
Abstract
The co-transporter activity of Na+-K+-2Cl− 1 (NKCC1) is dependent on phosphorylation. In this study we show the energy-sensing kinase AMPK inhibits NKCC1 activity. Three separate AMPK activators (AICAR, Phenformin and A-769662) inhibited NKCC1 flux in a variety of nucleated cells. Treatment with A-769662 resulted in a reduction of NKCC1T212/T217 phosphorylation, and this was reversed by treatment with the non-selective AMPK inhibitor Compound C. AMPK dependence was confirmed by treatment of AMPK null mouse embryonic fibroblasts, where A-769662 had no effect on NKCC1 mediated transport. AMPK was found to directly phosphorylate a recombinant human-NKCC1 N-terminal fragment (1–293) with the phosphorylated site identified as S77. Mutation of Serine 77 to Alanine partially prevented the inhibitory effect of A-769662 on NKCC1 activity. In conclusion, AMPK can act to reduce NKCC1-mediated transport. While the exact mechanism is still unclear there is evidence for both a direct effect on phosphorylation of S77 and reduced phosphorylation of T212/217.
Introduction
The electroneutral cation-chloride-coupled co-transporter family comprises the bumetanide-sensitive co-transporter Na+/K+/Cl−(NKCC), the thiazide-sensitive co-transporter Na+/Cl− (NCC), and the K+/Cl− (KCC) co-transporters. The two isoforms of NKCC, NKCC1 and NKCC2, regulate cell volume and maintain cellular homeostasis in response to osmotic and oxidative stress (Haas & Forbush, Citation1998). The ubiquitously expressed NKCC1 is thought to play roles in fluid secretion (Evans et al., Citation2000), salt balance (Kim et al., Citation2008), and neuronal development and signalling (Kahle et al., Citation2008). Disruption of the NKCC1 gene has been reported by a number of groups and is associated with reduced epithelial chloride secretion (Flagella et al., Citation1999, Grubb et al., Citation2000), male sterility (Pace et al., Citation2000), profound deafness, balance defects (Flagella et al., Citation1999) and a reduction in systolic blood pressure (Meyer et al., Citation2002).
NKCC1 can be acutely regulated by changes in phosphorylation and there are at least seven phosphorylation sites on the intracellular N-terminus (Darman & Forbush, Citation2002; Gimenez & Forbush, Citation2005; Richardson & Alessi, Citation2008; Vitari et al., Citation2006). The key site that increases co-transporter activity in human NKCC1 is T217 (Darman & Forbush, Citation2002). STE20 (sterile 20)/SPS1-related proline/alanine rich kinase (SPAK) and the closely related oxidative stress-responsive kinase-1 (OSR-1) phosphorylate this site and another three threonines (T203, 207, 212) (Vitari et al., Citation2006).
AMPK is a heterotrimeric (αβγ subunits) kinase that is responsive to changes in intracellular AMP levels, thereby functioning as a cellular energy sensor (Kahn et al., Citation2005; Steinberg & Kemp, Citation2009). Generally, AMPK has been studied in the context of cellular energy metabolism, appetite, and energy disorders such as diabetes and obesity. In addition to its metabolic functions, there is increasing evidence that AMPK has a role in the regulation of epithelial ion transport (reviewed in Pastor-Soler & Hallows, Citation2012).
AMPK is activated allosterically by a rise in the AMP/ATP or ADP/ATP ratio (Oakhill et al., Citation2011; Xiao et al., Citation2011), reflecting a state of energy deficiency within the cell. Furthermore, full activation of AMPK requires phosphorylation of the Thr172 on the α subunit by one of three, and possibly more, upstream kinases. When activated, AMPK phosphorylates a wide number of downstream targets, whose effect is to inhibit energy consuming pathways and activate those that produce energy. Solute transport consumes a large proportion of total cellular energy, so it is not surprising that AMPK regulates many epithelial ion transporters and channels (Alesutan et al., Citation2011a, Citationb; Bhalla & Hallows, Citation2008; Fraser et al., Citation2007; Hallows et al., Citation2003; Sopjani et al., Citation2010a,Citationb). We have previously reported that the kidney-specific sodium co-transporter NKCC2 was associated with AMPK in cultured and transfected cells, and phosphorylated by AMPK on Ser130 in its amino terminus (Fraser et al., Citation2007). Phosphorylation of NKCC2 on Ser130 is associated with activation of NKCC2 (Fraser et al., Citation2007; Richardson et al., Citation2011). While phosphorylation of the analogous site in the closely related family member NKCC1 has been demonstrated, there is no apparent functional effect of AMPK activation in red blood cells (Sid et al., Citation2010). The effect of AMPK activation on NKCC1 in epithelial cells has not been determined. In the current study we determined whether AMPK regulates NKCC1 in nucleated cells.
Materials and methods
Antibodies and Western blot analysis
The antibody directed against pT212/217, the major activating site in NKCC1, has previously been described (Fraser et al., Citation2013). This antibody was raised against a peptide also expressing the phosphosite pT212, as previously described by Flemmer et al. (Citation2002). Mouse monoclonal antibody against β-tubulin (Sigma Aldrich, St. Louis, MO), AMPK pT172, and ACC1 pS79 were obtained from Cell Signalling Technologies (Danvers, MA).
Cells were lysed with ice cold cell lysis buffer containing 50 mM Tris.HCl, pH7.5, 1 mM EGTA, 1 mM EDTA, 1 mM Na3VO4, 50 mM NaF, 5 mM Na4P2O7, 0.27 M sucrose, 1% Triton-X100, 1 mM benzamidine, 0.1 mM PMSF and 1 mM DTT. The lysates were clarified by centrifugation at 4 °C for 5 min at 14 000 g. Supernatants were stored at −80 °C until used. Protein concentration was quantified using the Bradford Assay (BioRad, Hercules, CA). Samples were separated by SDS-PAGE and electrically transferred to a polyvinylidene difluoride membrane (Immobilon-P; Millipore, Bedford, MA) using a Transblot Turbo apparatus (BioRad). The membrane was blocked in 10% BSA in Tris-buffered saline (TBS) and then incubated in primary antibody. Antibody conjugates were formed with FITC conjugated secondary antibody (Dako, Glostrup, Denmark) and anti-FITC POD (Roche Diagnostics, Basil, Switzerland). Immunoreactive proteins were detected by enhanced chemiluminescence with the Western Lightning system (PerkinElmer, Waltham, MA). If the membrane was to be probed with another primary antibody, antibody bound to the membrane was stripped by incubation in Reblot stripping solution (Chemicon, Billerica, MA) for 15 min. Quantification of Western blots was performed by densitometry with analysis using Image J software (NIH, Bethesda, MD).
Cell culture, stable cell line generation and cell treatments
The kidney cell lines HEK-293, MDCK and mouse inner medullary collecting duct cells (mIMCD3) were obtained from the ATCC (Manassas, VA). The murine macula densa derived cell line MMDD1 was a generous gift from Prof. J. Schnermann (National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD). Bovine aortic endothelial cells (BAEC) were prepared and cultured as previously described (Mount et al., Citation2008). Mouse embryonic fibroblasts (MEF) were isolated from mice bearing inactivating mutations of the β1 and β2 subunits of AMPK (AMPK β1floxβ2KO mice) that removes all AMPK activity (MEF-KO) and compared with MEF from WT mice (MEF-WT). Western blots showed that all cells used expressed NKCC1, while MMDD1 also expressed detectable NKCC2 as has previously been reported (Yang et al., Citation2000). Cells were grown in complete media with 10% FCS. Treatment with activators of AMPK was performed on cells that had been cultured in serum free media overnight. A-769662 was dissolved in DMSO at a concentration of 100 mM and used at a final concentration of 100 μM for 30 min. Phenformin and AICAR were dissolved in DMSO at a concentration of 1 M and used at a final concentration of 1 mM for 60 min. Compound C in DMSO was used at a final concentration of 10 μM for 30 min. The appropriate volume of DMSO was added to vehicle-treated cells.
HEK293 cells were transfected with plasmids containing human Myc-NKCC1 or Myc-NKCC1S77A using Effectene (Qiagen; Limburg, Netherlands). Repeated passaging in the presence of G418 to generate stable cell lines. Expression levels of NKCC1 were monitored by Western blot analysis.
86Rb-uptake assay
NKCC1 activity was measured using an 86Rb-uptake assay. Cells were plated on 24-well plates and 86Rb-uptake assay was performed on cells that were 95% confluent, as previously described (Fraser et al., Citation2007). Briefly, growth medium was removed and the cells were incubated overnight in serum-free media. Cells were then stimulated with AMPK activators for the optimal time and the media then changed to Earles Balanced Salt Solution (EBSS) also containing the AMPK activator, 1 mM Ouabain and 10 μM Bumetanide as an inhibitor of NKCC in some cases for a further 15 min. Media was then replaced with EBSS with inhibitors containing 2 μCi/ml 86Rb for 15 min. Cells were washed three times 100 mM MgCl2 in HEPES pH7.4 and lysed in 300 μl of 1 N NaOH/0.1% SDS. Radioactivity was measured by liquid scintillation counting (Tri-Carb 2000; Perkin Elmer).
AMPK activity
AMPK α1 was immunoprecipitated from 150 μg of lysates overnight at 4 °C using anti-AMPK α1 antibody bound to Protein A Sepharose beads and AMPK activity measured as previously described (Fraser et al., Citation2005). Briefly, immunoprecipitates were resuspended in assay buffer containing 50 mM HEPES, 1 mM DTT, 5% glycerol, 0.05% TritonX-100. Activity was determined by phosphorylation of SAMS peptide in a reaction containing 100 μM SAMS, 200 μM [γ-32P] ATP, 100 μM AMP and 10 mM MgCl2 at 30 °C in a 40 μl volume. Reactions were terminated after 8 min by spotting 25 μl of the reaction mixture onto P81 phosphocellulose paper (Whatman) and washing in 1% phosphoric acid (Glass et al., Citation1978). Radioactivity was measured by liquid scintillation counting (Tri-Carb 2000; Perkin Elmer).
In vitro phosphorylation
Baculovirus expressed NKCC1 (1–293) was prepared in kinase assay buffer (50 mM Hepes (pH 7.5), 10 mM MgCl2, 5% glycerol, 1 mM DTT (dithiothrietol), 0.05% Triton X-100, 250 μM [γ-32P]ATP (5000 c.p.m./pmole) and 100 μM 5′-AMP). The reactions were initiated through the addition of 1 μl of purified rat liver AMPK diluted in 50 mM Tris/HCl (pH 7.5) and incubated at 30 °C for 1 h.
Phosphorylation site identification by mass spectrometry
FLAG-NKCC1 (1–293) was expressed in baculovirus, purified and phosphorylated using AMPK. In solution tryptic digest was performed and digests were dried under vacuum, resuspended in 60% (v/v) acetonitrile, 5% (v/v) trifluoroacetic acid, and purified on TiO2 beads (GL Sciences) using micro-columns. The TiO2 beads were washed twice with 60 μl of 60% (v/v) acetonitrile, 5% (v/v) trifluoroacetic acid and eluted with 120 μl of 1.25% (w/v) NH4OH (BDH). Purified phosphopeptides were dried under vacuum, resuspended in 20 μl of 2% (v/v) formic acid (Sigma), and analyzed by LC/MS. Chromatography was performed on a Dionex Ultimate 3000 nano-LC at a flow rate of 500 nl/min. Peptides were resolved on a 20-cm × 75-μm C18 column (LC Packings Pepmap100 C18 resin) using a 3–35% acetonitrile gradient. Mass spectrometry was performed on a QSTAR-pulsar i (Applied Biosystems, Foster City, CA) using a micro-ion spray source (Applied Biosystems). Data were acquired using a data-dependent acquisition program and analyzed by MASCOT software. MS/MS spectra were manually validated to confirm the accuracy of assigned phosphorylation sites.
Statistics
Statistics were performed using Instat Version 3.05 (GraphPad Software, San Diego, CA). Data are presented as means ±1 standard deviation. Multiple group means were compared by ANOVA followed by a post-hoc test. Comparison of means from two groups was performed by unpaired t-test. p Values <0.05 were considered significant.
Results
Effect of AMPK activators on 86Rb uptake in nucleated mammalian cells
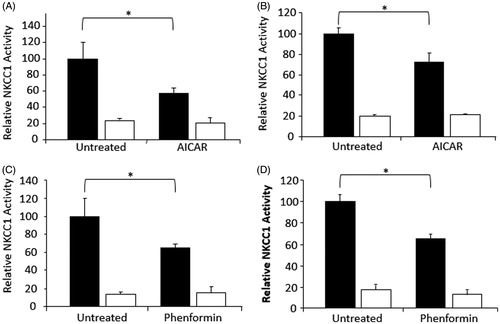
The effect of AMPK activators on bumetanide-sensitive co-transporter activity was determined in MDCK cells, the mouse inner medullary collecting duct cell line mIMCD3, and primary cultures of bovine aortic endothelial cells (BAEC). All of these cells express NKCC1 (data not shown). Cells were stimulated with 1 mM AICAR or 1 mM Phenformin to stimulate AMPK activity. Activators used were those demonstrated to consistently increase AMPK activity in each cell line (data not shown). In each cell type there was a reduction in bumetanide-sensitive 86Rb uptake, which varied from 25–45% (p < 0.001) ().
Figure 1. Activation of AMPK reduces NKCC1-mediated flux. Rubidium flux was measured in cells stimulated with either 1 mM AICAR or 1 mM phenformin for 1 hour. Cell types were; (A) MDCK, (B) MMDD1, (C) mIMCD3, and (D) BAEC. NKCC1 (or NKCC1/2 in the case of MMDD1 cells) mediated flux was determined as vehicle-treated flux (black bars) above the presence of 5 μM bumetanide (white bars). Fluxes were performed with n = 6 on at least three separate occasions. Counts were standardized against untreated control and then pooled. *p < 0.001.
The direct small molecule AMPK activator A-769662 was used to activate AMPK in MDCK cells (100 uM for 30 min). A-769662 increased AMPK activity in MDCK cells, approximately 5-fold () and this activator was used, therefore, in subsequent activity and phosphorylation experiments. Pre-incubation with A-769662 for 30 min reduced bumetinide-sensitive rubidium flux in MDCK to basal levels (), and the effect was maintained out to 60 min ().
Figure 2. Activation of AMPK with A-769662 reduces NKCC1-mediated flux and phosphorylation in MDCK cells. (A) MDCK cells were stimulated with 100 μM A-769662 for 30 minutes prior to rubidium flux. NKCC1-mediated flux was determined as vehicle treated flux (black bars) above the presence of 5 μM bumetanide (white bars) (n = 24). (B) Cells stimulated with A-769662 showed marked AMPK activation (n = 3). (C) Following pre-treatment with A-769662, NKCC1-mediated flux was measured at time points for up to 60 minutes (n = 6 at each time-point). (D) Phosphorylation of NKCC1T212/217 was measured by Western blot analysis relative to β-tubulin and measured by densitometry (n = 10). *p < 0.001, **p < 0.0001 relative to vehicle-treated cells at the equivalent time-point.
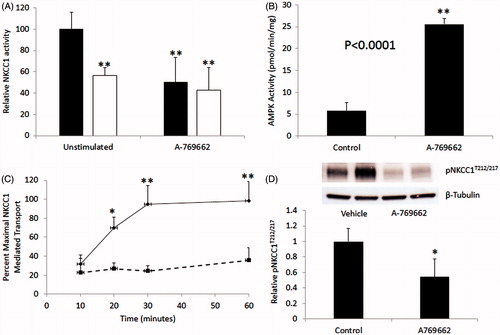
NKCC1 activity is dependent on phosphorylation on T217 by SPAK/OSR1 (Darman & Forbush, Citation2002). Treatment of MDCK cells with A769662 resulted in a 50% reduction in phosphorylated NKCC1 at this site (p < 0.0001) ().
In vitro phosphorylation of NKCC1 by AMPK and phosphorylation site identification
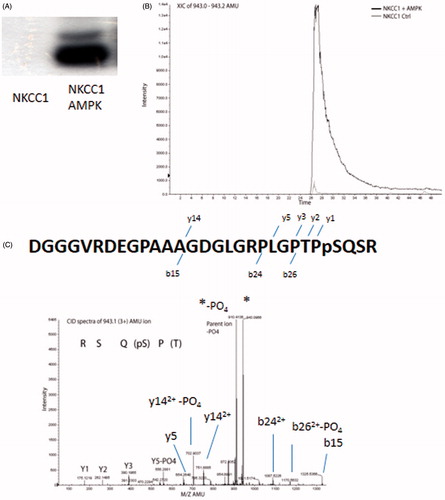
We have previously reported that AMPK phosphorylates NKCC2 (Fraser et al., Citation2007). Sid et al. (Citation2010) have reported AMPK phosphorylation of dogfish NKCC1 N-terminal (1–260) on residues S38 and S214 corresponding to human S77 and S242. To confirm that AMPK phosphorylates the N-terminus of NKCC1, the N-terminal domain of human NKCC1 (amino acids 1–293) was PCR-amplified and cloned with an N-terminal FLAG tag epitope and expressed in baculovirus. AMPK purified from rat liver extract was incubated with the NKCC1 N-terminus and separated by SDS-PAGE, which demonstrated phosphorylation (). The phosphorylated N-terminus of NKCC1 (1–293) was then analyzed by tandem mass spectrometry to identify the AMPK phosphorylation sites. One peptide was identified in high abundance and was essentially absent in control reactions (). The CID spectra identified this peptide as 52DGGGVRDEGPAAAGDGLGRPLGPTPPSQSR80 with a theoretical mass of 2826.30 mass units and an observed peptide mass of 2826.39 (). The peptide corresponds to the human NKCC1 amino acids 52–80 with a phosphate group on S77.
Figure 3. AMPK phosphorylates NKCC1 on S77. (A) In vitro phosphorylation of the N-terminal fragment of human NKCC1 (1–293) by purified AMPK. (B) Identification of AMPK phosphorylation site on NKCC1 by tandem mass spectrometry showing increase in the amount of 943.1(3+) AMU ion after treatment with AMPK. (C) CID spectra of the 943.1 AMU ion showing y ions and the corresponding sequence. Y5 shows loss of PO4 (18 AMU), indicating that y4 (not observed) was phosphorylated. Spectra also shows predominate neutral loss of 98 AMU (PO4) from the parent ion.
S77 is partially responsible for the inhibition of NKCC1 activity with AMPK
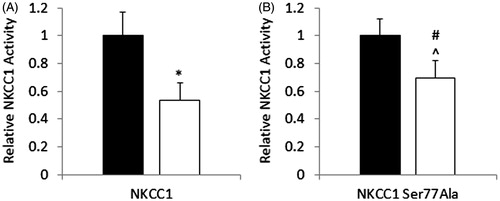
To determine whether the S77 site that was identified as an AMPK site in NKCC1 was functional, Myc tagged human NKCC1 or Myc tagged human NKCC1 with a point mutation of S77A was stably expressed in HEK293 cells. Rubidium flux was measured following pre-incubation with 1 mM phenformin. In HEK-293 cells expressing NKCC1-WT there was a 47% reduction in bumetanide-sensitive flux relative to vehicle treated cells. In HEK-293 cells expressing the NKCC1-S77A mutant the magnitude of the phenformin-mediated inhibition was reduced (31% reduction) ().
Figure 4. Mutation of NKCC1S77A results in a partial protection against AMPK-mediated reduction of NKCC1 activity. HEK293 cells stably expressing NKCC1 WT (A) or NKCC1S77A (B) were treated with 1 mM phenformin for 60 minutes (white bars) or vehicle (black bars) and then subjected to rubidium flux. NKCC1 mediated flask was considered to be the bumetanide sensitive flux. n = 12, *p < 0.001, #p < 0.05.
Inhibition of NKCC1 co-transporter activity by A-769662 is AMPK-dependant
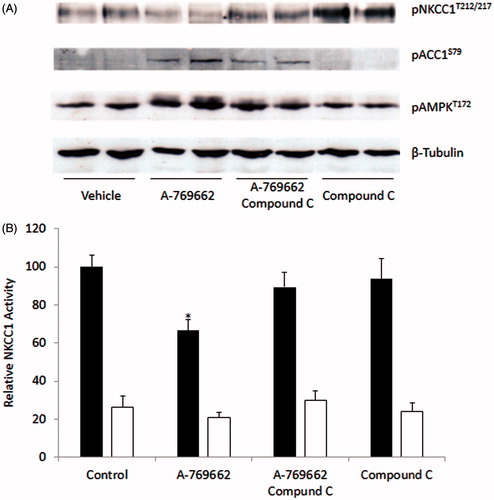
To demonstrate that the effect of A-769662 on NKCC1 co-transporter activity was dependent on AMPK activation, Compound C was used to inhibit AMPK activation. MDCK cells pre-treated with A-769662 were also treated with Compound C or vehicle. Activation of AMPK was measured through phosphorylation of the downstream target ACC1S79 and phosphorylation of AMPKT172. There was a reduction in AMPK activity in cells treated with Compound C. This was associated with increased NKCC1T212/T217 phosphorylation () and a restoration of NKCC1-mediated flux (). MDCK cells treated with Compound C alone had an elevated NKCC1T212/T217 phosphorylation however NKCC1 activity was not increased beyond vehicle-treated control levels.
Figure 5. A-769662 mediated reduction in NKCC1 is AMPK-dependant. MDCK cells were treated with A-769662, with or without the AMPK inhibitor Compound C. (A) Western blot analysis demonstrated that A7-69662 dephosphorylation of NKCC1T212/217 was prevented by Compound C. AMPK activity was demonstrated by phosphorylation of the downstream target ACCS79 and phosphorylation of AMPKT172. β-tubulin was used as a loading control. (B) NKCC1 activity was measured as bumetanide sensitive 86Rb flux in MDCK cells (vehicle black bars, bumetanide white bars). n = 10, *p < 0.001.
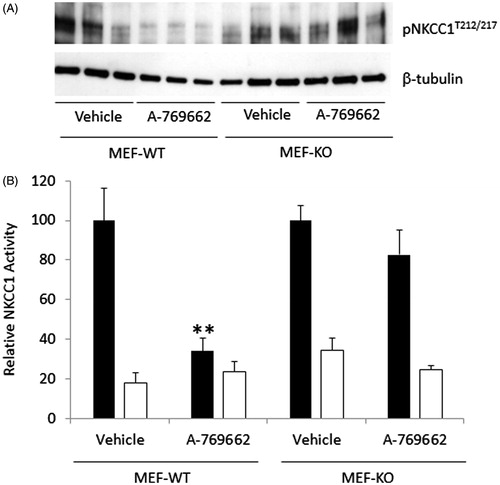
To further determine whether the effect of A-769662 on NKCC1 is AMPK-dependent, MEF derived from either WT (MEF-WT) or AMPK β1floxedβ2KO mice (MEF-KO) were treated with A-769662. The MEF-KO have very little AMPK activity (submitted manuscript). MEF-WT incubated with A-769662 had reduced phosphorylation of NKCC1T212/217 and a highly significant reduction in NKCC1 mediated rubidium flux. In contrast, MEF-KO pre-incubated with A769662 had no significant reduction in NKCC1 mediated rubidium flux or phosphorylation of NKCC1T212/217 ().
Figure 6. A-769662 mediated reduction in NKCC1-mediated flux in MEF cells is AMPK-dependent. (A) WT and AMPK null MEF cells (MEF-AMPKβ1floxβ2KO) treated with A-769662 were analyzed by Western blot to determine the phosphorylation state of NKCC1T212/217. (B) MEF cells were analyzed for NKCC1-mediated 86Rb flux (vehicle black bars, bumetanide white bars). n = 12, **p < 0.0001.
Discussion
It has long been known that the co-transporter activity of NKCC1 is regulated by phosphorylation (reviewed in Russell, Citation2000), but more recently the importance of phosphorylation by the Ste-20 kinases SPAK and OSR-1 has been increasingly recognized. We and others have previously reported that AMPK phosphorylates NKCC2 on Ser130 (Fraser et al., Citation2007; Richardson et al., Citation2011) and, in the present study, have investigated whether AMPK phosphorylates NKCC1 to regulate its function.
Sequence alignment of NKCC1 and NKCC2 shows strong conservation around Ser242, analogous to Ser130 in NKCC2, suggesting that NKCC1 may also be an AMPK substrate. In vitro, AMPK was able to phosphorylate NKCC1, however, when analyzed by LC-MS the most abundant phosphorylated peptide corresponded to S77 residue. AMPK has previously been shown to phosphorylate the corresponding residue in dogfish NKCC1, Ser38, (Sid et al., Citation2010) and pS77 has been detected in multiple phosphoproteomic studies including in mitosis of HeLa cells (Dephoure et al., Citation2008). Interestingly, we were not able to identify the Ser242 site as an AMPK phosphorylation site although Sid et al. (Citation2010) have reported AMPK mediated phosphorylation in dogfish NKCC1 on the equivalent site, Ser214. S77 is a predicted phosphorylation site by virtue of hydrophobic residues at the +4 and −5 positions but notably does not have a basic residue between −1 and −3 typical of many AMPK phosphorylation sites (reviewed in Steinberg & Kemp, Citation2009).
Phosphorylation on Ser130 by AMPK and other kinases in NKCC2 has been associated with an increase in NKCC2 activity (Richardson et al., Citation2011) and mutation of this site also results in a marked reduction of co-transporter activity in Xenopus oocytes (Fraser et al., Citation2007). The effect of AMPK activation on NKCC1 activity has previously been examined in red blood cells and demonstrated to be unaltered (Sid et al., Citation2010). However, as compared to nucleated cells, red blood cells are atypical cell type with reduced metabolic activity compared to nucleated cells. All of the nucleated cells we studied showed a significant reduction in NKCC1 mediated transport using a variety of AMPK activating drugs. Pre-incubation with the specific AMPK activator A-769662 resulted in a significant loss of NKCC1 mediated transport in MDCK cells associated with a 5-fold activation of AMPK.
The A-769662 at the concentration used in this study has been demonstrated to have off target effects including effects on the Na-K-ATPase which lead to a reduction in 86Rb flux in L6 myotubules (Benziane et al., Citation2009). Importantly, however, the activity studies presented here were all performed in the presence of ouabain to block Na-K-ATPase activity. Additionally, the reduction in NKCC1 activity was observed using three separate AMPK activators with different modes of activation. Phenformin is a mitochondrial uncoupler increases AMP (Zhang et al., Citation2007), whereas AICAR is phosphorylated by adenylate kinase to ZMP (Corton et al., Citation1995). A-769662, by contrast, allosterically activates AMPK through binding to its regulatory β1 subunit (Scott et al., Citation2008).
AMPK dependence of the effect of A-769662 was supported by use of the AMPK inhibitor Compound C, which partially but significantly reversed the reduction in NKCC1 mediated transport. Importantly when MEF cells devoid of AMPK activity were stimulated with A-769662 there was no significant reduction in NKCC1 activity contrary to MEF cells derived from wild-type mice.
As the Ser130 site of NKCC2 has been demonstrated to be important in activation of NKCC2 it is unlikely that the analogous NKCC1 site, Ser242, would be responsible for the inhibition of NKCC1 activity observed. We were also unable to identify this site by in vitro phosphorylation in human NKCC1. Consequently, we examined the predominant AMPK phosphorylation site identified, S77. Mutation of this site to an alanine resulted in a partial abrogation of the AMPK-mediated reduction of NKCC1 activity, however, this was not complete suggesting the presence of other pathways.
Activation of NKCC1 mediated transport is absolutely dependent on phosphorylation of T217 (Darman & Forbush, Citation2002), and so it was not surprising that the reduced NKCC1 activity observed with A-769662 treatment led to a significant reduction in T212/217 phosphorylation. The reduction in T212/217 phosphorylation could be reversed by blocking AMPK activity using Compound C. Moreover, it did not occur in the MEF cells devoid of AMPK activity suggesting that AMPK may be involved in either reducing phosphorylation of NKCC1 by SPAK/OSR-1 or promoting phosphatase activity.
In conclusion, these results confirm that AMPK phosphorylates NKCC1 on S77 in vitro and reduces co-transporter activity in nucleated cells. They differ from studies performed in red blood cells. The reason for this is unclear but could relate to the limited metabolic activity of that cell type. Regulation of the co-transporter activity of NKCC1 by AMPK emphasizes the important role of this kinase in modifying energy-sensitive processes in nucleated cells.
Declaration of interest
This work was supported by grants and fellowships from the Australian Research Council (BEK – DP130104548) and the National Health and Medical Research Council (DAP – APP1006517) (BEK, SG – APP1028254, APP1068798) and supported in part by the Victorian Government's Operational Infrastructure Support Program.
References
- Alesutan I, Foller M, Sopjani M, Dermaku-Sopjani M, Zelenak C, Frohlich H, et al. 2011a. Inhibition of the heterotetrameric K+ channel KCNQ1/KCNE1 by the AMP-activated protein kinase. Mol Membr Biol 28:79–89
- Alesutan I, Munoz C, Sopjani M, Dermaku-Sopjani M, Michael D, Fraser S, et al. 2011b. Inhibition of Kir2.1 (KCNJ2) by the AMP-activated protein kinase. Biochem Biophys Res Commun 408:505–510
- Benziane B, Bjornholm M, Lantier L, Viollet B, Zierath JR, Chibalin AV. 2009. AMP-activated protein kinase activator A-769662 is an inhibitor of the Na(+)-K(+)-ATPase. Am J Physiol Cell Physiol 297:C1554–566
- Bhalla V, Hallows KR. 2008. Mechanisms of ENaC regulation and clinical implications. J Am Soc Nephrol 19:1845–1854
- Corton JM, Gillespie JG, Hawley SA, Hardie DG. 1995. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem 229:558–565
- Darman RB, Forbush B. 2002. A regulatory locus of phosphorylation in the N terminus of the Na-K-Cl cotransporter, NKCC1. J Biol Chem 277:37542–37550
- Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. 2008. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci USA 105:10762–10767
- Evans RL, Park K, Turner RJ, Watson GE, Nguyen HV, Dennett MR, et al. 2000. Severe impairment of salivation in Na+/K+/2Cl− cotransporter (NKCC1)-deficient mice. J Biol Chem 275:26720–26726
- Flagella M, Clarke LL, Miller ML, Erway LC, Giannella RA, Andringa A, et al. 1999. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J Biol Chem 274:26946–26955
- Flemmer AW, Gimenez I, Dowd BF, Darman RB, Forbush B. 2002. Activation of the Na-K-Cl cotransporter NKCC1 detected with a phospho-specific antibody. J Biol Chem 277:37551–37558
- Fraser S, Mount P, Hill R, Levidiotis V, Katsis F, Stapleton D, et al. 2005. Regulation of the energy sensor AMP-activated protein kinase in the kidney by dietary salt intake and osmolality. Am J Physiol Renal Physiol 288:F578–586
- Fraser SA, Choy SW, Pastor-Soler NM, Li H, Davies MR, Cook N, et al. 2013. AMPK couples plasma renin to cellular metabolism by phosphorylation of ACC1. Am J Physiol Renal Physiol 305:F679–690
- Fraser SA, Gimenez I, Cook N, Jennings I, Katerelos M, Katsis F, et al. 2007. Regulation of the renal-specific Na+-K+-2Cl− co-transporter NKCC2 by AMP-activated protein kinase (AMPK). Biochem J 405:85–93
- Gimenez I, Forbush B. 2005. Regulatory phosphorylation sites in the NH2 terminus of the renal Na-K-Cl cotransporter (NKCC2). Am J Physiol Renal Physiol 289:F1341–1345
- Glass DB, Masaracchia RA, Feramisco JR, Kemp BE. 1978. Isolation of phosphorylated peptides and proteins on ion exchange papers. Anal Biochem 87:566–575
- Grubb BR, Lee E, Pace AJ, Koller BH, Boucher RC. 2000. Intestinal ion transport in NKCC1-deficient mice. Am J Physiol Gastrointest Liver Physiol 279:G707–718
- Haas M, Forbush B 3rd. 1998. The Na-K-Cl cotransporters. J Bioenerg Biomembr 30:161–172
- Hallows KR, Kobinger GP, Wilson JM, Witters LA, Foskett JK. 2003. Physiological modulation of CFTR activity by AMP-activated protein kinase in polarized T84 cells. Am J Physiol Cell Physiol 284:C1297–1308
- Kahle KT, Staley KJ, Nahed BV, Gamba G, Hebert SC, Lifton RP, Mount DB. 2008. Roles of the cation-chloride cotransporters in neurological disease. Nat Clin Pract Neurol 4:490–503
- Kahn BB, Alquier T, Carling D, Hardie DG. 2005. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1:15–25
- Kim SM, Eisner C, Faulhaber-Walter R, Mizel D, Wall SM, Briggs JP, Schnermann J. 2008. Salt sensitivity of blood pressure in NKCC1-deficient mice. Am J Physiol Renal Physiol 295:F1230–1238
- Meyer JW, Flagella M, Sutliff RL, Lorenz JN, Nieman ML, Weber CS, et al. 2002. Decreased blood pressure and vascular smooth muscle tone in mice lacking basolateral Na(+)-K(+)-2Cl(−) cotransporter. Am J Physiol Heart Circ Physiol 283:H1846–1855
- Mount PF, Lane N, Venkatesan S, Steinberg GR, Fraser SA, Kemp BE, Power DA. 2008. Bradykinin stimulates endothelial cell fatty acid oxidation by CaMKK-dependent activation of AMPK. Atherosclerosis 200:28–36
- Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S, Kemp BE. 2011. AMPK is a direct adenylate charge-regulated protein kinase. Science 332:1433–1435
- Pace AJ, Lee E, Athirakul K, Coffman TM, O’Brien DA, Koller BH. 2000. Failure of spermatogenesis in mouse lines deficient in the Na(+)-K(+)-2Cl(−) cotransporter. J Clin Invest 105:441–450
- Pastor-Soler NM, Hallows KR. 2012. AMP-activated protein kinase regulation of kidney tubular transport. Curr Opin Nephrol Hypertens 21:523–533
- Richardson C, Alessi DR. 2008. The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signalling pathway. J Cell Sci 121:3293–3304
- Richardson C, Sakamoto K, De Los Heros P, Deak M, Campbell DG, Prescott AR, Alessi DR. 2011. Regulation of the NKCC2 ion cotransporter by SPAK-OSR1-dependent and -independent pathways. J Cell Sci 124:789–800
- Russell JM. 2000. Sodium-potassium-chloride cotransport. Physiol Rev 80:211–276
- Scott JW, Van Denderen BJ, Jorgensen SB, Honeyman JE, Steinberg GR, Oakhill JS, et al. 2008. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem Biol 15:1220–1230
- Sid B, Miranda L, Vertommen D, Viollet B, Rider MH. 2010. Stimulation of human and mouse erythrocyte Na(+)-K(+)-2Cl(−) cotransport by osmotic shrinkage does not involve AMP-activated protein kinase, but is associated with STE20/SPS1-related proline/alanine-rich kinase activation. J Physiol 588:2315–2328
- Sopjani M, Alesutan I, Dermaku-Sopjani M, Fraser S, Kemp BE, Foller M, Lang F. 2010a. Down-regulation of Na+-coupled glutamate transporter EAAT3 and EAAT4 by AMP-activated protein kinase. J Neurochem 113:1426–1435
- Sopjani M, Bhavsar SK, Fraser S, Kemp BE, Foller M, Lang F. 2010b. Regulation of Na+-coupled glucose carrier SGLT1 by AMP-activated protein kinase. Mol Membr Biol, 27:137–144
- Steinberg GR, Kemp BE. 2009. AMPK in health and disease. Physiol Rev 89:1025–1078
- Vitari AC, Thastrup J, Rafiqi FH, Deak M, Morrice NA, Karlsson HK, Alessi DR. 2006. Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem J 397:223–231
- Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, et al. 2011. Structure of mammalian AMPK and its regulation by ADP. Nature 472:230–233
- Yang T, Park JM, Arend L, Huang Y, Topaloglu R, Pasumarthy A, et al. 2000. Low chloride stimulation of prostaglandin E2 release and cyclooxygenase-2 expression in a mouse macula densa cell line. J Biol Chem 275:37922–37929
- Zhang L, He H, Balschi JA. 2007. Metformin and phenformin activate AMP-activated protein kinase in the heart by increasing cytosolic AMP concentration. Am J Physiol Heart Circ Physiol 293:H457–466