
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Solid-state NMR is unique for its ability to obtain three-dimensional structures and to measure atomic-resolution structural and dynamic information for membrane proteins in native lipid bilayers. An increasing number and complexity of integral membrane protein structures have been determined by solid-state NMR using two main methods. Oriented sample solid-state NMR uses macroscopically aligned lipid bilayers to obtain orientational restraints that define secondary structure and global fold of embedded peptides and proteins and their orientation and topology in lipid bilayers. Magic angle spinning (MAS) solid-state NMR uses unoriented rapidly spinning samples to obtain distance and torsion angle restraints that define tertiary structure and helix packing arrangements. Details of all current protein structures are described, highlighting developments in experimental strategy and other technological advancements. Some structures originate from combining solid- and solution-state NMR information and some have used solid-state NMR to refine X-ray crystal structures. Solid-state NMR has also validated the structures of proteins determined in different membrane mimetics by solution-state NMR and X-ray crystallography and is therefore complementary to other structural biology techniques. By continuing efforts in identifying membrane protein targets and developing expression, isotope labelling and sample preparation strategies, probe technology, NMR experiments, calculation and modelling methods and combination with other techniques, it should be feasible to determine the structures of many more membrane proteins of biological and biomedical importance using solid-state NMR. This will provide three-dimensional structures and atomic-resolution structural information for characterising ligand and drug interactions, dynamics and molecular mechanisms of membrane proteins under physiological lipid bilayer conditions.
Introduction
Membrane proteins are coded by up to 30% of the open reading frames in known genomes (Fagerberg et al., Citation2010; Liu & Rost, Citation2001; Wallin & von Heijne, Citation1998) and typical biological membranes consist of up to 50% mass fraction of proteins (Johansson & Lindahl, Citation2009). They have important roles in many biological processes (e.g., transport of ions and molecules, control of transmembrane potential, generation and transduction of energy, signal recognition and transduction, catalysis of chemical reactions) and mutations in membrane proteins have been linked with a number of human diseases (Klepper & Voit, Citation2002; Kurze et al., Citation2010; Partridge et al., Citation2002; Patching, Citation2015; Quadri et al., Citation2012; Ragona et al., Citation2014; Rosenbaum et al., Citation2009; Sanders & Myers, Citation2004; Shukla et al., Citation2012; Striano et al., Citation2012; Suls et al., Citation2009; von Heijne, Citation2007; Watanabe et al., Citation2008; Weber et al., Citation2008). The molecular targets for around 50–60% of current validated medicines are membrane proteins and they remain the principal target for new drug discovery (Bahar et al., Citation2010; Bakheet & Doig, Citation2009; Drews, Citation2000; Hopkins & Groom, Citation2002; Lundstrom, Citation2006; Overington et al., Citation2006; Rask-Andersen et al., Citation2011) with many more likely to be identified following efforts to map the tissue-specific human proteome (Lindskog, Citation2015; Uhlén et al., Citation2015). Owing to the difficulties in applying the main biophysical techniques for atomic-resolution protein structure determination, X-ray crystallography, single particle electron microscopy and NMR spectroscopy, the number of structures of membrane proteins is still relatively few. They contribute only around 2.5% of entries in the Protein Data Bank (PDB) (http://www.rcsb.org/pdb/home/home.do), thus limiting the amount of information available for traditional structure-based drug design. There is an almost infinite amount of information yet to be obtained about the structures, ligand interactions, molecular mechanisms, dynamics and multidimensional relationships of membrane proteins along with scope for development and application of a wide range of chemical, biochemical, biophysical, molecular imaging and computational techniques to achieve this. Solid-state NMR spectroscopy is one such technique with significant recent developments and huge future potential for investigating integral membrane proteins such as receptors, channels and transporters, including their roles as molecular targets in the discovery of new drugs.
Solid-state NMR spectroscopy is unique for its ability to obtain three-dimensional structures and to measure atomic-resolution distance and orientational structural restraints for membrane proteins in their native membranes or reconstituted under near-native conditions in lipid bilayers, proteoliposomes or in bicelles. Since sample environment has a significant effect on membrane protein folding and structure (Chen et al., Citation2014; Cross et al., Citation2014; Murray et al., Citation2014a), structural information obtained using solid-state NMR can be of the highest physiological relevance. Developments in membrane protein expression, isotope labelling, reconstitution and NMR sample preparation strategies and in NMR pulse sequences, probe design, higher field magnets and computational methods have led to an increasing number and complexity of integral membrane protein structures determined by solid-state NMR (http://www.drorlist.com/nmr/SPNMR.html). A number of recent review articles on this theme are available (Brown & Ladizhansky, Citation2015; Cross et al., Citation2014; Franks et al., Citation2012; Hong et al., Citation2012; Judge & Watts, Citation2011; Judge et al., Citation2015; Opella, Citation2013a, Citation2014, Citation2015; Radoicic et al., Citation2014; Wang & Ladizhansky, Citation2014; Ward et al., Citation2015; Yao et al., Citation2013; Zhao, Citation2012). In some cases, structural information from solid-state NMR has been combined with information from other techniques such as X-ray crystallography and solution-state NMR, which can enhance structural validity and provide a more comprehensive understanding of the protein under investigation. Solid-state NMR is therefore complementary to other structural biology techniques.
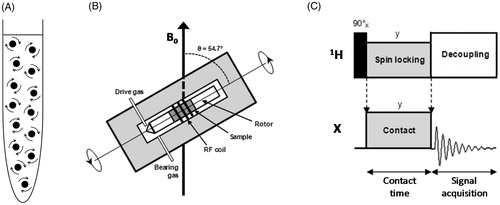
Obtaining structural information from membrane proteins by NMR requires the acquisition of high-resolution and high sensitivity NMR spectra of the isotopically-labelled (13C, 15N-labelled) protein. In the case of solution-state NMR, narrow spectral lines are principally achieved through the intrinsic rapid isotropic tumbling of the purified and solubilized protein in the NMR sample (). There is a limit to the size of the particles that can be analysed, however, and elevated sample temperatures (20–50 °C) usually have to be used to accelerate particle tumbling, which can introduce issues of sample stability. Detergent micelles are most often used for solubilization (Arora, Citation2013; Hiruma-Shimizu et al., Citation2014, Citation2015; Kim et al., Citation2009; Klammt et al., Citation2012; Maslennikov & Choe, Citation2013; Nietlispach & Gautier, Citation2011; Page et al., Citation2006; Patching, Citation2011; Reckel & Hiller, Citation2013) as well as other membrane mimetics such as organic solvents, lipids, bicelles, nanodiscs, fluorinated surfactants and amphipols (Bayburt & Sligar, Citation2010; Dürr et al., Citation2012, Citation2013; Inagaki et al., Citation2013; Popot, Citation2010; Warschawski et al., Citation2011; Zhou & Cross, Citation2013a; Zoonens & Popot, Citation2014). For larger membrane proteins that tumble more slowly and also have higher spectral crowding, deuteration of the protein and detergent, selective isotope labelling and TROSY-type NMR experiments are also used to achieve NMR spectra of the required quality (Patching, Citation2011; Hiruma-Shimizu et al., Citation2014, Citation2015; Kalverda et al., Citation2014). In the case of solid-state NMR, where the NMR sample is solid or semi-solid, there is no limit to the size of the complex that can be analysed and alternative methods have to be used to achieve narrow spectral lines. Uniformly or selectively isotope labelled membrane proteins can be analysed in their native membranes, reconstituted in lipid bilayers, proteoliposomes or bicelles, solubilized and frozen in detergent micelles or analyzed as crystals or microcrystals. Depending on the methods used, the sample temperature can be below 4 °C, which favours membrane protein stability. Removal of the line broadening effects of dipole-dipole coupling and chemical shift anisotropy interactions in solids is achieved by applying a dipolar decoupling pulse sequence and by rapid mechanical rotation of the NMR sample (typically at frequencies of 1–35 kHz) held at an angle of 54.74° relative to the applied magnetic field, which is known as magic-angle spinning (MAS) (Andrew et al., Citation1958, Citation1959; Andrew, Citation2010; Hennel & Klinowski, Citation2005; Lowe, Citation1959) (). The signal-to-noise ratio for observing dilute spin low gyromagnetic ratio nuclei (e.g., 13C, 15N) is enhanced by transferring magnetization from abundant high gyromagnetic ratio nuclei (usually 1H) using the cross-polarization (CP) experiment (Pines et al., Citation1973) (). These methods are combined to give the basic CP-MAS experiment for achieving high-resolution solid-state NMR spectra (Stejskal & Memory, Citation1994; Taylor, Citation2004; Yannoni, Citation1982). With membrane protein samples, further NMR experiments of varying complexity are then employed to obtain appropriate spectra for extracting assignment and coupling information, distance and orientational restraints and relaxation and dynamic parameters (Astrof & Griffin, Citation2002; Das et al., Citation2013; Gopinath & Veglia, Citation2015; Higman et al., Citation2009; Huber et al., Citation2012; Ladizhansky, Citation2009; Lalli et al., Citation2011; Lu & Opella, Citation2014; Miao et al., Citation2013; Mote et al., Citation2011, Citation2013; Shi & Ladizhansky, Citation2012; Sperling et al., Citation2010; Tang et al., Citation2013a).
Figure 1. Methods for acquiring high-resolution NMR spectra. (A) Rapid and random tumbling of molecules in a solution-state NMR sample. (B) The solid-state NMR magic-angle spinning (MAS) experiment. A solid or semi-solid sample packed into the centre of a cylindrical rotor (outside diameter 1.3–15 mm) is rapidly spun (typically at frequencies of 1–35 kHz) in the NMR magnet about an angle of 54.7° relative to the applied magnetic field (B0) (Andrew et al., Citation1958, Citation1959; Lowe, Citation1959). (C) The solid-state NMR cross-polarization (CP) experiment. The basic CP pulse sequence transfers magnetization excited on abundant 1H nuclei to dilute low gyromagnetic ratio nuclei (X = 13C, 15N, etc) during a ‘contact’ period (Pines et al., Citation1973). During the contact time, radiofrequency fields for both 1H and X are turned on and the ratio of power levels between 1H and X are made equal to the ratio of their gyromagnetic ratios () under Hartmann-Hahn matching conditions (Hartmann & Hahn, Citation1962). Following the contact time, the enhanced magnetization on the X nuclei is detected whilst the abundant protons are decoupled. Recycling of the pulse sequence is dependent on T1H and not T1X, which allows accumulation of many more repetitions in a given time. The pictures in B and C were modified from Middleton and Patching (Citation2013).
This review article first gives an overview of membrane protein structure determination by solid-state NMR using two main methods, those that use oriented samples without MAS and those that use unoriented samples with MAS. Isotope labelling strategies, sample preparation procedures and NMR experiments are also considered. Structures of integral membrane proteins determined by solid-state NMR are then described and compared, highlighting important structures and breakthroughs in experimental strategy and technology. Future perspectives for using solid-state NMR to determine structures of membrane proteins are also considered.
Membrane protein structure determination by solid-state NMR
A majority of the membrane protein structures determined by solid-state NMR have used macroscopically oriented samples of the protein reconstituted in lipid bilayers. MAS solid-state NMR methods have been used to obtain structural information by measurement of accurate distances between specific atoms and by measurement of dihedral torsion angles with the protein reconstituted in lipid bilayers or as microcrystals. In some cases a combination of oriented sample and MAS methods have been used for full structure determination. It should be emphasized that rational and efficient isotope labelling of the protein and robust and high quality sample preparation are essential in all cases. Each of these methods will now be considered in turn.
Oriented sample solid-state NMR
Alignment of a solid sample in a single specific orientation in the static field of an NMR magnet (rather than the random distribution in powder samples) eliminates much of the line broadening interactions. It is fortunate that lipid bilayers have a natural tendancy to self-assemble in an aligned manner such that membrane proteins in highly oriented lipid bilayers provide very useful samples for membrane protein structure determination. Macroscopic orientation of membrane protein samples in the NMR magnet is achieved by two main techniques: Mechanical alignment of lipid bilayers between glass plates and magnetic alignment of bicelles (). Mechanical alignment typically requires 5–20 mg of isotopically-labelled protein reconstituted into lipids with complete removal of detergent (Bechinger et al., Citation1996; Das et al., Citation2013; Middleton et al., Citation2000; Nevzorov et al., Citation2004). The alignment procedure usually involves transfer of proteoliposomes onto thin glass plates (typically 35–40 in number), partial dehydration under controlled temperature and relative humidity, stacking of the slides, rehydration under controlled temperature and relative humidity followed by sealing of the final sample (Das et al., Citation2013; Hansen et al., Citation2015). The resultant sample has lipid layers containing embedded proteins with the bilayer normal aligned parallel to the direction of the magnetic field (). Magnetic alignment of samples is made possible by exploiting the unique phase behaviour of bicelles, which are typically composed of a long-chain (C12–C18) phospholipid (e.g., DMPC, DMPG, POPC, POPG) and a short-chain detergent (e.g., DHPC, 6-O-PC, DH7PC, DPC) (De Angelis & Opella, Citation2007; Diller et al., Citation2009; Kim, Citation2006; Marcotte & Auger, Citation2005; Nolandt et al., Citation2012). The long-chain component constitutes the planar bilayer and the short-chain component forms the edges of the bicelle. Under favourable conditions of lipid composition, concentration and temperature, bicelles spontaneously align in the static field of an NMR magnet with their bilayer normal perpendicular to the direction of the magnetic field () (Dürr et al., Citation2013; Mote et al., Citation2011; Nolandt et al., Citation2012; Whiles et al., Citation2002). Magnetic alignment typically occurs for a concentrated lipid mixture with a q value (ratio of long-chain to short chain component) of greater than 2.5 and at temperatures above the gel to liquid crystalline transition temperature of the long-chain component (Kim, Citation2006). Uniform alignment of the long-chain lipid component into bilayers in the magnetic field is confirmed by observing sharp upfield signals in 31P NMR spectra (Sanders & Schwonek, Citation1992; Triba et al., Citation2006). Bicelles can be flipped to a parallel alignment with the magnetic field by addition of paramagnetic ions (Prosser et al., Citation1996) or by the use of diphenyl lipids (Loudet et al., Citation2007; Park et al., Citation2008) ().
Figure 2. Oriented samples of a membrane protein in a mechanically aligned lipid bilayer and in magnetically aligned bicelles. Orientation of a membrane protein mechanically aligned in a lipid bilayer on glass plates (A), magnetically aligned in bicelles with the bilayer normal perpendicular to the direction of the magnetic field (B) and magnetically aligned in flipped bicelles with the bilayer normal parallel to the direction of the magnetic field (C). The arrow indicates the direction of the magnetic field. This picture was modified with permission from Kim (Citation2006), which was originally published in Bull Korean Chem Soc [Kim Y. Citation2006. Solid-state NMR studies of membrane proteins using phospholipid bicelles. Bull Korean Chem Soc 27:386–388], copyright by Korean Chemical Society 2006.
![Figure 2. Oriented samples of a membrane protein in a mechanically aligned lipid bilayer and in magnetically aligned bicelles. Orientation of a membrane protein mechanically aligned in a lipid bilayer on glass plates (A), magnetically aligned in bicelles with the bilayer normal perpendicular to the direction of the magnetic field (B) and magnetically aligned in flipped bicelles with the bilayer normal parallel to the direction of the magnetic field (C). The arrow indicates the direction of the magnetic field. This picture was modified with permission from Kim (Citation2006), which was originally published in Bull Korean Chem Soc [Kim Y. Citation2006. Solid-state NMR studies of membrane proteins using phospholipid bicelles. Bull Korean Chem Soc 27:386–388], copyright by Korean Chemical Society 2006.](/cms/asset/0a03a959-ebc6-4a25-9f5a-889435f38f68/imbc_a_1139754_f0002_b.jpg)
Challenges associated with both mechanical and magnetic alignment procedures include production of sufficient quantities of isotopically-labelled protein, achieving a protein:lipid ratio and concentration that favours successful alignment and gives enough protein in the NMR sample for signal detection. This is important for avoiding poor signal to noise and lengthy spectrometer time. Such parameters usually have to be considered on a case by case basis with each protein under investigation. Furthermore, the success of the experiment depends on an extremely high degree of macroscopic sample order, which has only been achieved in relatively few cases. Many of these cases involved mechanical alignment of single-span systems from organic solvents, which is not compatible with the large majority of membrane proteins. Unless the use of organic solvents can be avoided, the magnetic alignment approach appears to be more promising in this respect. Since magnetically aligned samples usually involve the introduction of a non-native short-chain lipid or detergent in order to form bicelles, mechanically aligned samples may be the closest to physiological conditions, however. Analysis of the reconstituted protein by other biochemical and biophysical techniques is also useful for confirming structural and functional integrity of the protein used in NMR samples.
Oriented sample solid-state NMR mainly uses 15N detection for deriving structural restraints to avoid the large homonuclear interactions present in uniformly 13C-labelled proteins and it can derive orientation restraints directly from measuring frequencies of single-line resonances. Measurement of 1H-15N dipolar couplings and 15N anisotropic chemical shifts provides tilt and azimuthal (rotational) angles of the protein domains to determine the orientations of contiguous peptide planes relative to the lipid bilayer. This provides information that defines the secondary structure and global fold of the protein and also the orientation and topology of the protein within the lipid bilayer. Separated local field (SLF) experiments (Lopez et al., Citation2007) are used to provide spectral resolution and pulse sequences such as PISEMA (Wu et al., Citation1994), SAMMY (Nevzorov & Opella, Citation2003), SAMPI-4 (Nevzorov & Opella, Citation2003), HIMSELF (Dvinskikh et al., Citation2006) and PELF (Gopinath et al., Citation2011) are used to correlate backbone dipolar couplings with anisotropic chemical shifts of uniformly 15N labelled proteins. Such anisotropic parameters are used as orientation restraints for calculation of membrane protein structure and topology. For example, application of the PISEMA experiment for membrane protein structure determination was demonstrated for the membrane bound form of the 50-residue fd bacteriophage pVIII coat protein oriented in POPC/POPG bilayers (Marassi & Opella, Citation2003). Two-dimensional experimental PISEMA spectra were obtained from one uniformly 15N-labelled sample and from four selectively 15N-labelled samples (Ala, Gly, Leu and Val) of the protein (). Pisa wheel PISEMA spectra (Marassi & Opella, Citation2000) were also calculated from a protein model based on the amino acid sequence with forced helical and rotational parameters (). Matching of calculated and experimental PISEMA spectra helped to achieve complete sequential resonance assignments, a high-resolution three-dimensional structure of the protein and to define its orientation in the lipid bilayer. The structure has a transmembrane helix (residues 21–45) with a tilt angle of 26° up to residue 40, where it changes to 16° (). Tilting allows this 35-Å-long helix to be accommodated in the lipid bilayer, which in the case of POPC/POPG has a thickness of around 31 Å. A short turn sequence (residues 19–20) links the transmembrane helix to a 16-Å-long N-terminal amphipathic helix (residues 8–18) that lies on the surface of the lipid bilayer, in which non-polar residues face the hydrophobic lipid core and polar residues face the aqueous environment (Marassi & Opella, Citation2003).
Figure 3. Structure determination of the fd bacteriophage pVIII coat protein using oriented samples and PISEMA solid-state NMR. (A) Sequence of the 50-residue fd bacteriophage pVIII coat protein indicating residues that comprise an amphipathic helix (8–18) and a transmembrane helix (21–45). Residues selectively 15N-labelled in separate samples are highlighted with colours: Ala (red), Gly (yellow), Leu (green), Val (blue). (B) Experimental PISEMA spectrum obtained from uniformly 15N-labelled protein in POPC/POPG bilayers. (C) Pisa wheel PISEMA spectrum calculated from a protein model with the amphipathic helix parallel to within 3° of the membrane surface and the transmembrane helix with a tilt angle of 30°. The helical and Pisa wheel rotations are set to match the resonances in the experimental PISEMA spectra from the Ala (red), Gly (yellow), Leu (green), and Val (blue) residues. (D) Structure of the fd bacteriophage pVIII coat protein in POPC/POPG bilayers. The structure is coloured with the N-terminus in blue (top left) and the C-terminus in red (bottom). The dashed grey line represents the membrane-water interface and the arrow represents the direction of the magnetic field. The pictures in A–C were reproduced with permission from Marassi and Opella (Citation2003), which were originally published in Prot Sci [Marassi FM, Opella SJ. Citation2003. Simultaneous assignment and structure determination of a membrane protein from NMR orientational restraints. Protein Sci 12:403–411.], copyright by The Protein Society 2003. The structure in D was drawn from the PDB file 1MZT using Jmol: An open-source Java viewer for chemical structures in 3D (http://www.jmol.org/) (Herráez, Citation2006). This Figure is reproduced in colour in Molecular Membrane Biology online.
![Figure 3. Structure determination of the fd bacteriophage pVIII coat protein using oriented samples and PISEMA solid-state NMR. (A) Sequence of the 50-residue fd bacteriophage pVIII coat protein indicating residues that comprise an amphipathic helix (8–18) and a transmembrane helix (21–45). Residues selectively 15N-labelled in separate samples are highlighted with colours: Ala (red), Gly (yellow), Leu (green), Val (blue). (B) Experimental PISEMA spectrum obtained from uniformly 15N-labelled protein in POPC/POPG bilayers. (C) Pisa wheel PISEMA spectrum calculated from a protein model with the amphipathic helix parallel to within 3° of the membrane surface and the transmembrane helix with a tilt angle of 30°. The helical and Pisa wheel rotations are set to match the resonances in the experimental PISEMA spectra from the Ala (red), Gly (yellow), Leu (green), and Val (blue) residues. (D) Structure of the fd bacteriophage pVIII coat protein in POPC/POPG bilayers. The structure is coloured with the N-terminus in blue (top left) and the C-terminus in red (bottom). The dashed grey line represents the membrane-water interface and the arrow represents the direction of the magnetic field. The pictures in A–C were reproduced with permission from Marassi and Opella (Citation2003), which were originally published in Prot Sci [Marassi FM, Opella SJ. Citation2003. Simultaneous assignment and structure determination of a membrane protein from NMR orientational restraints. Protein Sci 12:403–411.], copyright by The Protein Society 2003. The structure in D was drawn from the PDB file 1MZT using Jmol: An open-source Java viewer for chemical structures in 3D (http://www.jmol.org/) (Herráez, Citation2006). This Figure is reproduced in colour in Molecular Membrane Biology online.](/cms/asset/a37c4b96-34d3-4607-8ac2-bdfb06f6d916/imbc_a_1139754_f0003_c.jpg)
Sequential resonance assignments are ideally performed for full structure elucidation, which are assisted by performing multidimensional NMR experiments (Astrof & Griffin, Citation2002; Gopinath et al., Citation2013, Citation2015; Lu & Opella, Citation2014; Mote et al., Citation2011) and by employing selective, reverse and sparse isotope labelling strategies (Esteban-Martín et al., Citation2010; Filipp et al., Citation2009; Higman et al., Citation2009; Lin et al., Citation2011; Verardi et al., Citation2012). The overlap of signals in NMR spectra of helical proteins is a widespread problem and overcoming spectral crowding becomes more challenging as the protein under investigation becomes larger and more complex with multiple unique transmembrane helices. In oriented samples of 13C/15N-labelled proteins, the strong 13C-13C homonuclear dipolar couplings generally make 13C-detection and use of resonance assignment experiments intractable. Furthermore, full characterization of proteins with multiple helices requires measurement of distances between the helices to define the helix packing arrangement. Hence, oriented sample solid-state NMR has been successful in determining the structures of many membrane proteins with a single transmembrane spanning helix, but other complementary methods usually have to be used for achieving structures of proteins with two or more unique transmembrane spanning helices.
MAS solid-state NMR
MAS of the sample () provides high-resolution spectra containing information on isotropic chemical shifts and interatomic distances and torsional angles. Since MAS removes 13C-13C homonuclear dipolar couplings in uniformly 13C/15N-labelled proteins, it allows 13C-detection and use of an array of experiments for sequential resonance assignments. Under MAS conditions the recoupling of dipolar interactions between labelled atoms allows measurement of through-space accurate distances (≤0.1 Å of up to around 5 Å using 1H, 2H, 13C, 15N) between specific sites in different residues of the protein that fix one site relative to the other and can therefore provide extremely high-resolution distance restraints for structure calculation. For example, the 2D experiments proton-driven spin diffusion (PDSD) (Bloembergen, Citation1949; Grommek et al., Citation2006) and dipolar-assisted rotational resonance (DARR) (Takegoshi et al., Citation2001, Citation2003) recouple 13C-13C interactions and the experiments rotational-echo double resonance (REDOR) (Gullion & Schaefer, Citation1989) and transferred-echo double resonance (TEDOR) (Hing et al., Citation1992) recouple heteronuclear interactions such as 13C-15N. Longer range heteronuclear distances can be measured using these experiments if higher gyromagnetic ratio nuclei such as 19F are introduced at specific sites, increasing the distance limit to around 7, 10 and 15 Å for 15N-19F, 13C-19F, and 19F-19F, respectively. For a transmembrane protein that already has a well-defined backbone structure based on orientational restraints from aligned samples, only a few distance restraints are required to define tertiary and oligomeric structures of the transmembrane domains. In this way, a combination of restraints from oriented sample and MAS solid-state NMR can provide atomic-resolution structures of membrane proteins (Murray et al., Citation2013). Such measurements are assisted by employing selective, reverse and sparse isotope labelling strategies to target specific sites. For example, a recent structure of the cell division structural and regulatory protein CrgA from Mycobacterium tuberculosis was determined from PISEMA and CP-MAS experiments on uniformly 13C,15N-labelled and amino acid specific 15N-labelled samples and on samples produced by reverse labelling (Das et al., Citation2015a) (see below).
Using MAS solid-state NMR methods on their own it is challenging to obtain a sufficient number of unambiguous distance and torsional restraints for full three-dimensional structural characterization. Hence, relatively few structures of integral membrane proteins have been determined using MAS solid-state NMR methods alone. Impressively though, structures determined exclusively by MAS methods include that of the seven transmembrane spanning Anabaena sensory rhodopsin in a trimeric form (Wang et al., Citation2013) (see below). MAS solid-state NMR is also very useful for elucidating the atomic-resolution structures, molecular conformations, positions and orientations of ligands and drug compounds in the binding sites of membrane proteins from measurement of accurate interatomic distances and torsion angles (Cady et al., Citation2010; Edwards et al., Citation2010; Lakatos et al., Citation2012; Lopez et al., Citation2008; Middleton et al., Citation2011; Patching et al., Citation2008, Citation2013; Whittaker et al., Citation2015; Williamson et al., Citation2007). Such measurements can contribute important information for the design and discovery of drugs involving membrane-embedded targets (Ding et al., Citation2013a; Tapaneeyakorn et al., Citation2011; Watts, Citation2005; Williamson, Citation2009).
A form of MAS solid-state NMR referred to as ‘rotational alignment’ has been introduced by Opella and colleagues that merges the capabalities of oriented sample and MAS solid-state NMR for membrane protein structure determination (Park et al., Citation2010a). The ‘rotational alignment’ concept is based on an equivalence of structural restraints from oriented and unoriented (powder) samples of membrane proteins. The alignment of membrane proteins is defined by the direction of the bilayer normal relative to the magnetic field and they also undergo fast rotational diffusion about the same bilayer normal in liquid crystalline membranes (Das et al., Citation2015b). This rotational diffusion averages the chemical shift anisotropy and heteronuclear dipolar coupling powder patterns to axially symmetric powder patterns with reduced frequency spans. Importantly, it is found that the frequencies associated with the parallel edges of the motionally averaged powder patterns can be used to measure the angle between the principal axis of the spin-interaction tensor and the bilayer normal. Furthermore, the parallel edge of a rotationally averaged powder pattern has a frequency exactly matching that of the single-line resonance obtained in the spectrum of a static sample aligned parallel to the magnetic field (Opella Citation2013a, Citation2013b; Park et al., Citation2010a). In favourable cases, it is therefore possible to measure additional orientational restraints from 13C/15N-labelled samples in unoriented lipid bilayers. Under MAS conditions, it is then possible to apply an array of multidimensional experiments for measuring orientation-dependent frequencies from the motionally averaged powder patterns and distance restraints, including those from 13C-detection, along with systematic resonance assignment schemes for complete three-dimensional structure determination (Das et al., Citation2014; Marassi et al., Citation2011). The concept of ‘rotational alignment’ was first demonstrated using two proteins with a single transmembrane α-helix, the HIV Vpu channel and the full length membrane-bound form of fd bacteriophage pVIII coat protein in phospholipid bilayers (Park et al., Citation2010a). For example, experiments on 15N-leucine site-specific labelled Vpu transmembrane domain showed how it is feasible to measure parallel and perpendicular edges of motionally averaged powder patterns from MAS spectra of unoriented samples (). The experimental spectrum and sideband intensities ‘back calculated’ from the powder pattern were in close agreement with each other under static conditions (low temperature) and whilst the protein was undergoing rapid rotational diffusion (high temperature). Essentially identical results were obtained using two different MAS frequencies. These results were also in agreement with 15N measurements on the same 15N-leucine labelled Vpu transmembrane domain in static aligned and unoriented samples (Park et al., Citation2010a). ‘Rotational alignment’ has since been used to assist structure determination of a number of integral membrane proteins (Opella, Citation2013a, Citation2013b), including that of the seven transmembrane spanning human chemokine receptor CXCR1 (Park et al., Citation2012) (see below). The concept may be useful in further cases where membrane proteins rotate about their long axis and the dipole N-H couplings project on to this long axis such that they can be interpreted in terms of additional angle constraints.
Figure 4. Measurement of parallel and perpendicular edges of motionally averaged powder patterns from MAS spectra of unoriented samples. MAS solid-state 15N NMR spectra of 15N-leucine-11 labelled transmembrane domain of HIV Vpu channel in unoriented DMPC lipid bilayers. In each panel (A–D) the three spectra represent a powder pattern calculated from experimental sideband intensities (top), sideband intensities ‘back calculated’ from the powder pattern (middle) and the experimental NMR spectrum (bottom). Experimental spectra were obtained at high temperature (30 °C, A and B) and at low temperature (5 °C, C and D) and using MAS frequencies of 2.2 kHz (A and C) and 3.0 kHz (B and D). This Figure was reproduced with permission from Park et al. (Citation2010a), which was originally published in J Phys Chem B [Park SH, Das BB, De Angelis AA, Scrima M, Opella SJ Citation2010a. Mechanically, magnetically, and “rotationally aligned” membrane proteins in phospholipid bilayers give equivalent angular constraints for NMR structure determination. J Phys Chem B 114:13995–14003], copyright by American Chemical Society 2010.
![Figure 4. Measurement of parallel and perpendicular edges of motionally averaged powder patterns from MAS spectra of unoriented samples. MAS solid-state 15N NMR spectra of 15N-leucine-11 labelled transmembrane domain of HIV Vpu channel in unoriented DMPC lipid bilayers. In each panel (A–D) the three spectra represent a powder pattern calculated from experimental sideband intensities (top), sideband intensities ‘back calculated’ from the powder pattern (middle) and the experimental NMR spectrum (bottom). Experimental spectra were obtained at high temperature (30 °C, A and B) and at low temperature (5 °C, C and D) and using MAS frequencies of 2.2 kHz (A and C) and 3.0 kHz (B and D). This Figure was reproduced with permission from Park et al. (Citation2010a), which was originally published in J Phys Chem B [Park SH, Das BB, De Angelis AA, Scrima M, Opella SJ Citation2010a. Mechanically, magnetically, and “rotationally aligned” membrane proteins in phospholipid bilayers give equivalent angular constraints for NMR structure determination. J Phys Chem B 114:13995–14003], copyright by American Chemical Society 2010.](/cms/asset/70751f93-e7ca-472d-b3d0-490478c8ed07/imbc_a_1139754_f0004_b.jpg)
Structures of integral membrane proteins
As of October 2015 there were thirty six entries in the Protein Data Bank (PDB) for integral membrane protein structures determined by solid-state NMR ( and ). These represented 20 unique proteins coming from a number of different organisms and with a range of functions. Of the 20 different proteins, two had a β-strand transmembrane domain, 13 had a single transmembrane α-helix or existed as oligomers thereof, two had two unique transmembrane α-helices, one had four unique transmembrane α-helices and two had seven unique transmembrane α-helices. The information given in confirms that a majority of the structures were determined from oriented samples of the protein reconstituted in lipid bilayers and also from unoriented samples using MAS methods and, in some cases, combinations of these. The gallery in demonstrates that challenges are continuously being overcome to enable structure determination of integral membrane proteins of increasing size and complexity using solid-state NMR. The following sections will consider the structures and the solid-state NMR methods used to achieve them for proteins with a β-strand transmembrane domain and then those with an α-helical transmembrane domain in order of increasing size and complexity.
Figure 5. Gallery of solid-state NMR structures of integral membrane proteins. Backbone structures are shown for integral membrane proteins that have been determined by solid-state NMR or by solid-state NMR in combination with another technique (e.g., crystallography, solution-state NMR) and that have a PDB entry and have been published in a peer-reviewed journal (up to October 2015). Proteins are arranged (β-strand then α-helical transmembrane domain) in order of increasing size and complexity and they correspond with the details given in . Structures are coloured with the N-terminus in blue and the C-terminus in red. Structures were drawn with the given PDB file using Jmol: an open-source Java viewer for chemical structures in 3D (http://www.jmol.org/) (Herráez, Citation2006). This Figure is reproduced in colour in Molecular Membrane Biology online.
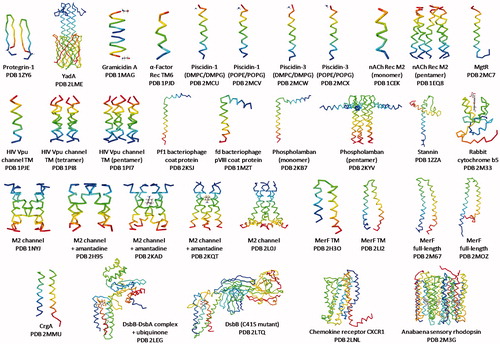
Table 1. Details of solid-state NMR structures of integral membrane proteins. Information is provided for structures of integral membrane proteins that have been determined by solid-state NMR or by solid-state NMR in combination with another technique (e.g., solution-state NMR, X-ray crystallography) and that have a PDB entry and have been published in a peer-reviewed journal (up to October 2015). Proteins are listed (β-strand then α-helical transmembrane domain) in order of increasing size and complexity and they correspond with the structures shown in .
Proteins with a β-strand transmembrane domain
There are two PDB entries for structures of proteins with a β-strand transmembrane domain that have been determined by solid-state NMR, protegrin-1 and autotransporter adhesion protein YadA.
Protegrin-1
Protegrin-1 is a porcine 19-residue cysteine/arginine-rich antimicrobial cationic peptide, which induces pore formation in microbial membranes. The structure of protegrin-1 was determined using MAS methods exclusively. REDOR distance measurements (13C-19F, 1H-13C and 15N-13C) on site-specifically labelled protegrin-1 in POPC lipid bilayers revealed a β-hairpin structure with two molecules aligned in parallel with the C-terminal strand of the hairpin forming the dimer interface (, PDB 1ZY6) (Mani et al., Citation2006). The dimer interface is stabilized by six hydrogen bonds and it was noted that this parallel arrangement determined in lipids differs from an antiparallel dimer structure of the peptide determined in DPC micelles by solution-state NMR (Roumestand et al., Citation1998). Subsequent molecular dynamics simulations showed that the parallel arrangement of the protegrin-1 dimer induces the greatest membrane disruption effect at the amphipathic interface of the lipid bilayer (Jang et al., Citation2007).
Autotransporter adhesion protein YadA
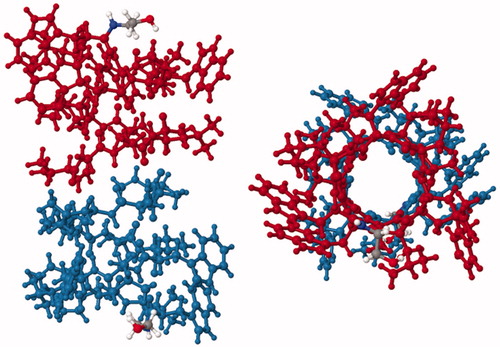
More recently, a structure of the membrane anchor domain of the Trimeric Autotransporter Adhesin (TAA) family protein YadA from the outer membrane of the bacterium Yersinia enterocolitica has been determined by solid-state NMR. TAA proteins are important pathogenicity factors that mediate adhesion to host cells and tissues in conditions such as diarrhea, urinary tract infections and and airway infections. The structure was solved exclusively by MAS methods on microcrystals of the protein using a single uniformly 13C,15N-labelled sample, thus demonstrating that complex labelling techniques are not always necessary for complete structure determination (Shahid et al., Citation2012). A range of two-dimensional homonuclear and heteronuclear correlation NMR spectra (13C-13C PAR, 13C-13C DARR, TEDOR, NHHC, CHHC) were obtained from which 1192 non-redundant distance restraints were manually assigned for structure calculation using an inferential structure determination (ISD) approach (Bayrhuber et al., Citation2008). The 105-residue protomer comprises an N-terminal helix (residues 14–43) followed four β-strands (residues 53–63, 66–76, 81–90, 94–103) that insert into the membrane. This oligomerizes to form a trimeric β-barrel through which the helical domain passes in the form of a trimeric coiled coil (, PDB 2LME and ). The barrel is stabilized by hydrogen bonds between strands β1 and β4 of neighbouring protomers and its interior comprises mostly small amino acids (alanine, asparagine, glycine, serine, threonine) in order to accommodate the helical bundle. Only one bulky hydrophobic residue (methionine-96) faces into the pore and its sulphur atom is coordinated by the side chains of serine-38 and serine-39 in the helix. Dynamic measurements also obtained by solid-state NMR provided information on the flexibility and mobility of parts of the structure and insights into the autotransport mechanism of YadA. This structure of YadA demonstrated the potential for using MAS solid-state NMR methods with microcrystalline samples of membrane proteins for structure determination.
Figure 6. Solid-state NMR structure of the autotransporter adhesion protein YadA. This backbone structure is coloured with the N-terminus in blue (top) and the C-terminus in red. The structure was drawn with PDB file 2LME using Jmol: An open-source Java viewer for chemical structures in 3D (http://www.jmol.org/) (Herráez, Citation2006). This Figure is reproduced in colour in Molecular Membrane Biology online.

Proteins with a single transmembrane α-helix and oligomers thereof
A large majority of the solid-state NMR structures of proteins with a single transmembrane α-helix or oligomers thereof have been determined using oriented methods. Although a matter of definition, it should be mentioned here that some may consider many of these single transmembrane helix systems to be peptides rather than proteins. A few have had contributions from MAS methods or been determined by MAS methods exclusively. In some cases, solid-state NMR measurements have been combined with information from solution-state NMR to calculate the structures.
Gramicidin A ion channel
The first complete high-resolution structure of a protein in a lipid bilayer determined by solid-state NMR was that of the 15-residue ion channel gramicidin A. The structure was derived entirely on the basis of the amino acid sequence and 144 orientational restraints for the protein mechanically aligned in DMPC bilayers within 0.5 Å of ideal geometry (Ketchem et al., Citation1993, Citation1996). The protein was revealed to have an unusual transmembrane conformation as a right-handed L-helix with 6.5 residues per turn, which results from the alternating L- and D-amino acids in the gramicidin A sequence. One of these helices is only capable of spanning a single lipid monolayer, so a head-to-head dimerization occurs to achieve a pore that spans the bilayer (, PDB 1MAG and ). The dynamics in the backbone were also characterized from powder pattern averaging and from anisotropic 15N T1 relaxation measurements (Nicholson et al., Citation1991, North & Cross, Citation1993, Citation1995), then intra- and intermolecular distance measurements by MAS solid-state NMR confirmed the structure of the monomer and the model of the momomer-monomer interface (Fu et al., Citation2000). This breakthrough structure of gramicidin A demonstrated the potential of solid-state NMR as a feasible technique for determining the complete atomic-resolution structures of helical membrane proteins in lipid bilayers.
Figure 7. Solid-state NMR structure of the ion channel gramicidin A. Structure of gramicidin A viewed from the side (left) and from the top (right) coloured to distinguish the two head-to-head helices of the dimer (red and blue). The structure was drawn with PDB file 1MAG using Jmol: An open-source Java viewer for chemical structures in 3D (http://www.jmol.org/) (Herráez, Citation2006). This Figure is reproduced in colour in Molecular Membrane Biology online.
Yeast α-factor receptor transmembrane 6
The structure of an 18-residue segment of the α-factor receptor 6th transmembrane domain from Saccharomyces cerevisiae was determined in DMPC bilayers by oriented solid-state NMR along with independent observations from attenuated total reflection FTIR spectroscopy (Valentine et al., Citation2001). Ten homologues of TM6(252-269, C252A) in which individual residues were labelled with 15N were used to obtain chemical shifts and 1H-15N dipolar couplings. These were used to calculate a structure in which the C-terminus (residues 9–14) was α-helical and oriented with an angle of around 8° in respect to the bilayer normal (, PDB 1PJD). Complementary FTIR analysis provided a helix tilt angle of around 12.5°.
Antimicrobial peptides piscidin-1 and piscidin-3
High-resolution structures (backbone rmsd 0.35–0.39 Å) and orientations of the homologous 22-residue antimicrobial cationic peptides piscidin-1 and piscidin-3 from striped sea bass were determined in both DMPC/DMPG bilayers and in POPE/POPG bilayers by oriented solid-state NMR along with all-atom molecular dynamics (MD) simulations (Perrin et al., Citation2014). 2D HETCOR experiments were employed to provide restraints for calculating the structures, which for all systems and analysis methods had a helical tilt angle of between 83° and 93° with respect to the bilayer normal and a slight kink at G13 separating two helical segments (, PDB 2MCU, 2MCV, 2MCW, 2MCX). These disruptions in α-helical structure are assumed to correct for different amphipathic characteristics of their N- and C-terminal ends. The more potent piscidin-1 is distinguished from piscidin-3 by a larger ρ angle and less dynamic fraying at the N-terminal end (Perrin et al., Citation2014). Along with potent antimicrobial activities against both gram-positive and gram-negative bacteria, piscidin-1 has also shown evidence of anticancer activity (Lin et al., Citation2012).
Nicotinic acetylcholine receptor M2 channel
Backbone structures of the 25-residue nicotinic acetylcholine receptor M2 channel lining segment determined by oriented solid-state NMR in DMPC bilayers and by solution-state NMR in DPC micelles were very similar with an rmsd of 0.6 Å. Oriented solid-state NMR measurements also allowed determination of the complete three-dimensional structure, topology and global orientation of the peptide in the membrane (Opella et al., Citation1999). The peptide exists as an amphipathic α-helix that inserts in the lipid bilayer with a tilt angle of 12° relative to the bilayer normal and is rotated about its long axis such that polar residues face the N-terminal side of the membrane (, PDB 1CEK). Using this solid-state NMR structure of the monomer, an oligomeric model was built assuming a symmetric pentameric arrangement of helices. This resulted in a funnel-like structure with the widest opening on the N-terminal intracellular side (, PDB 1EQ8).
Salmonella regulatory peptide MgtR
PISEMA solid-state NMR was used to determine the structure and membrane orientation of the 30-residue Salmonella regulatory peptide MgtR in DMPC bilayers (Jean-Francois et al., Citation2014). Measurements using selectively 15N-labelled peptides revealed a transmembrane helix with a tilt angle of 32° relative to the bilayer normal (, PDB 2MC7). MgtR inhibits growth in macrophages through binding to the membrane protein MgtC. This work also used oriented sample solid-state NMR and EPR spectroscopy to demonstrate the formation of a heterodimer between MgtR and transmembrane helix 4 of MgtC from M. tuberculosis. Based on these experimental measurements, a structural model for the MgtR/MgtC TM4 heterodimer in a DMPC bilayer was created using computational methods.
HIV Vpu channel transmembrane domain
PISEMA solid-state NMR (DOPC/DOPG bilayer samples aligned on glass plates) was used along with RDC and chemical shift anisotropy measurements from weakly aligned solution-state NMR samples (DHPC micelles) to determine the three-dimensional structure and membrane orientation of the HIV Vpu channel transmembrane domain with precision equivalent to an rmsd of 0.4 Å (Park et al., Citation2003). The 36-residue construct, comprising residues 2–30 from the N-terminus of Vpu and a six-residue tag at the C-terminus, has a transmembrane helix spanning residues 8–25 with an average tilt angle of 13° relative to the bilayer normal (, PDB 1PJE). The helix has a slight kink at Ile17 resulting in tilt angles of 12° and 15° for residues 8–16 and 17–25, respectively. Oligomerization of the peptide in lipid bilayers occurs, so this work also provided tetrameric and pentameric models of the functional channel (, PDB 1PI7 and PDB 1PI8). Later structures of the same Vpu construct were determined from PISEMA measurements on magnetically aligned samples in 14-O-PC/6-O-PC and 16-O-PC/6-O-PC bicelles (PDB 2GOF and 2GOH) (Park et al., Citation2006). Comparison of the structures demonstrated how the hydrophobic thickness of a lipid bilayer influences the properties of a transmembrane helix (). Notably, the tilt angle of the helix was increased to 30° and 21° in 14-O-PC and 16-O-PC bicelles, respectively, due to a decrease in depth of the lipid bilayer. Also, the kink observed at Ile17 of the helix in the longer-chain C18 mechanically aligned bilayers was not seen in either of the structures obtained in the magnetically oriented shorter-chain (C14 or C16) bilayers (Park et al., Citation2006).
Figure 8. Backbone structures of the HIV Vpu channel transmembrane domain. Structures were determined by solution-state NMR in DHPC micelles (A), by solid-state NMR mechanically aligned on glass plates in DOPC/DOPG bilayers (B) (PDB 1PJE) and by solid-state NMR magnetically aligned in 16-O-PC/6-O-PC bicelles (C) (PDB 2GOH) and in 14-O-PC/6-O-PC bicelles (D) (PDB 2GOF). The Cα of Ile17 is indicated to show the position of the kink and the grey boxes indicate the thickness of the hydrophobic region of the lipid bilayers. This Figure was reproduced with permission from Park et al. (Citation2006), which was originally published in Biophys J [Park SH, De Angelis AA, Nevzorov AA, Wu CH, Opella SJ Citation2006. Three-dimensional structure of the transmembrane domain of Vpu from HIV-1 in aligned phospholipid bicelles. Biophys J 91:3032–3042], copyright by The Biophysical Society 2006.
![Figure 8. Backbone structures of the HIV Vpu channel transmembrane domain. Structures were determined by solution-state NMR in DHPC micelles (A), by solid-state NMR mechanically aligned on glass plates in DOPC/DOPG bilayers (B) (PDB 1PJE) and by solid-state NMR magnetically aligned in 16-O-PC/6-O-PC bicelles (C) (PDB 2GOH) and in 14-O-PC/6-O-PC bicelles (D) (PDB 2GOF). The Cα of Ile17 is indicated to show the position of the kink and the grey boxes indicate the thickness of the hydrophobic region of the lipid bilayers. This Figure was reproduced with permission from Park et al. (Citation2006), which was originally published in Biophys J [Park SH, De Angelis AA, Nevzorov AA, Wu CH, Opella SJ Citation2006. Three-dimensional structure of the transmembrane domain of Vpu from HIV-1 in aligned phospholipid bicelles. Biophys J 91:3032–3042], copyright by The Biophysical Society 2006.](/cms/asset/a00fc5bb-101e-47e6-846b-57a9026fcff6/imbc_a_1139754_f0008_b.jpg)
fd bacteriophage pVIII coat protein and Pf1 bacteriophage major coat protein
Structure determination of the membrane-bound form of fd bacteriophage pVIII coat protein by oriented sample PISEMA solid-state NMR (, PDB 1MZT) (Marassi & Opella, Citation2003) has already been described above. More recently, the structure and orientation of the 46-residue membrane-bound form of Pf1 bacteriophage major coat protein was determined from solid-state NMR measurements in mechanically aligned (DOPC/DOPG bilayers) and magnetically aligned (14-O-PC/6-O-PC bicelles) samples and from solution-state NMR measurements on weakly aligned DHPC micelles (Park et al., Citation2010b). The structure comprises a transmembrane helix (residues 23–45) with a tilt angle of 30° relative to the bilayer normal, a linker sequence (residues 16–18) and an N-terminal amphipathic helix (residues 5–15). The amphipathic helix lies parallel to the surface of the lipid bilayer with hydrophobic residues facing the membrane and acidic and polar residues exposed to the aqueous environment (, PDB 2KSJ). This work also investigated dynamics of the membrane-bound form of Pf1, providing implications of its structural rearrangement for virus assembly. This includes rotation of the transmembrane helix by 160° to allow proper alignment for packing into virus particles and tilting of the amphipathic helix away from the surface of the membrane such that it becomes almost parallel with the transmembrane helix (Park et al., Citation2010b).
Phospholamban
The structure and membrane topology of the monomeric form of the 53-residue regulator of cardiac muscle contractility, phospholamban, was determined by combining measurements from oriented solid-state NMR in DOPC bilayers and solution-state NMR in DPC micelles (Traaseth et al., Citation2009). This work reported the structure of the resting state (or T-state) of phospholamban, which also exists in a more dynamic active state (or R state). Solution-state NMR angular and distance NOE restraints were combined with orientational restraints from PISEMA and SAMPI4 measurements on uniformly 15N-labelled and selective amino acid 15N-labelled phospholamban aligned on glass plates. The structure is comprised of a transmembrane helix (residues 31–52) with a tilt angle of 24° relative to the membrane normal, a linker sequence (residues 23–30) and an N-terminal amphipathic helix that rests on the surface of the membrane with its hydrophobic residues facing into lipid bilayer (, PDB 2KB7). The NMR structure along with molecular dynamics simulations provided some explanations for interaction of monomeric phospholamban with the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA). In native membranes, phospholamban exists as a homopentamer that had been proposed to function either as a reservoir for active monomers or as an ion channel. A structure and membrane topology of the pentameric T-state of phospholamban was determined using a combined solution- and solid-state NMR approach (Verardi et al., Citation2011) similar to that used for the structure of the monomer already described above. The structure of the pentamer showed a ‘pinwheel’ topology with a narrow hydrophobic pore, which excluded an ion transport function. In the pentamer, the transmembrane helix crosses the bilayer with a tilt angle of 11°, which differs from that of the monomer (, PDB 2KYV). The ‘pinwheel’ arrangement differed from a ‘bellflower’ topology model of the pentamer that had previously been determined by solution-state NMR methods alone (Oxenoid & Chou, Citation2005). Comparisons of the two topology arrangements (Verardi et al., Citation2011) highlighted the importance of using solid-state NMR restraints for defining the orientation and topology of membrane proteins in a lipid bilayer.
Stannin
A combined solution- and solid-state NMR approach was used to determine the structure, dynamics, and membrane topology of the 88-residue mitochondrial protein stannin, which mediates neuronal cell apoptosis induced by trimethyltin chloride (Buck-Koehntop et al., Citation2005). Solution-state NMR NOE and hydrogen bond distance restraints were measured with the protein in SDS micelles and solid-state NMR orientational restraints were measured with the protein aligned on glass plates. The structure is comprised of a transmembrane helix (residues 10-33) with a tilt angle of around 20° relative to the membrane normal, a 28-residue unstructured linker and a distorted cytoplasmic helix (residues 61–79) that is partially buried into the bilayer with a tilt angle of 80° (, PDB 1ZZA). The linker contains a conserved CXC metal-binding motif and a putative 14-3-3 zeta binding domain.
Rabbit cytochrome b5
A combined solution- and solid-state NMR approach was also used to determine the structure of full-length 105-residue rabbit ferric microsomal cytochrome b5 (Ahuja et al., Citation2013). This was achieved based on TROSY-type solution-state NMR experiments with the protein in DPC micelles or in isotropic DMPC/DHPC bicelles and HIMSELF solid-state NMR experiments with the protein magnetically-aligned in DMPC/DHPC bicelles. The structure is comprised of a large heme domain and a C-terminal transmembrane domain, which are connected by a 15-residue linker (, PDB 2M33). The transmembrane domain was not visible in the solution-state NMR spectra and could only be characterized from the solid-state NMR spectra. The transmembrane domain was also visible in a MAS 1H-15N-HMQC spectrum of selectively [15N]alanine-labelled protein in DPC micelles (Ahuja et al., Citation2013).
Influenza M2 proton channel
A number of structures of the tetrameric transmembrane domain of the M2 proton channel from influenza A virus have been determined using various solid-state NMR methods. The first used a 25-residue construct (residues 22–46) mechanically oriented in DMPC bilayers and REDOR solid-state NMR to measure a distance between 15N-labelled His37 and 13C-labelled Trp41 to be less than 3.9 Å. This distance along with orientational restraints defining the backbone structure allowed determination of side chain pairings, torsion angles and distance restraints to characterize the tetrameric helical bundle (Nishimura et al., Citation2002). This structure suggested that both His and Trp side chains are oriented so that they face into the pore and that the channel is closed due to proximity of the four Trp indole groups. The structure also showed an open cavity towards the C-terminus that appeared suitable for binding of the antiviral drugs amantadine and rimantadine (, PDB 1NYJ). PISEMA experiments on site-specifically-labelled samples of an 18-residue contruct (residues 26–43) with bound amantadine oriented in DMPC bilayers were subsequently used to obtain an amantadine-blocked model of the channel (Hu et al., Citation2007). Spectral changes observed on the binding of amantadine in both PISEMA spectra and in CP-MAS spectra of the protein in DMPC/DMPG liposomes also helped to define structural details. This structure had a tightening of the channel near the N-terminus and a widening of the channel near the C-terminus along with a kink in the transmembrane helix at Gly34 separating fragments that had tilt angles of 20° and 31° relative to the membrane normal (, PDB 2H95).
Using exclusively MAS solid-state NMR methods on unoriented samples of a 25-residue construct (residues 22–46) with bound amantadine in lipid bilayers, some further structures of the M2 channel were determined. Because single-site mutation of Ser31 had abolished the effectiveness of the channel for binding amantadine and rimantadine, MAS experiments were used to investigate the roles of this residue and other residues in drug binding and on channel structure (Cady et al., Citation2009). From measurements in DLPC bilayers, Ser31 was the site of largest chemical shift perturbation on the binding of amantadine. Chemical shift restraints provided a monomer structure with a kink in the helix at Gly34 consistent with the structure described above. Using tilt angles and interhelical distances measured by various MAS experiments on unoriented samples obtained in this work and previously (Cady et al., Citation2007; Cady & Hong, Citation2008a, Citation2008b; Luo & Hong, Citation2006; Luo et al., Citation2007), a tetrameric model for the channel with bound amantadine was constructed (, PDB 2KAD). This structure differed from a solution-state NMR structure [rimantadine-bound M2 (residues 18–60) in DHPC micelles, PDB 2RLF (Schnell & Chou, Citation2008)] and a crystal structure [amantadine-bound M2 (residues 22–46) in octyl-β-D-glucopyranoside, PDB 3C9J (Stouffer et al., Citation2008)] of the channel in terms of openness of the N-terminus, constriction at Ser31 and side-chain conformations of Trp41 (Cady et al., Citation2009). In the solution-state NMR structure, the drug binding site was also at a different location showing four rimantadine molecules bound to the C-terminal lipid-facing surface of the helices. Based on the MAS solid-state NMR structure in lipid bilayers and modelling of the distances between amantadine and Ser31, a binding site for amantadine inside the channel pore and towards the N-terminus was proposed (Cady et al., Citation2009). Further MAS solid-state NMR measurements on the channel in DMPC bilayers then confirmed that two amantadine binding sites do exist, a high affinity single occupancy site in the channel pore towards the N-terminus and low affinity external sites at the C-terminal surface that are only occupied when the drug reaches high concentrations in the bilayer (Cady et al., Citation2010). 2H MAS measurements on the perdeuterated amantadine allowed characterization of the different orientation and dynamics of the drug molecule in the two distinct binding sites. Then measurement of protein-amantadine distances from MAS 13C-2H REDOR experiments provided a 0.3 Å-resolution structure of the amantadine binding site and re-refinement of the amantadine-blocked structure (, PDB 2KQT and ). The helices were kinked at Gly34 with tilt angles of 30° and 19° for the N-terminal and C-terminal segments, respectively. The pore showed narrowing towards the N-terminus at Val27 and towards the C-terminus at His37 and Trp41, which are residues responsible for pH sensing and proton conduction. Amantadine fits into a hydrophobic cavity towards the N-terminus of the channel pore surrounded by residues, including Ser31, that when mutated provide drug resistance (Cady et al., Citation2010). This series of work on the M2 channel has especially highlighted the importance of performing solid-state NMR measurements in lipid bilayers to achieve native structures of membrane proteins and extremely high-resolution structures and conformations of drug molecules and their binding sites.
Figure 9. Solid-state NMR structure of the amantadine-bound M2 proton channel from influenza A virus in lipid bilayers. (A) Side view showing residues Val27, Ser31, Gly34, His37 and Trp41 and amantadine in the high-affinity binding site. (B) Top view showing the Val27 and Ser31 pore radii and amantadine in the high-affinity binding site. This picture was reproduced with permission from Cady et al. (Citation2010), which was originally published in Nature [Cady SD, Schmidt-Rohr K, Wang J, Soto CS, Degrado WF, Hong M. Citation2010. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature 463:689–692], copyright by Nature Publishing Group 2010. This Figure is reproduced in colour in Molecular Membrane Biology online.
![Figure 9. Solid-state NMR structure of the amantadine-bound M2 proton channel from influenza A virus in lipid bilayers. (A) Side view showing residues Val27, Ser31, Gly34, His37 and Trp41 and amantadine in the high-affinity binding site. (B) Top view showing the Val27 and Ser31 pore radii and amantadine in the high-affinity binding site. This picture was reproduced with permission from Cady et al. (Citation2010), which was originally published in Nature [Cady SD, Schmidt-Rohr K, Wang J, Soto CS, Degrado WF, Hong M. Citation2010. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature 463:689–692], copyright by Nature Publishing Group 2010. This Figure is reproduced in colour in Molecular Membrane Biology online.](/cms/asset/8851cbea-d892-47c0-a5d3-d606b10d202c/imbc_a_1139754_f0009_c.jpg)
A further solid-state NMR structure of the M2 channel using a longer 44-residue construct (residues 22–62) oriented in DOPC/DOPE bilayers provided insight into the mechanism of the protein (Sharma et al., Citation2010). The structure and orientation of the transmembrane helices (residues 26–46) were in agreement with other solid-state NMR structures showing a kink at Gly34 with tilt angles of 32° and 22° relative to the membrane normal for the N-terminal and C-terminal fragments, respectively (, PDB 2L0J). The pore in the transmembrane tetrameric bundle is lined by the residues Val27, Ser31, Gly34, His37, Trp41, Asp44 and Arg45 showing constrictions by Val27 at the N-terminal end and by Trp41 at the C-terminal end. Amphipathic helices formed by residues 48–58 had a tilt angle of 105° and lie at the interfacial region of the lipid bilayer. Non-polar residues of the amphipathic helices contribute further hydrophobic interactions to interlink the monomers, for example Phe48 interacts with Phe55 and Leu59 of an adjacent monomer and Phe54 interacts with Leu46 of another adjacent monomer. Charged residues in the amphipathic helices (Lys49, Arg53, His57, Lys60, Arg61) project outwardly to interact favorably with negatively charged lipids in native membranes. This structure was used to highlight the importance of the tetrameric His37-Trp41 cluster in protein function and to propose a detailed mechanism of acid activation and proton conductance for the M2 channel involving this cluster (Sharma et al., Citation2010).
A comparison of structures of the influenza A virus M2 channel determined by solid-state NMR, solution-state NMR and X-ray crystallography has highlighted significant backbone differences in the transmembrane domain that affect drug binding and the chemistry of the tetrameric cluster responsible for acid activation, proton selectivity and transport (Zhou & Cross, Citation2013b). These differences appear to originate predominantly from influences of the membrane mimetic environment, especially a sensitivity of helix tilt to the hydrophobic dimension of the environment. Structures of the M2 channel determined by solid-state NMR in lipid bilayers appear to best represent a feasible structure and mechanism of closest physiological relevance.
Proteins with two unique transmembrane α-helices
There are only two PDB entries for structures with two unique transmembrane spanning helices determined by solid-state NMR, those of mercury transporter MerF and of the cell division structural and regulatory protein CrgA. This demonstrates that significant challenges have had to be overcome in moving from a single-span to a bitopic helical membrane protein for determination of structure and topology by solid-state NMR.
Mercury transporter MerF
A number of structures of the mercury transporter MerF from Morganella morganii have been determined using solid-state NMR methods. The first of these used a 61-residue truncated form of MerF (full-length 81 residues) magnetically oriented in 14-O-PC/6-O-PC bicelles (De Angelis et al., Citation2006). PISEMA, SAMMY and other double-resonance experiments on uniformly and selectively 15N-labelled samples were performed to resolve and assign backbone amide resonances and to obtain orientational restraints. 1H/13C/15N triple-resonance experiments were also performed on double selectively 13C- and 15N-labelled samples for complete resonance assignments, including residues in non-helical regions of the protein. The resultant structure has two transmembrane helices (residues 27–41 and 52–68) connected by a 10-residue loop (residues 42–51) that lies in the interfacial region of the lipid bilayer. The helices are almost parallel with each other and have the same tilt angles relative to the membrane normal. The slightly longer second helix contains a sequence of hydrophilic residues (QRQRQA) at the C-terminus that extends beyond the hydrophobic part of the lipid bilayer (, PDB 2H3O). In calculating this structure, the structures of the helices and their tilt angles relative to the membrane normal were determined with good confidence, but their relative positions were dependent on the structure of the loop and on imposed hydrophobic and distance constraints given after the structural fitting. A single close set of loop structures was simultaneously consistent with the assigned data points, Ramachandran restraints and weak distance restraints (De Angelis et al., Citation2006). This first structure determined for MerF was a breakthrough in demonstrating the potential use of solid-state NMR for determining the structure and topology of bitopic and polytopic integral membrane proteins in a lipid bilayer. A more recent structure of the truncated form of MerF has been determined using MAS methods with ‘rotational alignment’ on the protein unoriented in DMPC liposomes (Das et al., Citation2012). A combination of homonuclear two-dimensional and triple-resonance two- and three-dimensional MAS experiments on uniformly 13C/15N labelled protein were used to achieve complete assignment of all backbone resonances. Three-dimensional SLF experiments were performed to characterize the rotationally averaged dipolar coupling and chemical shift anisotropy powder patterns associated with resolved isotropic resonances. All NMR measurements were performed on a single proteoliposome sample containing 3.5 mg of uniformly 13C/15N-labeled protein. The calculated structure was consistent with the helix-loop-helix arrangement seen in the previous structure determined in magnetically aligned bilayers, but provided further refinement of its three-dimensional orientation in the lipid bilayer (, PDB 2LJ2). This included details of the N-terminal amphipathic helix (residues15–24) that orients towards the membrane surface with hydrophobic residues facing the lipid bilayer and polar residues facing the aqueous environment (Das et al., Citation2012).
The structures of the truncated protein have been superceded by two recent structures of full-length (81-residue) MerF determined by solid-state NMR. Using MAS methods with ‘rotational alignment’ on the protein in 14-O-PC bilayers, the calculated structure had two long helices that extend well beyond the lipid bilayer connected by a well-defined loop (, PDB 2M67) (Lu et al., Citation2013). The N-terminal amphipathic helix aligns nearly parallel to the membrane normal and forms an extension of the first transmembrane helix. This is different to the helix orientation seen in the truncated protein where it has a perpendicular orientation with the membrane (). The core of the protein, including the two transmembrane helices and the loop, is almost identical in the full-length and truncated proteins, however. This structure of full-length MerF has also been improved by the inclusion of chemical shift anisotropy constraints with the protein in DMPC bilayers (, PDB 2MOZ) (Tian et al., Citation2014). In addition to using a lipid environment, this work has emphasized the importance of using full-length proteins for native membrane protein structure determination, thus providing information that can be more confidently related to protein function.
Figure 10. Comparison of truncated and full-length MerF solid-state NMR structures. The structure of the truncated 60-residue protein (magenta) is superimposed on the structure of the full-length 81-residue protein (aqua). A position of negligible conformational change (Ala52) and the position of large conformational change (Ala19) are indicated. This picture was reproduced with permission from Lu et al. (Citation2013), which was originally published in J Am Chem Soc [Lu GJ, Tian Y, Vora N, Marassi FM, Opella SJ Citation2013. The structure of the mercury transporter MerF in phospholipid bilayers: A large conformational rearrangement results from N-terminal truncation. J Am Chem Soc 135:9299–9302], copyright by American Chemical Society 2013. This Figure is reproduced in colour in Molecular Membrane Biology online.
![Figure 10. Comparison of truncated and full-length MerF solid-state NMR structures. The structure of the truncated 60-residue protein (magenta) is superimposed on the structure of the full-length 81-residue protein (aqua). A position of negligible conformational change (Ala52) and the position of large conformational change (Ala19) are indicated. This picture was reproduced with permission from Lu et al. (Citation2013), which was originally published in J Am Chem Soc [Lu GJ, Tian Y, Vora N, Marassi FM, Opella SJ Citation2013. The structure of the mercury transporter MerF in phospholipid bilayers: A large conformational rearrangement results from N-terminal truncation. J Am Chem Soc 135:9299–9302], copyright by American Chemical Society 2013. This Figure is reproduced in colour in Molecular Membrane Biology online.](/cms/asset/fb4706be-12a6-4e8c-85b6-e2fbb369720f/imbc_a_1139754_f0010_c.jpg)
Cell division structural and regulatory protein CrgA
A structure of the 93-residue cell division structural and regulatory protein CrgA from M. tuberculosis has been determined in lipid bilayers using both oriented sample and MAS solid-state NMR methods to obtain orientational and interhelical distance restraints, respectively (Das et al., Citation2015a). Two-dimensional PISEMA and DARR experiments and other three-dimensional experiments were performed on both uniformly 13C,15N-labelled and amino acid specific 15N-labelled samples and on samples produced by reverse labelling. A complete structure of the transmembrane domain was refined using restrained molecular dynamics simulations in an all-atom representation of the same lipid bilayer environment as in the NMR samples. The structure has two transmembrane helices in a left-handed packing arrangement that cross at an angle of 24° at a conserved Gly39 residue (, PDB 2MMU). The N-terminal and interhelical loop regions were partially disorded. The structure provided localization of conserved residues possibly involved in the binding of CrgA with partner proteins in regulating the cell division process (Das et al., Citation2015a).
Proteins with four unique transmembrane α-helices
Two structures coming from the Escherichia coli disulphide bond generating system DsbB-DsbA have been determined.
Disulphide bond generating system DsbB-DsbA
A structure of the 41 kDa DsbB-DsbA disulphide bond generating system from E. coli was determined using a method that combines calculations from solid-state NMR and X-ray crystallography data (, PDB 2LEG) (Tang et al., Citation2011). In this complex, DsbB (179 residues) is an integral membrane protein with four transmembrane helices that oxidizes DsbA (189 residues), a periplasmic domain that forms disulphide bonds in substrate proteins. Reoxidation of DsbA occurs through disulphide bond rearrangements among four conserved cysteines in DsbB (Cys41-Cys44 and Cys104-Cys130). Electrons are then transferred from DsbA cysteines to the cofactor ubiquinone and then to the respiratory chain. A 3.7 Å crystal structure of DsbB-DsbA with bound ubiquinone was refined by introducing restraints from MAS solid-state NMR measurements performed on samples of DsbB-DsbA in POPE bilayers. Chemical shift assignments from comprehensive three- and four-dimensional solid-state NMR experiments were used to obtain backbone angle restraints and distance restraints were extracted from two-dimensional 13C-13C spectra obtained on samples selectively labelled using [2-13C]- or [1, 3-13C] glycerol. Introduction of solid-state NMR restraints into the structure calculation improved the overall backbone precision from 1.70 Å–1.03 Å and that in the transmembrane region was improved from 2.20 Å–0.92 Å compared with the crystal structure alone. The overall quality of the structure was also improved since 22% of DsbB transmembrane residues were promoted into the most favoured regions of Ramachandran space compared with those in the crystal structure. Solid-state NMR chemical shifts from DsbB in lipids compared with solution-state NMR chemical shifts from DsbB in detergent micelles were closely similar for most residues in transmembrane helices except for a few outliers close to the membrane surface (Tang et al., Citation2011).
A further high-resolution structural model of DsbB (C41S mutant) was derived by combining experimental X-ray crystallography data from DsbB(Cys41Ser) bound to the monoclonal Fab antibody fragment and solid-state NMR data from DsbB in POPE lipids with molecular dynamics simulations (, PDB 2LTQ) (Tang et al., Citation2013b). Introduction of solid-state NMR restraints into the structure calculation by a method similar to that described above improved the overall backbone precision from 2.36 Å–1.35 Å compared with the crystal structure alone (). The experimental structure was embedded in a POPE bilayer and simulations were performed to propose a mechanism for DsbB in the membrane. Two-dimensional 13C-13C correlation experiments with selective proton diffusion from water or lipid acyl chains were used to describe the membrane topology of DsbB. Furthermore, two-dimensional 13C-13C correlation spectra with 15N REDOR dephasing and DARR mixing of uniformly 13C,15N-labelled DsbB(Cys41Ser) suggested the presence of a flexible periplasmic loop and an interhelical hydrogen bond between Glu26 and Tyr153 in the first and fourth transmembrane helices, respectively, which had not been seen in any previous crystal or solution-state NMR structures (Tang et al., Citation2013b). The approach described here for refining X-ray crystal structures of membrane proteins with solid-state NMR restraints combined with molecular dynamics simulations should be useful for describing structure-function relationships of further membrane proteins embedded in native lipid bilayers.
Figure 11. Solid-state NMR refinement of the DsbB (C41S mutant) transmembrane domain crystal structure. (A) Overlay of 10 lowest energy structures of DsbB (Cys41Ser) calculated using only X-ray reflections (PDB 2ZUQ) with a backbone RMSD of 2.36 Å. (B) Overlay of 10 lowest energy structures of DsbB (Cys41Ser) calculated using X-ray reflections and solid-state NMR restraints (PDB 2LTQ) with a backbone RMSD of 1.35 Å. The structure is coloured magenta, cyan and white to represent the secondary structure elements α-helix, coil and turn, respectively. This picture was modified with permission from Tang et al. (Citation2013b), which was originally published in J Mol Biol [Tang M, Nesbitt AE, Sperling LJ, Berthold DA, Schwieters CD, Gennis RB, Rienstra CM Citation2013b. Structure of the disulfide bond generating membrane protein DsbB in the lipid bilayer. J Mol Biol 425:1670–1682], copyright by Elsevier Ltd 2013. This Figure is reproduced in colour in Molecular Membrane Biology online.
![Figure 11. Solid-state NMR refinement of the DsbB (C41S mutant) transmembrane domain crystal structure. (A) Overlay of 10 lowest energy structures of DsbB (Cys41Ser) calculated using only X-ray reflections (PDB 2ZUQ) with a backbone RMSD of 2.36 Å. (B) Overlay of 10 lowest energy structures of DsbB (Cys41Ser) calculated using X-ray reflections and solid-state NMR restraints (PDB 2LTQ) with a backbone RMSD of 1.35 Å. The structure is coloured magenta, cyan and white to represent the secondary structure elements α-helix, coil and turn, respectively. This picture was modified with permission from Tang et al. (Citation2013b), which was originally published in J Mol Biol [Tang M, Nesbitt AE, Sperling LJ, Berthold DA, Schwieters CD, Gennis RB, Rienstra CM Citation2013b. Structure of the disulfide bond generating membrane protein DsbB in the lipid bilayer. J Mol Biol 425:1670–1682], copyright by Elsevier Ltd 2013. This Figure is reproduced in colour in Molecular Membrane Biology online.](/cms/asset/5780e973-b1fa-4855-a5ea-7de180259516/imbc_a_1139754_f0011_c.jpg)
Proteins with seven unique transmembrane α-helices
Two impressive structures of membrane proteins with seven unique transmembrane spanning α-helices have been determined by solid-state NMR, those of human chemokine receptor CXCR1 and Anabaena sensory rhodopsin.
Chemokine receptor CXCR1
The first three-dimensional structure of a membrane protein with seven unique transmembrane spanning helices to be determined by solid-state NMR was that of the 350-residue human chemokine receptor CXCR1 in DMPC bilayers using MAS methods with ‘rotational alignment’ (Park et al., Citation2012). CXCR1 is a class A rhodopsin-like G-protein-coupled receptor (GPCR) with high-affinity for the CXC chemokine interleukin-8 that mediates immune and inflammatory responses. CXCR1 was also the first GPCR to have its structure determined in liquid crystalline phospholipid bilayers and the first ligand-activated GPCR with its structure determined using an unmodified amino acid sequence. NMR measurements used samples containing 2-4 mg of uniformly 13C,15N-labelled, 2-13C-glycerol-labelled or 13C/15N-Phe labelled CXCR1 refolded in DMPC proteoliposomes. Under NMR sample conditions the receptor showed high affinity for binding interleukin-8 and coupling to its G-protein, thus confirming physiological relevance of the structure. Resonances were resolved and assigned using 13C-detected three-dimensional, triple-resonance experiments, which achieved 97% assignment of the backbone resonances for residues 20–325. 13C signals from the mobile N-and C-termini (residues 1–19 and 326–350) were not detected in the spectra. Three-dimensional 13C-detected SLF experiments were used to obtain the orientation restraints for structure determination and the backbone structure was calculated by a molecular fragment replacement approach using the programs CS-Rosetta (Shen et al., Citation2008), Rosetta (Das & Baker, Citation2008; Yarov-Yarovoy et al., Citation2006) and Xplor-NIH (Schwieters et al., Citation2003) using the structure of bovine rhodopsin as a topological template. In this respect, the structure could be considered to be more of a model verified by many different NMR constraints. Nonetheless, the resultant structure has the expected fold of a GPCR, with seven transmembrane helices connected by three extracellular loops and three intracellular loops followed by an amphipathic helix that aligns along the membrane surface (, PDB 2LNL and ). The average backbone pairwise RMSD for the structure was 1.7 Å and it is expected that the accuracy and precision will be improved by inclusion of side chain restraints and distance restraints. Interestingly, the dipolar coupling measurements provided similar values for the magnitude and symmetry of the molecular order tensor for residues in the helices and loops, suggesting that these regions of CXCR1 encounter similar degrees of order in the membrane. Amongst other details, the structure revealed the locations of two highly conserved disulphide bonds, one connecting the N-terminus to the extracellular start of the seventh transmembrane helix (Cys30–Cys277) and the other connecting the extracellular end of the third transmembrane helix to the second extracellular loop (Cys110–Cys187), which are important for ligand binding. Charged residues are mainly located near the membrane-water interface with negative charges clustered in the extracellular loops where they contribute to ligand binding and receptor activation (Park et al., Citation2012).
Figure 12. Solid-state NMR structure of the human chemokine receptor CXCR1 in lipid bilayers. (A) Side view. (B) Top view from the extracellular surface. (C) Bottom view from the intracellular surface. The structure is coloured with the N-terminus in blue and the C-terminus in red. These structures were drawn from the PDB file 2LNL using using Jmol: An open-source Java viewer for chemical structures in 3D (http://www.jmol.org/) (Herráez, Citation2006). This Figure is reproduced in colour in Molecular Membrane Biology online.
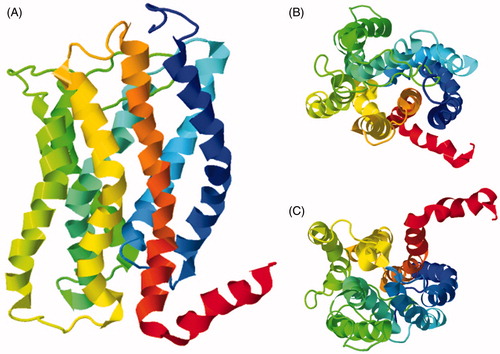
Anabaena sensory rhodopsin
The second three-dimensional structure of a membrane protein with seven unique transmembrane helices to be determined by solid-state NMR was that of the 235-residue homotrimeric Anabaena sensory rhodopsin in DMPC/DMPA bilayers using exclusively MAS methods (, PDB 2M3G and ) (Wang et al., Citation2013). This retinal-binding photoreceptor initiates a phototransduction cascade involving a soluble transducer that interacts with DNA and may regulate the expression of proteins responsible for photosynthesis and circadian clock in cyanobacterium Anabaena sp. PCC 7120 (Jung et al., Citation2003; Wang et al., Citation2011). NMR measurements were performed on samples containing approximately 8 mg of protein that was uniformly 13C,15N-labelled or sparsely labelled with 1,3- or 2-13C-glycerol to improve spectral resolution. Sparse labelling was especially important for allowing identification of long-range interhelical correlations that defined the interhelical packing. NMR experiments included two-dimensional 13C-13C proton driven spin diffusion and 13C-detected proton spin diffusion (CHHC) (Lange et al., Citation2002) and three-dimensional homogenously broadened rotational resonance (HBR2) (Peng et al., Citation2008) for resolving and assigning signals. A large number of torsional and internuclear distance restraints combined with paramagnetic relaxation enhancement (PRE) restraints were measured to define the backbone structure, arrangement of helices, side chain conformations and the position of the retinal cofactor in the monomer and also the oligomerization interface of the trimer. PRE restraints were measured on a uniformly 13C,15N-labelled S26C mutant with a covalently attached nitroxide paramagnetic tag. The structure was determined using a semiautomated structure calculation protocol without relying on a pre-existing structural template. The trimeric structure has an overall topology that closely resembles that of bacteriorhodopsin in which the oligomerization interface involves the second helix in one monomer and the fourth and fifth helices in the adjacent monomer. There are some conserved residues at this interface that are involved in the intermonomer packing. The helical arrangement in the transmembrane core of the monomer is in agreement with the general fold typical of microbial rhodopsins and with the crystal structure of the same protein (Vogeley et al., Citation2004), including positions of conserved aromatic and polar residues in the retinal-binding pocket and unique residues in the cytoplasmic polar cluster involving the second, third and seventh helices. The conformation of the solid-state NMR structure determined in lipids does show some notable differences compared with the crystal structure, however (Wang et al., Citation2013). The structure of Anabaena sensory rhodopsin further demonstrates the technical feasibility of using solid-state NMR for three-dimensional structure determination of polytopic integral membrane proteins. The authors emphasize that the key for transferring the technology to a greater number and range of membrane proteins is in the sample preparation. That is to say, production of a sufficient quantity of isotopically labelled protein which can be reconstituted at a high protein:lipid ratio in a functionally active form and that is stable for the duration of necessary NMR measurements. The structurally rigid transmembrane domain and relatively short extramembranous loops in Anabaena sensory rhodopsin are also favourable for producing high quality NMR spectra. Such structural properties are not found in the large majority of membrane proteins, however.
Figure 13. Solid-state NMR structure of Anabaena sensory rhodopsin in lipid bilayers. (A) Side view. (B) Top view from the extracellular surface. The structure is coloured with the N-terminus in blue and the C-terminus in red and the retinal cofactor molecule is coloured grey. These structures were drawn from the PDB file 2M3G using Jmol: an open-source Java viewer for chemical structures in 3D (http://www.jmol.org/) (Herráez, Citation2006). This Figure is reproduced in colour in Molecular Membrane Biology online.
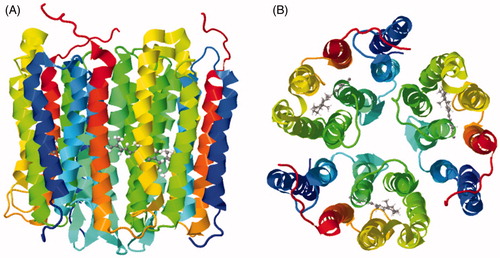
Conclusions and future perspectives
There has been an increasing number and complexity of integral membrane protein structures determined by solid-state NMR, which have a range in types of function and includes proteins with seven unique transmembrane spanning α-helices. Although there are various challenges that have to be overcome, a clear benefit of using solid-state NMR is its ability to determine three-dimensional structures and to obtain atomic-resolution structural information for integral membrane proteins in native or native-like lipid bilayers. As exemplified in this review, this is important for defining structures, ligand and drug interactions, dynamics and molecular mechanisms of membrane proteins under conditions of physiological relevance. The structures have also demonstrated that a full-length membrane protein should be used in solid-state NMR measurements where possible, since use of a truncated protein can have significant effects on structural conformation and topology and on derived models of mechanism and ligand binding. Whilst oriented sample solid-state NMR has provided mainly structures of membrane proteins with a single transmembrane spanning α-helix, combination with distance and torsion angle restraints from MAS solid-state NMR has been important for defining the structure and helical packing arrangement of homo-oligomers, bitopic membrane proteins and of some polytopic membrane proteins, including that of the seven transmembrane-spanning human chemokine receptor CXCR1 (Park et al., Citation2012) and (Wang et al., Citation2013). The structure of the seven-transmembrane spanning Anabaena sensory rhodopsin was determined using exclusively MAS methods (Wang et al., Citation2013).
Making solid-state NMR more routine for determining the structures of larger and more complex membrane proteins will require the challenges of spectral crowding and signal assignment to be overcome and this is an ongoing theme of development. For example, a strategy has been devised for signal assignment in all labelled transmembrane amino acids of the three-helix protein Rv1861 from M. tuberculosis using an oriented sample, amino acid specific 15N-labelling and SLF experiments (Murray et al., Citation2014b). A method that combines 15N reverse labelling and afterglow spectroscopy has been developed for assigning MAS solid-state NMR spectra of the four-helix multidrug resistance protein EmrE (Banigan et al., Citation2013). There have also been developments in resolving and assigning spectra of β-barrel membrane proteins, demonstrated with the eight-stranded β-barrel bacterial outer membrane proteins OmpX and Ail (Ding et al., Citation2013b; Yao et al., Citation2013). Some of the membrane protein structures have been determined by combining information from solid-state NMR and solution-state NMR measurements, for example those of the HIV Vpu channel transmembrane domain (Park et al., Citation2003), Pf1 bacteriophage major coat protein (Park et al., 2010) and phospholamban (Traaseth et al., Citation2009; Verardi et al., Citation2011). In certain cases, complementing solid-state NMR measurements with information from solution-state NMR, and vice versa, will likely be useful for defining the structures of further membrane proteins. The approach described for refining X-ray crystal structures of proteins from the DsbB-DsbA disulphide bond generating system from E. coli with solid-state NMR restraints combined with molecular dynamics simulations (Tang et al., Citation2011, Citation2013b) should be useful for refining further crystal structures and for describing structure-function relationships of further membrane proteins in native lipid bilayers. Structures and specific structural restraints obtained from solid-state NMR measurements on membrane proteins in lipid bilayers can be used to validate structures of the same proteins determined in different membrane mimetic environments by solution-state NMR or by X-ray crystallography. A general validation strategy using minimal data from oriented sample solid-state NMR has been demonstrated using the homotrimeric membrane-embedded enzyme diacylglycerol kinase, which has three transmembrane spanning α-helices and an amphipathic α-helix (Murray et al., Citation2014a). There have also been some recent developments in solid-state NMR approaches to obtain dynamic information for membrane proteins, demonstrated with the fd bacteriophage pVIII coat protein (Cheng et al., Citation2015) and with Anabaena sensory rhodopsin (Ward et al., Citation2015).
All solid-state NMR methods with membrane proteins require rational and efficient isotope labelling of the protein along with robust and high quality sample preparation. Common sample preparation challenges that have to be overcome include production of sufficient quantities of isotopically-labelled protein, achieving a protein:lipid ratio and concentration that favours successful alignment (for oriented methods) and that gives enough protein in the NMR sample for signal detection. Such parameters usually have to be considered on a case by case basis with each protein under investigation. A further challenge is identification of suitable membrane protein targets for solid-state NMR structure determination based on current technical capabilities and sample requirements. Membrane proteins that can only be obtained in relatively small quantities (hundreds of micrograms or less) will benefit from technical developments that improve resolution and sensitivity. Recent such developments include microcoil MAS and microsamples (Takeda, Citation2012), fast MAS at 100 kHz and above (Agarwal et al., Citation2014; André et al., Citation2015; Böckmann et al., Citation2015), MAS with the sample at cryogenic temperatures (Barnes et al., Citation2009; Concistrè et al., Citation2013; Thurber & Tycko, Citation2008; Tycko, Citation2013), DNP-enhanced MAS (Akbey et al., Citation2013; Koers et al., Citation2014; Maciejko et al., Citation2015; Smith et al., Citation2015; Su et al., Citation2015) and higher field (1 GHz plus) magnets (Hashi et al., Citation2015). Deuteration of the protein and sample allows acquisition of high-resolution solid-state NMR proton spectra that are useful for membrane protein structure determination (Reif, Citation2012), it also improves T2 relaxation times and resolution that facilitate sequential backbone assignments using 13C-detected experiments (Tang et al., Citation2010). Furthermore, fractional deuteration can assist structural analysis by reducing spectral congestion in 1H,13C,15N-correlation experiments and, since residual protons are predominantly found at amino acid side chain positions, enhance the prospects for obtaining side chain signal assignments and for detecting medium to long range contacts (Nand et al., Citation2012). Development of NMR methods for measuring long range 13C-13C distances will be useful to assist membrane protein structure determination, for example, a relaxation-compensated difference PDSD approach has been described that allows resolution of clean multi-bond, inter-residue and intermolecular long range correlations (Wang et al., Citation2015).
With continued efforts in membrane protein target identification and in developments of protein expression, purification, isotope labelling and sample preparation strategies, solid-state NMR probe technology, NMR experiments, structure calculation and modelling methods and their combination with other techniques, it should be feasible to determine the structures of many more membrane proteins of biological and biomedical importance. This will provide three-dimensional structures and atomic-resolution structural information for characterizing ligand and drug interactions, dynamics and molecular mechanisms of membrane proteins under physiological lipid bilayer conditions. The progress in X-ray crystallography and in other structural biology techniques such as single particle electron microscopy will also create a significant demand for additional and complementary spectroscopic studies of membrane proteins, including solid-state NMR.
Declaration of interest
The author reports no conflicts of interest. The author alone is responsible for the content and writing of the paper.
Correction Statement
This article was originally published with errors, which have now been corrected in the online version. Please see Correction (http://dx.doi.org/10.1080/09687688.2020.1829809)
References
- Agarwal V, Penzel S, Szekely K, Cadalbert R, Testori E, Oss A, et al. 2014. De novo 3D structure determination from sub-milligram protein samples by solid-state 100 kHz MAS NMR spectroscopy. Angew Chem Int Ed 53:12253–12256
- Ahuja S, Jahr N, Im SC, Vivekanandan S, Popovych N, Le Clair SV, et al. 2013. A model of the membrane-bound cytochrome b5-cytochrome P450 complex from NMR and mutagenesis data. J Biol Chem 288:22080–22095
- Akbey U, Franks WT, Linden A, Orwick-Rydmark M, Lange S, Oschkinat H. 2013. Dynamic nuclear polarization enhanced NMR in the solid-state. Top Curr Chem 338:181–228
- André M, Piotto M, Caldarelli S, Dumez JN. 2015. Ultrafast high-resolution magic-angle-spinning NMR spectroscopy. Analyst 140:3942–3946
- Andrew ER, Bradbury A, Eades RG. 1958. Nuclear magnetic resonance spectra from a crystal rotated at high speed. Nature 182:1659
- Andrew ER, Bradbury A, Eades RG. 1959. Removal of dipolar broadening of nuclear magnetic resonance spectra of solids by specimen rotation. Nature 183:1802
- Andrew ER. 2010. Magic angle spinning. In McDermott A, ed. Solid state NMR studies of biopolymers. Chichester, UK: John Wiley & Sons, 83–97
- Arora A. 2013. Solution NMR spectroscopy for the determination of structures of membrane proteins in a lipid environment. Methods Mol Biol 974:389–413
- Astrof NS, Griffin RG. 2002. Soft-triple resonance solid-state NMR experiments for assignments of U-13C, 15N labeled peptides and proteins. J Magn Reson 158:157–163
- Bahar I, Lezon TR, Bakan A, Shrivastava IH. 2010. Normal mode analysis of biomolecular structures: functional mechanisms of membrane proteins. Chem Rev 110:1463–1497
- Bakheet TM, Doig AJ. 2009. Properties and identification of human protein drug targets. Bioinformatics 25:451–457
- Banigan JR, Gayen A, Traaseth NJ. 2013. Combination of 15N reverse labeling and afterglow spectroscopy for assigning membrane protein spectra by magic-angle-spinning solid-state NMR: application to the multidrug resistance protein EmrE. J Biomol NMR 55:391–399
- Barnes AB, Mak-Jurkauskas ML, Matsuki Y, Bajaj VS, van der Wel PC, Derocher R, et al. 2009. Cryogenic sample exchange NMR probe for magic angle spinning dynamic nuclear polarization. J Magn Reson 198:261–270
- Bayburt TH, Sligar SG. 2010. Membrane protein assembly into nanodiscs. FEBS Lett 584:1721–1727
- Bayrhuber M, Meins T, Habeck M, Becker S, Giller K, Villinger S, et al. 2008. Structure of the human voltage-dependent anion channel. Proc Natl Acad Sci USA 105:15370–15375
- Bechinger B, Gierasch LM, Montal M, Zasloff M, Opella SJ. 1996. Orientations of helical peptides in membrane bilayers by solid state NMR spectroscopy. Solid State Nucl Magn Reson 7:185–191
- Bloembergen N. 1949. On the interaction of nuclear spins in a crystalline lattice. Physica 15:386–426
- Böckmann A, Ernst M, Meier BH. 2015. Spinning proteins, the faster, the better? J Magn Reson 253:71–79
- Brown LS, Ladizhansky V. 2015. Membrane proteins in their native habitat as seen by solid-state NMR spectroscopy. Protein Sci 24:1333–1346
- Buck-Koehntop BA, Mascioni A, Buffy JJ, Veglia G. 2005. Structure, dynamics, and membrane topology of stannin: a mediator of neuronal cell apoptosis induced by trimethyltin chloride. J Mol Biol 354:652–665
- Cady SD, Goodman C, Tatko CD, DeGrado WF, Hong M. 2007. Determining the orientation of uniaxially rotating membrane proteins using unoriented samples: A 2H, 13C, and 15N solid-state NMR investigation of the dynamics and orientation of a transmembrane helical bundle. J Am Chem Soc 129:5719–5729
- Cady SD, Hong M. 2008a. Simultaneous extraction of multiple orientational constraints of membrane proteins by 13C-detected N-H dipolar couplings under magic angle spinning. J Magn Reson 191:219–225
- Cady SD, Hong M. 2008b. Amantadine-induced conformational and dynamical changes of the influenza M2 transmembrane proton channel. Proc Natl Acad Sci USA 105:1483–1488
- Cady SD, Mishanina TV, Hong M. 2009. Structure of amantadine-bound M2 transmembrane peptide of influenza A in lipid bilayers from magic-angle-spinning solid-state NMR: the role of Ser31 in amantadine binding. J Mol Biol 385:1127–1141
- Cady SD, Schmidt-Rohr K, Wang J, Soto CS, Degrado WF, Hong M. 2010. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature 463:689–692
- Chen Y, Zhang Z, Tang X, Li J, Glaubitz C, Yang J. 2014. Conformation and topology of diacylglycerol kinase in E. coli membranes revealed by solid-state NMR spectroscopy. Angew Chem Int Ed Engl 53:5624–5628
- Cheng X, Jo S1, Qi Y, Marassi FM, Im W. 2015. Solid-state NMR-restrained ensemble dynamics of a membrane protein in explicit membranes. Biophys J 108:1954–1962
- Concistrè M, Johannessen OG, Carignani E, Geppi M, Levitt MH. 2013. Magic-angle spinning NMR of cold samples. Acc Chem Res 46:1914–1922
- Cross TA, Ekanayake V, Paulino J, Wright A. 2014. Solid state NMR: the essential technology for helical membrane protein structural characterization. J Magn Reson 239:100–109
- Das R, Baker D. 2008. Macromolecular modeling with rosetta. Annu Rev Biochem 77:363–382
- Das BB, Nothnagel HJ, Lu GJ, Son WS, Tian Y, Marassi FM, Opella SJ. 2012. Structure determination of a membrane protein in proteoliposomes. J Am Chem Soc 134:2047–2056
- Das BB, Lin EC, Opella SJ. 2013. Experiments optimized for magic angle spinning and oriented sample solid-state NMR of proteins. J Phys Chem B 117:12422–12431
- Das BB, Zhang H, Opella SJ. 2014. Dipolar Assisted Assignment Protocol (DAAP) for MAS solid-state NMR of rotationally aligned membrane proteins in phospholipid bilayers. J Magn Reson 242:224–232
- Das N, Dai J, Hung I, Rajagopalan MR, Zhou HX, Cross TA. 2015a. Structure of CrgA, a cell division structural and regulatory protein from Mycobacterium tuberculosis, in lipid bilayers. Proc Natl Acad Sci USA 112:E119–126
- Das BB, Park SH, Opella SJ. 2015b. Membrane protein structure from rotational diffusion. Biochim Biophys Acta 1848(1PtB):229–245
- De Angelis AA, Howell SC, Nevzorov AA, Opella SJ. 2006. Structure determination of a membrane protein with two trans-membrane helices in aligned phospholipid bicelles by solid-state NMR spectroscopy. J Am Chem Soc 128:12256–12267
- De Angelis AA, Opella SJ. 2007. Bicelle samples for solid-state NMR of membrane proteins. Nat Protoc 2:2332–2338
- Diller A, Loudet C, Aussenac F, Raffard G, Fournier S, Laguerre M, et al. 2009. Bicelles: a natural ‘molecular goniometer’ for structural, dynamical and topological studies of molecules in membranes. Biochimie 91:744–751
- Ding X, Zhao X, Watts A. 2013a. G-protein-coupled receptor structure, ligand binding and activation as studied by solid-state NMR spectroscopy. Biochem J 450:443–457
- Ding Y, Yao Y, Marassi FM. 2013b. Membrane protein structure determination in membrana. Acc Chem Res 46:2182–2190
- Drews J. 2000. Drug discovery: a historical perspective. Science 287:1960–1964
- Dürr UH, Gildenberg M, Ramamoorthy A. 2012. The magic of bicelles lights up membrane protein structure. Chem Rev 112:6054–6074
- Dürr UH, Soong R, Ramamoorthy A. 2013. When detergent meets bilayer: birth and coming of age of lipid bicelles. Prog Nucl Magn Reson Spectrosc 69:1–22
- Dvinskikh SV, Yamamoto K, Ramamoorthy A. 2006. Heteronuclear isotropic mixing separated local field NMR spectroscopy. J Chem Phys 125:34507
- Edwards R, Madine J, Fielding L, Middleton DA. 2010. Measurement of multiple torsional angles from one-dimensional solid-state NMR spectra: application to the conformational analysis of a ligand in its biological receptor site. Phys Chem Chem Phys 12:13999–14008
- Esteban-Martín S, Strandberg E, Salgado J, Ulrich AS. 2010. Solid state NMR analysis of peptides in membranes: influence of dynamics and labeling scheme. Biochim Biophys Acta 1798:252–257
- Fagerberg L, Jonasson K, von Heijne G, Uhlén M, Berglund L. 2010. Prediction of the human membrane proteome. Proteomics 10:1141–1149
- Filipp FV, Sinha N, Jairam L, Bradley J, Opella SJ. 2009. Labeling strategies for 13C-detected aligned-sample solid-state NMR of proteins. J Magn Reson 201:121–130
- Franks WT, Linden AH, Kunert B, van Rossum BJ, Oschkinat H. 2012. Solid-state magic-angle spinning NMR of membrane proteins and protein-ligand interactions. Eur J Cell Biol 91:340–348
- Fu R, Cotten M, Cross TA. 2000. Inter- and intramolecular distance measurements by solid-state MAS NMR: determination of gramicidin A channel dimer structure in hydrated phospholipid bilayers. J Biomol NMR 16:261–268
- Gopinath T, Mote KR, Veglia G. 2011. Proton evolved local field solid-state nuclear magnetic resonance using Hadamard encoding: theory and application to membrane proteins. J Chem Phys 135:074503
- Gopinath T, Mote KR, Veglia G. 2013. Sensitivity and resolution enhancement of oriented solid-state NMR: application to membrane proteins. Prog Nucl Magn Reson Spectrosc 75:50–68
- Gopinath T, Mote KR, Veglia G. 2015. Simultaneous acquisition of 2D and 3D solid-state NMR experiments for sequential assignment of oriented membrane protein samples. J Biomol NMR 62:53–61
- Gopinath T, Veglia G. 2015. Multiple acquisition of magic angle spinning solid-state NMR experiments using one receiver: application to microcrystalline and membrane protein preparations. J Magn Reson 253:143–153
- Grommek A, Meier BH, Ernst M. 2006. Distance information from proton-driven spin diffusion under MAS. Chem Phys Lett 427:404–409
- Gullion T, Schaefer J. 1989. Rotational-echo double-resonance NMR. J Magn Reson 81:196–200
- Hansen SK, Bertelsen K, Paaske B, Nielsen NC, Vosegaard T. 2015. Solid-state NMR methods for oriented membrane proteins. Prog Nucl Magn Reson Spectrosc 88–89:48–85
- Hartmann SR, Hahn EL. 1962. Nuclear double resonance in the rotating frame. Phys Rev 128:2042
- Hashi K, Ohki S, Matsumoto S, Nishijima G, Goto A, Deguchi K, et al. 2015. Achievement of 1020MHz NMR. J Magn Reson 256:30–33
- Hennel JW, Klinowski J. 2005. Magic angle spinning: A historical perspective. In Klinowski J, ed. New techniques in solid-state NMR. Berlin: Springer, 1–14
- Herráez A. 2006. Biomolecules in the computer: Jmol to the rescue. Biochem Mol Biol Educ 34:255–261
- Higman VA, Flinders J, Hiller M, Jehle S, Markovic S, Fiedler S, et al. 2009. Assigning large proteins in the solid state: a MAS NMR resonance assignment strategy using selectively and extensively 13C-labelled proteins. J Biomol NMR 44:245–260
- Hing AW, Vega S, Schaefer J. 1992. Transferred-echo double-resonance NMR. J Magn Reson 96:205–209
- Hiruma-Shimizu K, Kalverda AP, Henderson PJ, Homans SW, Patching SG. 2014. Synthesis of uniformly deuterated n-dodecyl-β-D-maltoside (d39-DDM) for solubilization of membrane proteins in TROSY NMR experiments. J Labelled Comp Radiopharm 57:737–743
- Hiruma-Shimizu K, Shimizu H, Thompson GS, Kalverda AP, Patching SG. 2015. Deuterated detergents for structural and functional studies of membrane proteins: properties, chemical synthesis and applications. Mol Membr Biol. 2015. [Epub ahead of print]. DOI: 10.3109/09687688.2015.1125536
- Hong M, Zhang Y, Hu F. 2012. Membrane protein structure and dynamics from NMR spectroscopy. Annu Rev Phys Chem 63:1–24
- Hopkins AL, Groom CR. 2002. The druggable genome. Nat Rev Drug Discov 1:727–730
- Hu J, Asbury T, Achuthan S, Li C, Bertram R, Quine JR, et al. 2007. Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from Influenza A virus. Biophys J 92:4335–4343
- Huber M, Böckmann A, Hiller S, Meier BH. 2012. 4D solid-state NMR for protein structure determination. Phys Chem Chem Phys 14:5239–5246
- Inagaki S, Ghirlando R, Grisshammer R. 2013. Biophysical characterization of membrane proteins in nanodiscs. Methods 59:287–300
- Jang H, Ma B, Nussinov R. 2007. Conformational study of the protegrin-1 (PG-1) dimer interaction with lipid bilayers and its effect. BMC Struct Biol 7:21
- Jean-Francois FL, Dai J, Yu L, Myrick A, Rubin E, Fajer PG, et al. 2014. Binding of MgtR, a Salmonella transmembrane regulatory peptide, to MgtC, a Mycobacterium tuberculosis virulence factor: a structural study. J Mol Biol 426:436–446
- Johansson AC, Lindahl E. 2009. Protein contents in biological membranes can explain abnormal solvation of charged and polar residues. Proc Natl Acad Sci USA 106:15684–15689
- Judge PJ, Watts A. 2011. Recent contributions from solid-state NMR to the understanding of membrane protein structure and function. Curr Opin Chem Biol 15:690–695
- Judge PJ, Taylor GF, Dannatt HR, Watts A. 2015. Solid-state nuclear magnetic resonance spectroscopy for membrane protein structure determination. Methods Mol Biol 1261:331–347
- Jung KH, Trivedi VD, Spudich JL. 2003. Demonstration of a sensory rhodopsin in eubacteria. Mol Microbiol 47:1513–1522
- Kalverda AP, Gowdy J, Thompson GS, Homans SW, Henderson PJ, Patching SG. 2014. TROSY NMR with a 52 kDa sugar transport protein and the binding of a small-molecule inhibitor. Mol Membr Biol 31:131–140
- Ketchem RR, Hu W, Cross TA. 1993. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science 261:1457–1460
- Ketchem RR, Lee KC, Huo S, Cross TA. 1996. Macromolecular structural elucidation with solid-state NMR-derived orientational constraints. J Biomol NMR 8:1–14
- Kim Y. 2006. Solid-state NMR studies of membrane proteins using phospholipid bicelles. Bull Korean Chem Soc 27:386–388
- Kim HJ, Howell SC, Van Horn WD, Jeon YH, Sanders CR. 2009. Recent advances in the application of solution NMR spectroscopy to multi-span integral membrane proteins. Prog Nucl Magn Reson Spectrosc 55:335–360
- Klammt C, Maslennikov I, Bayrhuber M, Eichmann C, Vajpai N, Chiu EJ, et al. 2012. Facile backbone structure determination of human membrane proteins by NMR spectroscopy. Nat Methods 9:834–839
- Klepper J, Voit T. 2002. Facilitated glucose transporter protein type 1 (GLUT1) deficiency syndrome: impaired glucose transport into brain – a review. Eur J Pediatr 161:295–304
- Koers EJ, van der Cruijsen EA, Rosay M, Weingarth M, Prokofyev A, Sauvée C, et al. 2014. NMR-based structural biology enhanced by dynamic nuclear polarization at high magnetic field. J Biomol NMR 60:157–168
- Kurze AK, Galliciotti G, Heine C, Mole SE, Quitsch A, Braulke T. 2010. Pathogenic mutations cause rapid degradation of lysosomal storage disease-related membrane protein CLN6. Hum Mutat 31:E1163–E1174
- Ladizhansky V. 2009. Homonuclear dipolar recoupling techniques for structure determination in uniformly 13C-labeled proteins. Solid State Nucl Magn Reson 36:119–128
- Lakatos A, Mörs K, Glaubitz C. 2012. How to investigate interactions between membrane proteins and ligands by solid-state NMR. Methods Mol Biol 914:65–86
- Lalli D, Schanda P, Chowdhury A, Retel J, Hiller M, Higman VA, et al. 2011. Three-dimensional deuterium-carbon correlation experiments for high-resolution solid-state MAS NMR spectroscopy of large proteins. J Biomol NMR 51:477–485
- Lange A, Luca S, Baldus M. 2002. Structural constraints from proton-mediated rare-spin correlation spectroscopy in rotating solids. J Am Chem Soc 124:9704–9705
- Lin MT, Sperling LJ, Frericks Schmidt HL, Tang M, Samoilova RI, Kumasaka T, et al. 2011. A rapid and robust method for selective isotope labeling of proteins. Methods 55:370–378
- Lin HJ, Huang TC, Muthusamy S, Lee JF, Duann YF, Lin CH. 2012. Piscidin-1, an antimicrobial peptide from fish (hybrid striped bass Morone saxatilis × M. chrysops), induces apoptotic and necrotic activity in HT1080 cells. Zoolog Sci 29:327–332
- Lindskog C. 2015. The potential clinical impact of the tissue-based map of the human proteome. Expert Rev Proteomics 12:213–215
- Liu J, Rost B. 2001. Comparing function and structure between entire proteomes. Prot Sci 10:1970–1979
- Lopez JJ, Mason AJ, Kaiser C, Glaubitz C. 2007. Separated local field NMR experiments on oriented samples rotating at the magic angle. J Biomol NMR 37:97–111
- Lopez JJ, Shukla AK, Reinhart C, Schwalbe H, Michel H, Glaubitz C. 2008. The structure of the neuropeptide bradykinin bound to the human G-protein coupled receptor bradykinin B2 as determined by solid-state NMR spectroscopy. Angew Chem Int Ed 47:1668–1671
- Loudet C, Manet S, Gineste S, Oda R, Achard MF, Dufourc EJ. 2007. Biphenyl bicelle disks align perpendicular to magnetic fields on large temperature scales: a study combining synthesis, solid-state NMR, TEM, and SAXS. Biophys J 92:3949–3959
- Lowe IJ. 1959. Free induction decays of rotating solids. Phys Rev Lett 2:285–287
- Lu GJ, Tian Y, Vora N, Marassi FM, Opella SJ. 2013. The structure of the mercury transporter MerF in phospholipid bilayers: a large conformational rearrangement results from N-terminal truncation. J Am Chem Soc 135:9299–9302
- Lu GJ, Opella SJ. 2014. Resonance assignments of a membrane protein in phospholipid bilayers by combining multiple strategies of oriented sample solid-state NMR. J Biomol NMR 58:69–81
- Lundstrom K. 2006. Latest development in drug discovery on G protein-coupled receptors. Curr Protein Pept Sci 7:465–470
- Luo W, Hong M. 2006. Determination of the oligomeric number and intermolecular distances of membrane protein assemblies by anisotropic 1H-driven spin diffusion NMR spectroscopy. J Am Chem Soc 128:7242–7251
- Luo W, Mani R, Hong M. 2007. Sidechain conformation and gating of the M2 transmembrane peptide proton channel of influenza A virus from solid-state NMR. J Phys Chem 111:10825–10832
- Maciejko J, Mehler M, Kaur J, Lieblein T, Morgner N, Ouari O, et al. 2015. Visualizing specific cross-protomer interactions in the homo-oligomeric membrane protein proteorhodopsin by DNP-enhanced solid-state NMR. J Am Chem Soc 137:9032–9043
- Mani R, Tang M, Wu X, Buffy JJ, Waring AJ, Sherman MA, Hong M. 2006. Membrane-bound dimer structure of a beta-hairpin antimicrobial peptide from rotational-echo double-resonance solid-state NMR. Biochemistry 45:8341–8349
- Marassi FM, Opella SJ. 2000. A solid-state NMR index of helical membrane protein structure and topology. J Magn Reson 144:150–155
- Marassi FM, Opella SJ. 2003. Simultaneous assignment and structure determination of a membrane protein from NMR orientational restraints. Protein Sci 12:403–411
- Marassi FM, Das BB, Lu GJ, Nothnagel HJ, Park SH, Son WS, et al. 2011. Structure determination of membrane proteins in five easy pieces. Methods 55:363–369
- Marcotte I, Auger M. 2005. Bicelles as model membranes for solid- and solution-state NMR studies of membrane peptides and proteins. Concepts in Magnetic Resonance Part A 24A:17–37
- Maslennikov I, Choe S. 2013. Advances in NMR structures of integral membrane proteins. Curr Opin Struct Biol 23:555–562
- Miao Y, Cross TA, Fu R. 2013. Identifying inter-residue resonances in crowded 2D (13)C- (13)C chemical shift correlation spectra of membrane proteins by solid-state MAS NMR difference spectroscopy. J Biomol NMR 56:265–273
- Middleton DA, Ahmed Z, Glaubitz C, Watts A. 2000. REDOR NMR on a hydrophobic peptide in oriented membranes. J Magn Reson 147:366–370
- Middleton DA, Hughes E, Esmann M. 2011. The conformation of ATP within the Na, K-ATPase nucleotide site: a statistically constrained analysis of REDOR solid-state NMR data. Angew Chem Int Ed 50:7041–7044
- Middleton DA, Patching SG. 2013. Solid-state NMR spectroscopy in drug design and discovery. In Andrushko V & Andrushko N, eds. Stereoselective synthesis of drugs and natural products. Chichester, UK: John Wiley & Sons, Ltd (Chapter 51)
- Mote KR, Gopinath T, Traaseth NJ, Kitchen J, Gor'kov PL, Brey WW, Veglia G. 2011. Multidimensional oriented solid-state NMR experiments enable the sequential assignment of uniformly 15N labeled integral membrane proteins in magnetically aligned lipid bilayers. J Biomol NMR 51:339–346
- Mote KR, Gopinath T, Veglia G. 2013. Determination of structural topology of a membrane protein in lipid bilayers using polarization optimized experiments (POE) for static and MAS solid state NMR spectroscopy. J Biomol NMR 57:91–102
- Murray DT, Das N, Cross TA. 2013. Solid state NMR strategy for characterizing native membrane protein structures. Acc Chem Res 46:2172–2181
- Murray DT, Li C, Gao FP, Qin H, Cross TA. 2014a. Membrane protein structural validation by oriented sample solid-state NMR: diacylglycerol kinase. Biophys J 106:1559–1569
- Murray DT, Hung I, Cross TA. 2014b. Assignment of oriented sample NMR resonances from a three transmembrane helix protein. J Magn Reson 240:34–44
- Nand D, Cukkemane A, Becker S, Baldus M. 2012. Fractional deuteration applied to biomolecular solid-state NMR spectroscopy. J Biomol NMR 52:91–101
- Nevzorov AA, Opella SJ. 2003. A “magic sandwich” pulse sequence with reduced offset dependence for high-resolution separated local field spectroscopy. J Magn Reson 164:182–186
- Nevzorov AA, Mesleh MF, Opella SJ. 2004. Structure determination of aligned samples of membrane proteins by NMR spectroscopy. Magn Reson Chem 42:162–171
- Nevzorov AA, Opella SJ. 2007. Selective averaging for high-resolution solid-state NMR spectroscopy of aligned samples. J Magn Reson 185:59–70
- Nicholson LK, Teng Q, Cross TA. 1991. Solid-state nuclear magnetic resonance derived model for dynamics in the polypeptide backbone of the gramicidin A channel. J Mol Biol 218:621–637
- Nietlispach D, Gautier A. 2011. Solution NMR studies of polytopic α-helical membrane proteins. Curr Opin Struct Biol 21:497–508
- Nishimura K, Kim S, Zhang L, Cross TA. 2002. The closed state of a H+ channel helical bundle combining precise orientational and distance restraints from solid state NMR. Biochemistry 41:13170–13177
- Nolandt OV, Walther TH, Grage SL, Ulrich AS. 2012. Magnetically oriented dodecylphosphocholine bicelles for solid-state NMR structure analysis. Biochim Biophys Acta 1818:1142–1147
- North CL, Cross TA. 1993. Analysis of polypeptide backbone T1 relaxation data using an experimentally derived model. J Magn Reson 101B:35–43
- North CL, Cross TA. 1995. Correlations between function and dynamics: time scale coincidence for ion translocation and molecular dynamics in the gramicidin channel backbone. Biochemistry 34:5883–5895
- Opella SJ, Marassi FM, Gesell JJ, Valente AP, Kim Y, Oblatt-Montal M, Montal M. 1999. Structures of the M2 channel-lining segments from nicotinic acetylcholine and NMDA receptors by NMR spectroscopy. Nat Struct Biol 6:374–379
- Opella SJ. 2013a. Structure determination of membrane proteins by nuclear magnetic resonance spectroscopy. Annu Rev Anal Chem (Palo Alto Calif) 6:305–328
- Opella SJ. 2013b. Structure determination of membrane proteins in their native phospholipid bilayer environment by rotationally aligned solid-state NMR spectroscopy. Acc Chem Res 46:2145–2153
- Opella SJ. 2014. The development of solid-state NMR of membrane proteins. Biomed Spectrosc Imaging 3:81–105
- Opella SJ. 2015. Solid-state NMR and membrane proteins. J Magn Reson 253:129–137
- Overington JP, Al-Lazikani B, Hopkins AL. 2006. How many drug targets are there? Nat Rev Drug Discov 5:993–996
- Oxenoid K, Chou JJ. 2005. The structure of phospholamban pentamer reveals a channel-like architecture in membranes. Proc Natl Acad Sci USA 102:10870–10875
- Page RC, Moore JD, Nguyen HB, Sharma M, Chase R, Gao FP, et al. 2006. Comprehensive evaluation of solution nuclear magnetic resonance spectroscopy sample preparation for helical integral membrane proteins. J Struct Funct Genomics 7:51–64
- Park SH, Mrse AA, Nevzorov AA, Mesleh MF, Oblatt-Montal M, Montal M, Opella SJ. 2003. Three-dimensional structure of the channel-forming trans-membrane domain of virus protein “u” (Vpu) from HIV-1. J Mol Biol 333:409–424
- Park SH, De Angelis AA, Nevzorov AA, Wu CH, Opella SJ. 2006. Three-dimensional structure of the transmembrane domain of Vpu from HIV-1 in aligned phospholipid bicelles. Biophys J 91:3032–3042
- Park SH, Loudet C, Marassi FM, Dufourc EJ, Opella SJ. 2008. Solid-state NMR spectroscopy of a membrane protein in biphenyl phospholipid bicelles with the bilayer normal parallel to the magnetic field. J Magn Reson 193:133–138
- Park SH, Das BB, De Angelis AA, Scrima M, Opella SJ. 2010a. Mechanically, magnetically, and “rotationally aligned” membrane proteins in phospholipid bilayers give equivalent angular constraints for NMR structure determination. J Phys Chem B 114:13995–14003
- Park SH, Marassi FM, Black D, Opella SJ. 2010b. Structure and dynamics of the membrane-bound form of Pf1 coat protein: implications of structural rearrangement for virus assembly. Biophys J 99:1465–1474
- Park SH1, Das BB, Casagrande F, Tian Y, Nothnagel HJ, Chu M, et al. 2012. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature 491:779–783
- Partridge AW, Therien AG, Deber CM. 2002. Polar mutations in membrane proteins as a biophysical basis for disease. Biopolymers 66:350–358
- Patching SG, Henderson PJ, Herbert RB, Middleton DA. 2008. Solid-state NMR spectroscopy detects interactions between tryptophan residues of the E. coli sugar transporter GalP and the alpha-anomer of the D-glucose substrate. J Am Chem Soc 130:1236–1244
- Patching SG. 2011. NMR structures of polytopic integral membrane proteins. Mol Membr Biol 28:370–397
- Patching SG, Henderson PJ, Sharples DJ, Middleton DA. 2013. Probing the contacts of a low-affinity substrate with a membrane-embedded transport protein using 1H-13C cross-polarisation magic-angle spinning solid-state NMR. Mol Membr Biol 30:129–137
- Patching SG. 2015. Roles of facilitative glucose transporter GLUT1 in [18F]FDG positron emission tomography (PET) imaging of human diseases. J Diagnostic Imaging Ther 2:30–102
- Peng X, Libich D, Janik R, Harauz G, Ladizhansky V. 2008. Dipolar chemical shift correlation spectroscopy for homonuclear carbon distance measurements in proteins in the solid state: application to structure determination and refinement. J Am Chem Soc 130:359–369
- Perrin BS Jr, Tian Y, Fu R, Grant CV, Chekmenev EY, Wieczorek WE, et al. 2014. High-resolution structures and orientations of antimicrobial peptides piscidin 1 and piscidin 3 in fluid bilayers reveal tilting, kinking, and bilayer immersion. J Am Chem Soc 136:3491–3504
- Pines A, Gibby MG, Waugh JS. 1973. Proton-enhanced NMR of dilute spins in solids. J Chem Phys 59:569–590
- Popot JL. 2010. Amphipols, nanodiscs, and fluorinated surfactants: three nonconventional approaches to studying membrane proteins in aqueous solutions. Annu Rev Biochem 79:737–775
- Prosser RS, Hunt SA, DiNatale JA, Vold RR. 1996. Magnetically aligned membrane model systems with positive order parameter: switching the sign of Szz with paramagnetic ions. J Am Chem Soc 118:269–270
- Quadri M, Federico A, Zhao T, Breedveld GJ, Battisti C, Delnooz C, et al. 2012. Mutations in SLC30A10 cause Parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet 90:467–477
- Radoicic J, Lu GJ, Opella SJ. 2014. NMR structures of membrane proteins in phospholipid bilayers. Q Rev Biophys 47:249–283
- Ragona F, Matricardi S, Castellotti B, Patrini M, Freri E, Binelli S, Granata T. 2014. Refractory absence epilepsy and glut1 deficiency syndrome: a new case report and literature review. Neuropediatrics 45:328–332
- Rask-Andersen M, Almén MS, Schiöth HB. 2011. Trends in the exploitation of novel drug targets. Nat Rev Drug Discov 10:579–590
- Reckel S, Hiller S. 2013. Perspectives of solution NMR spectroscopy for structural and functional studies of integral membrane proteins. Mol Phys 111:843–849
- Reif B. 2012. Ultra-high resolution in MAS solid-state NMR of perdeuterated proteins: implications for structure and dynamics. J Magn Reson 216:1–12
- Rosenbaum DM, Rasmussen SG, Kobilka BK. 2009. The structure and function of G-protein-coupled receptors. Nature 459:356–363
- Roumestand C, Louis V, Aumelas A, Grassy G, Calas B, Chavanieu A. 1998. Oligomerization of protegrin-1 in the presence of DPC micelles. A proton high-resolution NMR study. FEBS Lett 421:263–267
- Sanders CR, Schwonek JP. 1992. Characterization of magnetically orientable bilayers in mixtures of dihexanoylphosphatidylcholine and dimyristoylphosphatidylcholine by solid-state NMR. Biochemistry 31:8898–8905
- Sanders CR, Myers JK. 2004. Disease-related misassembly of membrane proteins. Annu Rev Biophys Biomol Struct 33:25–51
- Schnell JR, Chou JJ. 2008. Structure and mechanism of the M2 proton channel of influenza A virus. Nature 451:591–595
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. 2003. The Xplor-NIH NMR molecular structure determination package. J Magn Reson 160:65–73
- Shahid SA, Bardiaux B, Franks WT, Krabben L, Habeck M, van Rossum BJ, Linke D. 2012. Membrane-protein structure determination by solid-state NMR spectroscopy of microcrystals. Nat Methods 9:1212–1217
- Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath DD, et al. 2010. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science 330:509–512
- Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu G, et al. 2008. Consistent blind protein structure generation from NMR chemical shift data. Proc Natl Acad Sci USA 105:4685–4690
- Shi L, Ladizhansky V. 2012. Magic angle spinning solid-state NMR experiments for structural characterization of proteins. Methods Mol Biol 895:153–165
- Shukla HD, Vaitiekunas P, Cotter RJ. 2012. Advances in membrane proteomics and cancer biomarker discovery: current status and future perspective. Proteomics 12:3085–3104
- Smith AN, Caporini MA, Fanucci GE, Long JR. 2015. A method for dynamic nuclear polarization enhancement of membrane proteins. Angew Chem Int Ed 54:1542–1546
- Sperling LJ, Berthold DA, Sasser TL, Jeisy-Scott V, Rienstra CM. 2010. Assignment strategies for large proteins by magic-angle spinning NMR: the 21-kDa disulfide-bond-forming enzyme DsbA. J Mol Biol 399:268–282
- Stejskal EO, Memory JD. 1994. High resolution NMR in the solid state: fundamentals of CP/MAS. New York: Oxford University Press
- Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, et al. 2008. Structural basis for the function and inhibition of an influenza virus proton channel. Nature 451:596–599
- Striano P, Weber YG, Toliat MR, Schubert J, Leu C, Chaimana R, et al.; EPICURE Consortium. 2012. GLUT1 mutations are a rare cause of familial idiopathic generalized epilepsy. Neurology 78:557–562
- Su Y, Andreas L, Griffin RG. 2015. Magic angle spinning NMR of proteins: high-frequency dynamic nuclear polarization and 1H detection. Annu Rev Biochem 84:465–497
- Suls A, Mullen SA, Weber YG, Verhaert K, Ceulemans B, Guerrini R, et al. 2009. Early-onset absence epilepsy caused by mutations in the glucose transporter GLUT1. Ann Neurol 66:415–419
- Takeda K. 2012. Microcoils and microsamples in solid-state NMR. Solid State Nucl Magn Reson 47–48:1–9
- Takegoshi K, Nakamura S, Terao T. 2001. Dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem Phys Lett 344:631–637
- Takegoshi K, Nakamura S, Terao T. 2003. 13C-1H dipolar-driven 13C-13C recoupling without 13C rf irradiation in nuclear magnetic resonance of rotating solids. J Chem Phys 118:2325
- Tang M, Comellas G, Mueller LJ, Rienstra CM. 2010. High resolution 13C-detected solid-state NMR spectroscopy of a deuterated protein. J Biomol NMR 48:103–111
- Tang M, Sperling LJ, Berthold DA, Schwieters CD, Nesbitt AE, Nieuwkoop AJ, et al. 2011. High-resolution membrane protein structure by joint calculations with solid-state NMR and X-ray experimental data. J Biomol NMR 51:227–233
- Tang M, Comellas G, Rienstra CM. 2013a. Advanced solid-state NMR approaches for structure determination of membrane proteins and amyloid fibrils. Acc Chem Res 46:2080–2088
- Tang M, Nesbitt AE, Sperling LJ, Berthold DA, Schwieters CD, Gennis RB, Rienstra CM. 2013b. Structure of the disulfide bond generating membrane protein DsbB in the lipid bilayer. J Mol Biol 425:1670–1682
- Tapaneeyakorn S, Goddard AD, Oates J, Willis CL, Watts A. 2011. Solution- and solid-state NMR studies of GPCRs and their ligands. Biochim Biophys Acta 1808:1462–1475
- Taylor RE. 2004. Setting up 13C CP/MAS experiments. Concept Magn Reson A 22A:37–49
- Thurber KR, Tycko R. 2008. Biomolecular solid state NMR with magic-angle spinning at 25K. J Magn Reson 195:179–186
- Tian Y, Lu GJ, Marassi FM, Opella SJ. 2014. Structure of the membrane protein MerF, a bacterial mercury transporter, improved by the inclusion of chemical shift anisotropy constraints. J Biomol NMR 60:67–71
- Traaseth NJ, Shi L, Verardi R, Mullen DG, Barany G, Veglia G. 2009. Structure and topology of monomeric phospholamban in lipid membranes determined by a hybrid solution and solid-state NMR approach. Proc Natl Acad Sci USA 106:10165–10170
- Triba MN, Devaux PF, Warschawski DE. 2006. Effects of lipid chain length and unsaturation on bicelles stability. A phosphorus NMR study. Biophys J 91:1357–1367
- Tycko R. 2013. NMR at low and ultralow temperatures. Acc Chem Res 46:1923–1932
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. 2015. Proteomics. Tissue-based map of the human proteome. Science 347:1260419
- Valentine KG, Liu SF, Marassi FM, Veglia G, Opella SJ, Ding FX, et al. 2001. Structure and topology of a peptide segment of the 6th transmembrane domain of the Saccharomyces cerevisae alpha-factor receptor in phospholipid bilayers. Biopolymers 59:243–256
- Verardi R, Shi L, Traaseth NJ, Walsh N, Veglia G. 2011. Structural topology of phospholamban pentamer in lipid bilayers by a hybrid solution and solid-state NMR method. Proc Natl Acad Sci USA 108:9101–9106
- Verardi R, Traaseth NJ, Masterson LR, Vostrikov VV, Veglia G. 2012. Isotope labeling for solution and solid-state NMR spectroscopy of membrane proteins. Adv Exp Med Biol 992:35–62
- Vogeley L, Sineshchekov OA, Trivedi VD, Sasaki J, Spudich JL, Luecke H. 2004. Anabaena sensory rhodopsin: a photochromic color sensor at 2.0 A. Science 306:1390–1393
- von Heijne G. 2007. The membrane protein universe: what’s out there and why bother? J Int Med 261:543–557
- Wallin E, von Heijne G. 1998. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Prot Sci 7:1029–1038
- Wang S, Kim SY, Jung KH, Ladizhansky V, Brown LS. 2011. A eukaryotic-like interaction of soluble cyanobacterial sensory rhodopsin transducer with DNA. J Mol Biol 411:449–462
- Wang S, Munro RA, Shi L, Kawamura I, Okitsu T, Wada A, et al. 2013. Solid-state NMR spectroscopy structure determination of a lipid-embedded heptahelical membrane protein. Nat Methods 10:1007–1012
- Wang S, Ladizhansky V. 2014. Recent advances in magic angle spinning solid state NMR of membrane proteins. Prog Nucl Magn Reson Spectrosc 82:1–26
- Wang T, Williams JK, Schmidt-Rohr K, Hong M. 2015. Relaxation-compensated difference spin diffusion NMR for detecting 13C-13C long-range correlations in proteins and polysaccharides. J Biomol NMR 61:97–107
- Ward ME, Brown LS, Ladizhansky V. 2015. Advanced solid-state NMR techniques for characterization of membrane protein structure and dynamics: application to Anabaena sensory rhodopsin. J Magn Reson 253:119–128
- Warschawski DE, Arnold AA, Beaugrand M, Gravel A, Chartrand É, Marcotte I. 2011. Choosing membrane mimetics for NMR structural studies of transmembrane proteins. Biochim Biophys Acta 1808:1957–1974
- Watanabe H, Koopmann TT, Le Scouarnec S, Yang T, Ingram CR, Schott JJ, et al. 2008. Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest 118:2260–2268
- Watts A. 2005. Solid-state NMR in drug design and discovery for membrane-embedded targets. Nat Rev Drug Discov 4:555–568
- Weber YG, Storch A, Wuttke TV, Brockmann K, Kempfle J, Maljevic S, et al. 2008. GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak. J Clin Invest 118:2157–2168
- Whiles JA, Deems R, Vold RR, Dennis EA. 2002. Bicelles in structure-function studies of membrane-associated proteins. Bioorg Chem 30:431–442
- Whittaker CA, Patching SG, Esmann M, Middleton DA. 2015. Ligand orientation in a membrane-embedded receptor site revealed by solid-state NMR with paramagnetic relaxation enhancement. Org Biomol Chem 13:2664–2668
- Williamson PT, Verhoeven A, Miller KW, Meier BH, Watts A. 2007. The conformation of acetylcholine at its target site in the membrane-embedded nicotinic acetylcholine receptor. Proc Natl Acad Sci USA 104:18031–18036
- Williamson PTF. 2009. Solid-state NMR for the analysis of high-affinity ligand/receptor interactions. Concept Magn Reson A 34A:144–172
- Wu CH, Ramamoorthy A, Opella SJ. 1994. High resolution heteronuclear dipolar solid-state NMR spectroscopy. J Magn Reson A 109:270–272
- Yannoni CS. 1982. High-resolution NMR in solids: the CPMAS experiment. Acc Chem Res 15:201–208
- Yao Y, Ding Y, Tian Y, Opella SJ, Marassi FM. 2013. Membrane protein structure determination: back to the membrane. Methods Mol Biol 1063:145–158
- Yarov-Yarovoy V, Schonbrun J, Baker D. 2006. Multipass membrane protein structure prediction using Rosetta. Proteins 62:1010–1025
- Zhao X. 2012. Protein structure determination by solid-state NMR. Top Curr Chem 326:187–213
- Zhou HX, Cross TA. 2013a. Influences of membrane mimetic environments on membrane protein structures. Annu Rev Biophys 42:361–392
- Zhou HX, Cross TA. 2013b. Modeling the membrane environment has implications for membrane protein structure and function: influenza A M2 protein. Protein Sci 22:381–394
- Zoonens M, Popot JL. 2014. Amphipols for each season. J Membr Biol 247:759–796