HDACs in metabolic regulation and adipose tissue functionality. These authors also detected that, in the absence of HDAC5, other class II HDACs, including HDAC4 and HDAC9, may exert a redundant function. Moreover they demonstrated that cAMP-dependent down-regulation of GLUT4 mRNA expression in vivo is mediated by nuclear localization of HDAC4 and HDAC5: The Glut4 promoter contains an LXRE-binding site, and the cAMP signalling increases the recruitment of class II HDACs in this region, ultimately reducing Glut4 expression.
HDACs also play a role in cardiac lipid metabolism and function. Recent evidence suggests that HDACs are important metabolic regulators in the heart. Postnatal inactivation of HDAC3 in both cardiac and skeletal muscle do not show cardiac dysfunction of mice fed with normal chow diet, whereas mice fed with high fed diet develop hypertrophic cardiomyopathy and heart failure. The reduced expression of genes encoding enzymes involved in fatty acid oxidation and lipid metabolism, typically observed in these mice, do not make them able to cope with the dietary lipid overload. These results demonstrate that efficient lipid catabolism is needed to maintain myocardial metabolic homeostasis and physiology, especially when fatty acids are the main fuel sources (Sun et al. Citation2011). These results demonstrate that HDAC3 is required for cardiac metabolic regulation in response to a lipid enriched diet and that loss of HDAC3 compromises the ability of cardiac mitochondria to respond to nutritional changes and lipid overload.
Histone deacetylases inhibitors
Nowadays several HDAC inhibitors have been described; usually they are classified according to their chemical structure as short-chain fatty acids, hydroxamic acids, benzamides, ketones, and cyclic peptides with a pendant functional group. Unfortunately, most HDAC inhibitors, such as suberoylanilide hydroxamic acid (SAHA) and trichostatin A (TSA), inhibit HDAC isoforms non-specifically. Others, such as MS-275, a benzamide, are more selective for class IHDACs.
The therapeutic applications of HDAC inhibitors are multiple: In 2006, the US FDA approved SAHA for cutaneous manifestations of cutaneous T-cell lymphoma. Many evidences suggest that HDAC inhibitors can be considered an interesting approach in cancer therapy. The DNA damage response is in fact modulated by the acetylation status of histone and non-histone proteins and HDACs protect cancer cells from genotoxic insults. Thus, HDAC inhibitors can silence DNA repair pathways, inactivate non-histone proteins that are required for DNA stability and induce reactive oxygen species and DNA double-strand breaks (reviewed in Rajendran et al. Citation2011).
Recently, since HDACs play an important role in control of several cardiac events such as hypertrophy (Antos et al. Citation2003), autophagy (Cao et al. Citation2011), contractility (Gupta et al. Citation2008), HDAC inhibitors could represent a novel and promising therapy in patients with heart failure (reviewed in McKinsey Citation2012). Moreover, other recent evidences underline the importance of tubulin acetylation mediated by HDAC6 in the development of Huntington (Dompierre et al. Citation2007) and Parkinson diseases (Outeiro et al. Citation2007). Based on these results, the HDAC class II selective inhibitors have been theorized as new avenues for therapeutic intervention in some neurodegenerative disorders (reviewed in Li G. et al. 2011). Given the role of different HDACs in metabolic regulation, it is possible that in the future some specific inhibitors may find some clinical applications in lipid metabolic disorders.
Conclusions
The results reviewed in this paper provide strong evidences of the fundamental role of epigenetics and HDACs on the control of lipid homeostasis as well as other metabolic pathways in different tissues. Enzymes catalyzing epigenetic modifications, especially histone acetylation/deacetylation, constitute important components of the machinery regulating gene expression and it has become clear that HDACs, together with other corepressors forming the transcriptional repressive complexes, play a crucial role in lipid, glucose and energy metabolism.
In the light of these evidences, future studies are warranted to deepen our understanding on epigenetics and, more specifically, on the function of individual HDACs. To this end, an interesting approach is the enzymatic inhibition of HDACs, as demonstrated by the increasing number of studies using HDACs inhibitors as valuable tools to investigate the relationship between epigenetic modifications and lipid metabolism. Loss-of-function approaches are providing useful information on the role of HDACs in metabolic regulation. However, the availability of selective inhibitors will aid the comprehension of the specific role of each HDAC and may be an opportunity to explore their potential as future therapeutic agents. New knowledge on the function of HDACs in metabolic regulation will elucidate their relevance in pathophysiology of lipid metabolic disorders and generate novel insights on how these mechanisms can be targeted for therapeutical purposes in several pathological conditions such as dyslipidemia, obesity and insulin resistance.
Acknowledgements
We apologize to the authors of the numerous excellent papers that were not cited in this review due to space limitation. We wish to thank Miss Elda Desiderio Pinto for administrative assistance and for her valuable help typing this review. This work was made possible by grants from EC FP6 LSHM-CT2006-037498-SOUTH to MC, CARIPLO Foundation 2008.2511 to MC and 2009.2727 to FG, PRIN2009K7R7NA to MC, Giovanni Armenise-Harvard Foundation Career Development Award to NM, PRIN2008 2008ZTN724 to EDF.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Alaynick WA. 2008. Nuclear receptors, mitochondria and lipid metabolism. Mitochondrion 8:329–337.
- Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, Zhang CL, Schreiber K, 2003. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J Biol Chem 278:28930–28937.
- Boulias K, Talianidis I. 2004. Functional role of G9a-induced histone methylation in small heterodimer partner-mediated transcriptional repression. Nucleic Acids Res 32:6096–6103.
- Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, 2011. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci USA 108:4123–4128.
- Cantó C, Auwerx J. 2010. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci 67:3407–3423.
- Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, 2010. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARγ by Cdk5. Nature 466:451–456.
- Czubryt MP, McAnally J, Fishman GI, Olson EN. 2003. Regulation of peroxisome proliferator activated receptor gamma coactivator 1α (PGC-1α) and mitochondrial function by MEF2 and HDAC5. Proc Natl Acad Sci USA 100:1711–1716.
- Dawson MA, Kouzarides T. 2012. Cancer Epigenetics: From mechanism to therapy. Cell 150:12–27.
- De Fabiani E, Mitro N, Anzulovich AC, Pinelli A, Galli G, Crestani M. 2001. The negative effects of bile acids and tumor necrosis factor-α on the transcription of cholesterol 7α-hydroxylase gene (CYP7A1) converge to hepatic nuclear factor-4: A novel mechanism of feedback regulation of bile acid synthesis mediated by nuclear receptors. J Biol Chem 276:30708–30716.
- De Fabiani E, Mitro N, Gilardi F, Caruso D, Galli G, Crestani M. 2003. Coordinated control of cholesterol catabolism to bile acids and of gluconeogenesis via a novel mechanism of transcription regulation linked to the fasted-to-fed cycle. J Biol Chem 278:39124–39132.
- Dompierre JP, Godin JD, Charrin BC, Cordelieres FP, King SJ, Humbert S, 2007. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J Neurosci 27:3571–3583.
- Donohoe DR, Garge N, Zhang X, Sun W, O'Connell TM, Bunger MK, 2011. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab 13:517–526.
- Evans RM, Barish GD, Wang YX. 2004. PPARs and the complex journey to obesity. Nat Med 10:355–361.
- Fang S, Miao J, Xiang L, Ponugoti B, Treuter E, Kemper JK. 2007. Coordinated recruitment of histone methyltransferase G9a and other chromatin-modifying enzymes in SHP-mediated regulation of hepatic bile acid metabolism. Mol Cell Biol 27:1407–1424.
- Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, 2011. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science 331:1315–1319.
- Finkel T, Deng CX, Mostoslavsky R. 2009. Recent progress in the biology and physiology of sirtuins. Nature 460:587–591.
- Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, 2009. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58:1509–1517.
- Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, 2000. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell 6:517–526.
- Gray SG, Ekstrom TJ. 2001. The human histone deacetylase family. Exp Cell Res 262:75–83.
- Guarente L. 2006. Sirtuins as potential targets for metabolic syndrome. Nature 444:868–874.
- Gupta MP, Samant SA, Smith SH, Shroff SG. 2008. HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. J Biol Chem 283:10135–10146.
- Haberland M, Montgomery RL, Olson EN. 2009. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat Rev Genet 10:32–42.
- Haeusler RA, Kaestner KH, Accili D. 2010. FoxOs function synergistically to promote glucose production. J Biol Chem 285:35245–35248.
- Handschin C, Spiegelman BM. 2006. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev 27:728–735.
- Imai T, Takakuwa R, Marchand S, Dentz E, Bornert JM, Messaddeq N, 2004. Peroxisome proliferator-activated receptor gamma is required in mature white and brown adipocytes for their survival in the mouse. Proc Natl Acad Sci USA 101:4543–4547.
- Kemper JK, Kim H, Miao J, Bhalla S, Bae Y. 2004. Role of an mSin3A-Swi/Snf chromatin remodeling complex in the feedback repression of bile acid biosynthesis by SHP. Mol Cell Biol 24:7707–7719.
- Kerr TA, Saeki S, Schneider M, Schaefer K, Berdy S, Redder T, 2002. Loss of nuclear receptor SHP impairs but does not eliminate negative feedback regulation of bile acid synthesis. Dev Cell 2:713–720.
- Knutson SK, Chyla BJ, Amann JM, Bhaskara S, Huppert SS, Hiebert SW. 2008. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J 27:1017–1028.
- Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, 2004. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci USA 104:17335–17340.
- Li G, Jiang H, Chang M, Xie H, Hu L. 2011. HDAC6 α-tubulin deacetylase: A potential therapeutic target in neurodegenerative diseases. J Neurol Sci 304:1–8.
- Li P, Fan W, Xu J, Lu M, Yamamoto H, Auwerx J, 2011. Adipocyte NCoR knockout decreases PPARg phosphorylation and enhances PPARγ activity and insulin sensitivity. Cell 147:815–826.
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, 2002. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature 418:797–801.
- López-Rodas G, Brosch G, Georgieva EI, Sendra R, Franco L, Loidl P. 1993. Histone deacetylase. A key enzyme for the binding of regulatory proteins to chromatin. FEBS Lett 317:175–180.
- Lu J, McKinsey TA, Nicol RL, Olson EN. 2000. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci USA 97:4070–4075.
- Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, 2000. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell 6:507–515.
- Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D. 2007. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab 6:208–216.
- McGee SL, van Denderen BJ, Howlett KF, Mollica J, Schertzer JD, Kemp BE, 2008. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes 57:860–867.
- McKinsey TA. 2012. Therapeutic potential for HDAC inhibitors in the heart. Annu Rev Pharmacol Toxicol 10:303–319.
- McKinsey TA, Zhang CL, Olson EN. 2001. Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol Cell Biol 21:6312–6321.
- McKinsey TA, Zhang CL, Lu J, Olson EN. 2000. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408:106–111.
- Mihaylova MM, Vasquez DS, Ravnskjaer K, Denechaud PD, Yu RT, Alvarez JG, 2011. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 145:607–621.
- Miremadi A, Oestergaard MZ, Pharoah PD, Caldas C. 2007. Cancer genetics of epigenetic genes. Hum Mol Genet 16:28–49.
- Mitro N, Godio C, De Fabiani E, Scotti E, Galmozzi A, Gilardi F, 2007. Insights in the regulation of cholesterol 7α-hydroxylase gene reveal a target for modulating bile acid synthesis. Hepatology 46:885–897.
- Nerup J, Pociot F. 2001. A genome wide scan for type 1-diabetes susceptibility in Scandinavian families: Identification of new loci with evidence of interactions. Am J Hum Genet 69:1301–1313.
- Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, 2007. Sirtuin 2 inhibitors rescue α-synuclein-mediated toxicity in models of Parkinson's disease. Science 317:516–519.
- Passier R, Zeng H, Frey N, Naya FJ, Nicol RL, McKinsey TA, 2000. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Invest 105:1395–1406.
- Perissi V, Jepsen K, Glass CK, Rosenfeld MG. 2010. Deconstructing repression evolving models of co-repressor action. Nat Rev Genet 11:109–123.
- Potthoff MJ, Wu H, Arnold MA, Shelton JM, Backs J, McAnally J, 2007. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J Clin Invest 117:2459–2467.
- Pullinger CR, Eng C, Salen G, Shefer S, Batta AK, Erickson SK, 2002. Human cholesterol 7α-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Invest 110:109–117.
- Rajendran P, Ho E, Williams DE, Dashwood RH. 2011. Dietary phytochemicals, HDAC inhibition, and DNA damage/repair defects in cancer cells. Clin Epigenetics 3:1–23.
- Rangwala SM, Lazar MA. 2004. Peroxisome proliferator-activated receptor gamma in diabetes and metabolism. Trends Pharmacol Sci 25:331–336.
- Richards EJ, Elgin SCR. 2002. Epigenetic codes for heterochromatin formation and silencing: Rounding up the usual suspects. Cell 108:489–500.
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. 2005. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 434:113–118.
- Rodgers JT, Puigserver P. 2007. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci USA 104:12861–12866.
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, 2005. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310:1642–1646.
- Sun Z, Singh N, Mullican SE, Everett LJ, Li L, Yuan L, 2011. Diet-induced lethality due to deletion of the Hdac3 gene in heart and skeletal muscle. J Biol Chem 286:33301–33309.
- Tontonoz P, Spiegelman BM. 2008. Fat and beyond: The diverse biology of PPARγ. Ann Rev Biochem 77:289–312.
- Vaillant I, Paszkowski J. 2007. Role of histone and DNA methylation in gene regulation. Curr Opin Plant Biol 10:528–533.
- Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, 2004. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol 24:8374–8385.
- Viollet B, Guigas B, Leclerc J, Hebrard S, Lantier L, Mounier R, 2009. AMP-activated protein kinase in the regulation of hepatic energy metabolism: From physiology to therapeutic perspectives. Acta Physiol 196:81–98.
- Wang L, Lee YK, Bundman D, Han Y, Thevananther S, Kim CS, 2002. Redundant pathways for negative feedback regulation of bile acid production. Dev Cell 2:721–731.
- Weems JC, Griesel BA, Olson AL. 2012. Class II histone deacetylases downregulate GLUT4 transcription in response to increased cAMP signaling in cultured adipocytes and fasting mice. Diabetes 61:1404–1414.
- Xiang K, Wang Y, Zheng T, Jia W, Li J, Chen L, 2004. Genome-wide search for type 2 diabetes/impaired glucose homeostasis susceptibility genes in the Chinese: Significant linkage to chromosome 6q21-q23 and chromosome 1q21-q24. Diabetes 53:228–234.
- Yamamoto H, Williams EG, Mouchiroud L, Cantó C, Fan W, Downes M, 2011. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell 147:827–839.
- Yin L, Lazar MA. 2005. The orphan nuclear receptor Rev-erbα recruits the N-CoR/histone deacetylase3 corepressor to regulate the circadian Bmal1 gene. Mol Endocrinol 19:1452–1459.
- Yu S, Matsusue K, Kashireddy P, Cao WQ, Yeldandi V, Yeldandi AV, 2003. Adipocyte-specific gene expression and adipogenicsteatosis in the mouse liver due to peroxisome proliferator-activated receptor γ1 (PPARγ1) overexpression. J Biol Chem 278:498–505.
- Yu C, Markan K, Temple KA, Deplewski D, Brady MJ, Cohen RN. 2005. The nuclear receptor corepressors NCoR and SMRT decrease peroxisome proliferator-activated receptor gamma transcriptional activity and repress 3T3-L1 adipogenesis. J Biol Chem 280:13600–13605.
- Zamir I, Harding HP, Atkins GB, Hörlein A, Glass CK, Rosenfeld MG, 1996. A nuclear hormone receptor corepressor mediates transcriptional silencing by receptors with distinct repression domains. Mol Cell Biol 16:5458–5465.
- Zhang YL, Hernandez-Ono A, Siri P, Weisberg S, Conlon D, Graham MJ, 2006. Aberrant hepatic expression of PPARγ2 stimulates hepatic lipogenesis in a mouse model of obesity, insulin resistance, dyslipidemia, and hepatic steatosis. J Biol Chem 281:37603–37615.
- Zhao X, Sternsdorf T, Bolger TA, Evans RN, Yao TP, 2005. Regulation of MEF2 by histone deacetylase 4- and SIRT deacetylase mediated lysine modifications. Mol Cell Biol 25:8456–8464.