Abstract
5′ adenosine monophosphate-activated protein kinase (AMPK) plays a prominent role as a metabolic stress sensor. The role of hypothalamic AMPK in response to restraint and surgical stress has not been previously investigated. It has been recently suggested that the renin–angiotensin system, in addition to its role in stress regulation, may play a significant role in regulating metabolic pathways including the regulation of the AMPK system. This study was thus aimed to evaluate the effects of candesartan, an angiotensin II AT1 receptor blocker drug, on hypothalamic AMPK activity under basal conditions and after restraint in conscious rats or after surgical stress under general anesthesia. Male Wistar rats were treated with 5 mg/kg/day candesartan in the drinking water for 2 weeks. The hypothalamic AMPK activity was determined under basal and stress conditions, using a kinase activity assay. Chronic administration of candesartan significantly increased hypothalamic AMPK activity. Hypothalamic AMPK activity was also increased by restraint stress whereas no change was observed during surgical stress under anesthesia. The high levels of hypothalamic AMPK activation observed in candesartan-treated rats were not changed by restraint stress but were reduced to control levels by anesthesia and surgery. In conclusion, chronic candesartan treatment and restraint stress in conscious rats stimulate the hypothalamic AMPK activity, whereas surgical stress under anesthesia inhibits pathways regulating the AMPK activity even in candesartan-treated rats.
Introduction
5′ adenosine monophosphate-activated protein kinase (AMPK) is a heterotrimeric kinase of the serine–threonine kinase family present in all eukaryotic cells (Hardie and Carling Citation1997; Hardie et al. Citation2006). It plays a prominent role as a metabolic and stress sensor, mainly in terms of activation in response to an increasing adenosine monophosphate (AMP)/adenosine-5′-triphosphate (ATP) ratio, thereby improving survival under metabolic stress by integrating nutritional and hormonal signals in peripheral tissues and the hypothalamus (Kahn et al. Citation2005). The activation of AMPK in vivo is complex and varies according to the magnitude and type of the stress as well as the organ studied (Pacak Citation2000). The brain has been considered to be a key organ that needs to be protected during stress, and AMPK is highly expressed in several brain areas, especially the hypothalamus, in which the role of AMPK has been previously investigated (Kola Citation2008; Kola et al. Citation2008; Ronnett et al. Citation2009).
Recently, it has been suggested that the renin–angiotensin system may play a significant role in regulating the AMPK system (Yoshida et al. Citation2009). Angiotensin receptor blockers (ARBs) are considered to play a protective role in patients with hypertension, heart disease, and diabetes mellitus in addition to their antihypertensive effect (Atmaca and Gedik Citation2006; Ribeiro-Oliveira et al. Citation2008). The number of patients chronically treated with ARBs who are scheduled for surgery is increasing (Comfere et al. Citation2005), and the effects of these agents in perioperative medicine have not been fully determined. It is known that AMPK can also be activated by certain drugs and hormones as well as by cellular stressors that do not alter the AMP/ATP ratio, such as osmotic changes. In addition, although AMPK has been extensively studied in terms of its response to metabolic fuel deprivation (McBride and Hardie Citation2009), few reports are available on the in vivo activation of AMPK in response to common stress scenarios, such as restraint and during surgical procedures.
We therefore sought to evaluate the hypothalamic AMPK activity under basal conditions and in response to restraint stress or major surgery in anesthetized rats, after chronical treatment with an ARB (candesartan) in order to obtain new insights into the role of the AMPK system in these stress procedures. Specifically, we aimed to assess hypothalamic AMPK in two different stress models, and we hypothesized that candesartan would increase the hypothalamic AMPK activity.
Methods
Animals
Male Wistar rats, 7–9 weeks old (provided by the Federal University of Minas Gerais Medical School Animal Facility), were maintained under temperature-controlled conditions (25 ± 2°C) with an artificial 12-h light–dark cycle, with lights on from 07:00 to 19:00 h, and given standard chow and water ad libitum. Rats were housed in individual cages and handled daily. A total of 64 rats were treated with candesartan cilexetil (AstraZeneca, Cotia, Brazil) 5 mg/kg/day in the drinking water for 2 weeks (n = 32) or no additions (vehicle) for the same period (n = 32), according to a well-established protocol (Jones et al. Citation2004; De Cavanagh et al. Citation2005), utilizing metered drinking bottles with air holes. Body weight was measured with an accurate balance, and fluid intake was recorded daily. The University Ethics Committee for Animal Experimentation approved all procedures (Protocol No. 110/2007).
Experimental design
Following the 2-week treatment with candesartan, rats were transferred in the early morning on the day of the experiment to the temperature-controlled experimental room and were left to rest for 60 min in their home cages. Noise was controlled to a minimum. Experiments were conducted between 08.00 and 12.00 h.
For the restraint stress, a well-established emotional stressor in rodents, experimental rats were placed for 20 min in a plastic polyethylene tube (21-cm length, 4.5-cm diameter), where they could not move, causing intense discomfort without apparent pain, for 20 min (Reis et al. Citation1996; Ribeiro-de-Oliveira et al. Citation2001). For the surgical stress, experimental rats were anesthetized with 0.075 mg/g ketamine (Cristália, Itapira, Brazil) and 0.01 mg/g xylazine (Schering-Plough Coopers, Cotia, Brazil) injected intraperitoneally, according to the protocol of the local Ethics Committee for Animal Research. After an average of 5 min, when the rats were deeply anesthetized, a median laparotomy was carried out, followed by evisceration and exposure of the bowels for 20 min (Reis et al. Citation1996) without removal of any organ, and the exposed tissues were kept warm and moist with 0.9% saline.
Non-stressed rats and those after 20 min of stress were moved to an adjacent room where they were killed by decapitation without anesthesia for the conscious rats, and the brain was removed. The hypothalamus was excised as a 2-mm thick coronal section, from 2 to 4 mm from bregma, and from the supraoptic decussation to the dorsal end of the lower part of the third ventricle. The procedure took less than 3 min. The hypothalamus was immediately placed in liquid nitrogen and stored at − 80°C. Blood from the trunk was collected with syringes containing 6% sodium citrate and kept in ice until centrifugation (900 g, 20 min, 4°C). Plasma was then separated and stored for subsequent glucose measurements.
Assays
Plasma glucose was assayed in duplicate by glucose oxidase, utilizing commercial kits from CENTERLAB (Santa Luzia, Minas Gerais, Brazil). Glucose was measured to confirm that the rats were stressed as its concentration in plasma increases as a result of acute actions of noradrenaline, adrenaline, and glucocorticoids secreted during stress, and to compare the hyperglycemic responses to the different stressors.
The AMPK activity of hypothalamic tissue was evaluated at both baselines and after 20 min of exposure to each stressor. We chose to evaluate the AMPK activity after 20 min of stress based on our previous experience and that of others, showing a peak of hyperglycemic- and stress hormone-induced response around 20 min after stress (Yamada et al. Citation1993; Reis et al. Citation1996; Ribeiro-de-Oliveira et al. Citation1999; Ribeiro-de-Oliveira et al. Citation2001). Each hypothalamus was weighed after dissection and homogenized with Precellys 24 using CK14 tubes containing ceramic beads (Stretton Scientific, Stretton, UK) at 3600g for one cycle of 20 s in lysis buffer containing 50 mM Tris–HCl, 50 mM NaF, 5 mM Na pyrophosphate, 1 mM ethylenediaminetetraacetic acid, 250 mM sucrose, 1% Triton X-100, 1 mM dithiothreitol, 1 mM benzamidine, 0.1 mM phenylmethane sulfonyl fluoride, and 5 μg/ml soybean trypsin inhibitor. Tissue protein content was determined using bicinchoninic acid assay (Pierce, Rockford, IL, USA). The AMPK activity was assayed as previously described (Hawley et al. Citation2003): briefly, AMPK was immunoprecipitated with an equal mixture of α1AMPK and α2AMPK antibodies (Hawley et al. Citation2003), and the AMPK activity was determined by the phosphorylation of SAMS peptide (Pepceuticals Ltd, Nottingham, UK), a synthetic peptide substrate of AMPK (Hawley et al. Citation2003). The AMPK activity was calculated using the difference of the counts between SAMS containing and SAMS negative samples and expressed as nanomoles of ATP incorporated per minute per milligram of sample of peptide. The AMPK activity was expressed as percentage of control. The sensitivity of the AMPK assay was 5 pmol/ATP/min/mg protein with intra- and inter-assay coefficients of variation of 10% and 15%, respectively.
Statistical analysis
The data were evaluated using unpaired Student's t-test or parametric tests (one-way ANOVA followed by Bonferroni test) for fluid intake, body weight, and glucose concentrations, or non-parametric tests (Kruskal–Wallis test followed by Conover-Inman comparison) for AMPK experiments. GraphPad Prism4 (San Diego, CA, USA) was utilized for statistical analysis. All data are expressed as means ± standard error of the means (SEM), and significance was taken at p < 0.05.
Results
Fluid intake and body weight
There was no significant difference in fluid intake during the 2-week candesartan treatment between the candesartan-treated group and controls (n = 20 in each, 21.6 ± 1.0 vs. 19.5 ± 1.3 ml/100 g/day, respectively, t (38) = 1.24, p>0.05). There was no difference between the 2-week fluid intake of rats which on the day of the stress tests were assigned to the stress or to the control groups, indicating that they had equal exposure to candesartan.
The initial body weight (n = 20 in each group candesartan 111.3 ± 4.0 g vs. no-candesartan 111.3 ± 3.5 g, t (38) = 1.03, p>0.05) or final body weight (n = 20 in each group, candesartan 178.5 ± 6.9 vs. no-candesartan 193.1 ± 7.6 g, t (38) = 1.43, p>0.05) of candesartan-treated rats did not differ significantly from controls after 2 weeks of treatment. However, when the weight gain was compared between the groups, the candesartan-treated group showed a significantly lower weight gain than no-candesartan controls (n = 20 in each group, 67.2 ± 5.0 vs. 81.9 ± 4.6 g, t (38) = 2.16, p < 0.05).
Plasma glucose concentrations
Plasma glucose concentrations were compared by one-way ANOVA (n = 7–9 for each group, F (7) = 22.16, overall p < 0.01, ). Bonferroni's multiple comparison test using a significance criterion of p < 0.05 to indicate differences between groups showed that there was no significant difference when comparing plasma glucose concentrations in candesartan-treated non-stressed rats with those in non-stressed controls (87.5 ± 4.2 vs. 88.9 ± 1.7 mg/dl, respectively, p>0.05, ). After 20-min restraint stress, plasma glucose concentrations increased significantly when compared with those of the respective non-stressed controls in both the no-candesartan (p < 0.01, ) and candesartan-treated groups (, p < 0.05). However, there were no significant differences between the no-candesartan stressed and candesartan-treated stressed groups.
Figure 1. Effects of restraint stress and surgical stress upon plasma glucose concentrations in rats treated with candesartan for 2 weeks. **p < 0.01 compared to control; p < 0.05,
p < 0.01 compared to candesartan;
p < 0.01 compared to control+restraint stress;
p < 0.01 compared to candesartan+restraint stress. Data are expressed as mean ± SEM; n = 7–9 for each group. ANOVA followed by Bonferroni test.
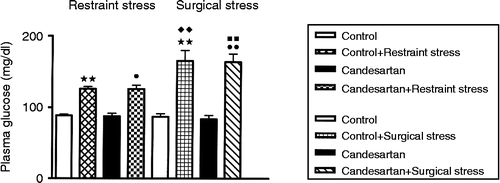
Similarly, in the surgical stress groups, plasma glucose concentrations increased significantly in both the no-candesartan stressed group vs. the no-candesartan non-stressed group (p < 0.01) and the candesartan stressed vs. the candesartan non-stressed group (p < 0.01, ). There was no difference in the plasma glucose concentrations between the candesartan-treated and no-candesartan non-stressed control groups or the candesartan-treated stressed and no-candesartan-stressed groups.
When the two different stresses were compared, the hyperglycemic responses were different (). Surgical stress under general anesthesia evoked a greater hyperglycemic stress response than restraint stress in both the candesartan-treated rats and the no-candesartan groups (p < 0.01 for both comparisons), confirming our previous findings (Reis et al. Citation1996).
Hypothalamic AMPK activity
In the restraint experiments, data were analyzed by the Kruskal–Wallis test (n = 7–9 for each group, T (3) = 14.4, overall p < 0.01). This was followed by the Conover-Inman comparison using a significance criterion of p < 0.05 to indicate differences between the groups. After 20 min restraint, the hypothalamic AMPK activity increased significantly in the hypothalamus of no-candesartan stressed rats when compared to that of no-candesartan non-stressed rats (p < 0.01, ). After 2 weeks of candesartan treatment, there was an increase in the hypothalamic AMPK activity in candesartan-treated non-stressed rats when compared to that in no-candesartan non-stressed controls (p < 0.01, ). However, the AMPK activity did not increase further in candesartan-treated stressed rats after 20-min restraint when compared to that in candesartan-treated non-stressed rats (p>0.05). The candesartan-treated stressed rats had higher AMPK values than no-candesartan non-stressed controls (p < 0.05, ).
Figure 2. Hypothalamic AMPK activity at baseline and after restraint and surgical stress in rats treated with candesartan for 2 weeks. *p < 0.05, **p < 0.01 compared to control; p < 0.05,
p < 0.01 compared to candesartan;
p < 0.01 compared to control+restraint stress;
p < 0.01 compared to candesartan+restraint stress. Data are expressed as mean ± SEM; n = 7–9 for each group. Kruskal–Wallis test followed by Conover-Inman comparison.
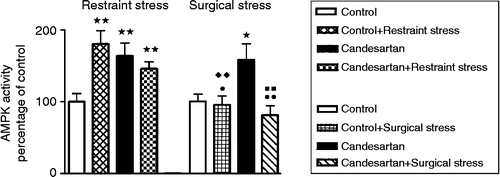
These data show that both the groups of restraint stressed rats had increased hypothalamic AMPK activity following restraint when compared to no-candesartan controls. Candesartan treatment may possibly have prevented further stress increases in hypothalamic AMPK during restraint.
In the surgical stress group, data were also analyzed by the Kruskal–Wallis test (n = 7–9 for each group, T (3) = 9.57, overall p < 0.05). This was followed by the Conover-Inman comparison using a significance criterion of p < 0.05 to indicate differences between the groups. After 20-min surgical stress under general anesthesia, no significant change could be observed in hypothalamic AMPK between no-candesartan stressed rats and the no-candesartan non-stressed control group (p>0.05, ). Candesartan treatment again elevated hypothalamic AMPK in the non-anesthetized, non-stressed rats (p < 0.05, ). No significant change was observed in the AMPK activity between the candesartan-treated anesthesia-surgical stressed group and the no-candesartan-treated rats (p>0.05 for all comparisons, ). However, there was a significant difference in the AMPK activity between the candesartan-treated anesthesia-surgical stressed group and candesartan-treated non-stressed rats (p < 0.01, ).
These data show that rats under general anesthesia subjected to surgical stress do not show change in hypothalamic AMPK when compared to no-candesartan controls, indicating that general anesthesia plus surgical stress blocks hypothalamic AMPK increases observed in the chronically candesartan-treated non-stressed group.
The level of AMPK activity under stress was different when two different stress models were compared (). The AMPK activity values in surgical stress under general anesthesia were significantly lower in both no-candesartan and candesartan-treated stressed groups than those in the same groups subjected to restraint, respectively (n = 7–9 for each group, T (7) = 30.6, overall p < 0.01, followed by the Conover-Inman test, p < 0.01 for both comparisons).
Discussion
The results of this study demonstrate that chronic candesartan treatment stimulated the hypothalamic AMPK activity. It also shows that the psychological stress caused by restraint stimulated the hypothalamic AMPK activity irrespective of the hyperglycemia induced by the restraint stress. The candesartan-induced increase in hypothalamic AMPK activity did not change further in restraint stress rats. Surgical stress under general anesthesia was, however, unable to increase hypothalamic AMPK levels, indicating that this procedure prevented a stress-induced or the candesartan-induced increase in hypothalamic AMPK activity. We consider that the combination of general anesthesia and surgery inhibited the pathways that lead to stress- or candesartan-induced AMPK activity elevations in the hypothalamus.
AMPK is a metabolic regulator sensor with extensive downstream targets involved in protein, fat, carbohydrate metabolism, and energy utilization. AMPK not only senses energy status, but also functions at the tissue and whole organism level to promote context-specific responses to physiological signals of metabolic status in response to ATP depletion (an increased AMP/ATP ratio) and related stimuli. Several stressors, such as exercise, hypoxia, and ischemic stroke, have been described as activators of AMPK (Ramamurthy and Ronnett Citation2006). Hypothalamic AMPK has an important role in energy-sensing circuits involved in energy homeostasis, and is thus positioned to function as a “master regulator” of energy balance (Kola Citation2008).
This study is the first, to the best of our knowledge, to address the role of hypothalamic AMPK in laboratory models of restraint and surgical stress. ARBs are widely used in clinical medicine, and little is known regarding their effects on the AMPK system (Feng et al. Citation2011). We have shown here that candesartan significantly increases the hypothalamic AMPK activity in the hypothalamus, which corresponds with recent data (Feng et al. Citation2011).
In addition to their therapeutic role in blood pressure regulation and vascular function, evidence has been accumulated regarding the beneficial effect of ARBs on the regulation of metabolic processes (Gilliam-Davis et al. Citation2007; Ecelbarger et al. Citation2010), although the mechanisms are yet to be clarified. Candesartan has been shown to increase the expression of peroxisome proliferator-activated receptor-gamma (PPAR-γ) and adiponectin (Zorad et al. Citation2006), which are both known to increase the hypothalamic AMPK activity (Yamauchi and Kadowaki Citation2008). Both PPAR-γ agonism and the effect on adiponectin might have contributed to the observed increase in basal hypothalamic AMPK activity (Erbe et al. Citation2006; Zorad et al. Citation2006; Fukuda et al. Citation2010). Similarly, it has been recently shown that another ARB, telmisartan, acts in skeletal muscle through the stimulation of the PPAR/AMPK pathway (Feng et al. Citation2011). Hypothalamic AMPK has emerged as an important player in the regulation of appetite, contributing to the control of energy metabolism balance at both the cell and whole body levels (Kola et al. Citation2005, Citation2008; Kola Citation2008). The stimulatory effect of candesartan on the hypothalamic AMPK activity could be due to PPAR-γ agonism (Aubert et al. Citation2010) and could also be the consequence of the effect of candesartan on body weight, or it may occur via a currently unrecognized mechanism. Recently, it has been shown that the body weight-reducing effect of high-dose ARB treatment depends on leptin signaling, as it was not observed in leptin-receptor mutant Zucker rats (Muller-Fielitz et al. Citation2011). However, leptin reduces hypothalamic AMPK levels, so the stimulation we have observed is unlikely to be due to increased leptin levels. We have also observed that candesartan-treatment increases the baseline AMPK activity in the liver (data not shown), in agreement with its PPAR-γ agonism peripherally, and its known beneficial effects on metabolism. These data suggest that candesartan-induced AMPK activity, involving the induction of endothelial nitric oxide synthase (e-NOS) and augmented bio-availability of nitric oxide, might explain its role in protection against cerebral ischemia in non-stressed conditions (Nishimura et al. Citation2000a; Liu et al. Citation2008; Zhang et al. Citation2008). Interestingly, hypothalamic AMPK was not further increased in candesartan-treated rats after restraint stress, and this could be possibly related to the prevention of deleterious over activation of the AMPK system (Ramamurthy and Ronnett Citation2006). Surgical stress under general anesthesia has been shown in other studies to switch off hypothalamic AMPK in the intra-operative period. It would thus be of interest to evaluate the levels of AMPK in the post-operative period in candesartan-treated individuals, and possibly correlate such data with surgical outcome in a population at risk for post-surgical neurologic complications.
Our previously described (Reis et al. Citation1996) different hyperglycemic responses to restraint and surgical stress have been reproduced in this study, corroborating the notion of a marked heterogeneity of neuroendocrine stress responses, as described by others (Gaillet et al. Citation1991; Palkovits et al. Citation1997; Pacak Citation2000). Likewise, hypothalamic AMPK activity was shown here to respond differentially to these stressors. After restraint, control rats showed increased hypothalamic AMPK levels, whereas no change was observed in hypothalamic AMPK levels after surgery under general anesthesia. Indeed, acute restraint typically activates neurons in the hypothalamic paraventricular nucleus (PVN) (Gibson et al. Citation1986), and changes in AMPK activity have been previously reported in the PVN in response to leptin (Minokoshi et al. Citation2004) and ghrelin (Kola et al. Citation2005). Interestingly, this effect was not overcome by the restraint-induced hyperglycemia. However, general anesthesia may prevent changes in activation in hypothalamic cells. The α2-adrenoceptor agonist xylazine utilized in these experiments acts centrally to decrease the efferent sympathetic activity and circulating norepinephrine concentrations, thus preserving energy consumption (Changmin et al. Citation2010). Ketamine, a N-methyl-d-aspartic acid receptor antagonist, has been recently demonstrated to lower brain AMPK activity when administered to rats before ischemia (Li et al. Citation2009). Thus, the anesthetic combination of ketamine and xylazine may have prevented an increase in AMPK during surgical stress. Indeed, surgical stress with this anesthetic combination was even able to attenuate hypothalamic AMPK activity in rats pre-treated with candesartan so that high baseline hypothalamic AMPK levels were reduced to levels as low as those found in no-candesartan-treated rats. Although these data could also be related to the marked hyperglycemia that was evoked by surgical stress under general anesthesia reducing AMPK activity values (Kola Citation2008), a direct effect of anesthesia plus surgery could turn off the pathways regulating hypothalamic AMPK activity.
The role of angiotensin II AT1 receptors in the regulation of stress responses has been previously investigated (Machado et al. Citation1998; Saavedra and Benicky Citation2007; Bregonzio et al. Citation2008). Interestingly, it has been shown that acute restraint increases AT1 mRNA in the PVN (Leong et al. Citation2002), whereas chronic peripheral administration of candesartan to block brain AT1 receptors (Nishimura et al. Citation2000b; Unger Citation2003; Pelisch et al. Citation2010) inhibits central responses to angiotensin II in rats (Nishimura et al. Citation2000a; Gohlke et al. Citation2002; Pelisch et al. Citation2010). Moreover, candesartan has been recently described as an anti-stress compound (Pavel et al. Citation2008), improving stress-related disorders (Pavlatou et al. Citation2008) and reducing cortisol responses to corticotrophin-releasing hormone stimulation (Pavlatou et al. Citation2008). Thus, the blockade of AT1 receptors in the PVN could be related to the absence of a further increase in hypothalamic AMPK activity in candesartan-treated rats subjected to restraint. The PVN is known to be specifically associated with changes in AMPK activity (Kola Citation2008), but in our study we did not study nuclei separately within the hypothalamus.
Interestingly, although hypothalamic AMPK activity was increased in candesartan-treated non-stressed controls, the treatment with this AT1 receptor blocker did not clearly interfere with the effect of anesthesia on hypothalamic AMPK activity. In the intra-operative period, general anesthesia reduced cerebral metabolism sparing hypothalamic AMPK, and the high preoperative hypothalamic AMPK levels found in the candesartan-treated rats were then reduced to levels observed in no-candesartan stressed rats. Although our observations of AMPK activity in the hypothalamus following candesartan and following two types of stress are novel, this study was not designed to specifically elucidate the underlying mechanisms. Our data demonstrate a picture of the in vivo variability of responses to common stressors, and the expanding roles of AMPK in stress and anesthesia. We did not include a group of rats with anesthesia alone (without surgery), as our hypothesis was based on the effects of combined anesthesia and surgery, mirroring a “real life” situation. Future studies should address the molecular pathways involved in AMPK changes following these common stress events, besides exploring AMPK changes in other brain areas as well as the hypothalamus in time course experiments.
In summary, this in vivo study has shown that restraint stress increased hypothalamic AMPK activity, whereas surgical stress under general anesthesia did not change its activity. These represent types of emotional stress and physical stress, respectively, involving different central pathways (Reis et al. Citation1996). We have also shown that candesartan treatment for 2 weeks increases basal hypothalamic AMPK activity and prevents a further restraint-induced hypothalamic AMPK increase, whereas in combination with anesthesia and surgery it reduces the high AMPK activity caused by candesartan pre-treatment.
Acknowledgments
This study was supported by the CNPq (National Counsel of Technological and Scientific Development), the Fapemig (Fundação para o Desenvolvimento da Pesquisa do Estado de Minas Gerais), and the Wellcome Trust.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Atmaca A, Gedik O. 2006. Effects of angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, and their combination on microalbuminuria in normotensive patients with type 2 diabetes. Adv Ther. 23:615–622.
- Aubert G, Burnier M, Dulloo A, Perregaux C, Mazzolai L, Pralong F, Zanchi A. 2010. Neuroendocrine characterization and anorexigenic effects of telmisartan in diet- and glitazone-induced weight gain. Metabolism. 59:25–32.
- Bregonzio C, Seltzer A, Armando I, Pavel J, Saavedra JM. 2008. Angiotensin II AT(1) receptor blockade selectively enhances brain AT(2) receptor expression, and abolishes the cold-restraint stress-induced increase in tyrosine hydroxylase mRNA in the locus coeruleus of spontaneously hypertensive rats. Stress. 11:457–466.
- Changmin H, Jianguo C, Dongming L, Guohong L, Mingxing D. 2010. Effects of xylazole alone and in combination with ketamine on the metabolic and neurohumoral responses in healthy dogs. Vet Anaesth Analg. 4:322–328.
- Comfere T, Sprung J, Kumar MM, Draper M, Wilson DP, Williams BA, Danielson DR, Liedl L, Warner DO. 2005. Angiotensin system inhibitors in a general surgical population. Anesth Analg. 100:636–644.
- De Cavanagh EM, Toblli JE, Ferder L, Piotrkowski B, Stella I, Fraga CG, Inserra F. 2005. Angiotensin II blockade improves mitochondrial function in spontaneously hypertensive rats. Cell Mol Biol. 51:573–578.
- Ecelbarger CM, Rash A, Sinha RK, Tiwari S. 2010. The effect of chronic candesartan therapy on the metabolic profile and renal tissue cytokine levels in the obese Zucker rat. Mediators Inflamm. 2010:1–12.
- Erbe DV, Gartrell K, Zhang YL, Suri V, Kirincich SJ, Will S, Perreault M, Wang S, Tobin JF. 2006. Molecular activation of PPARgamma by angiotensin II type 1-receptor antagonists. Vascul Pharmacol. 45:154–162.
- Feng X, Luo Z, Ma L, Ma S, Yang D, Zhao Z, Yan Z, He H, Cao T, Liu D, Zhu Z. 2011. Angiotensin II receptor blocker telmisartan enhances running endurance of skeletal muscle through activation of the PPAR-delta/AMPK pathway. J Cell Mol Med. 15:1572–1581.
- Fukuda M, Nakamura T, Kataoka K, Nako H, Tokutomi Y, Dong YF, Ogawa H, Kim-Mitsuyama S. 2010. Potentiation by candesartan of protective effects of pioglitazone against type 2 diabetic cardiovascular and renal complications in obese mice. J Hypertens. 28:340–352.
- Gaillet S, Lachuer J, Malaval F, Assenmacher I, Szafarczyk A. 1991. The involvement of noradrenergic ascending pathways in the stress-induced activation of ACTH and corticosterone secretions is dependent on the nature of stressors. Exp Brain Res. 87:173–180.
- Gibson A, Hart SL, Patel S. 1986. Effects of 6-hydroxydopamine-induced lesions of the paraventricular nucleus, and of prazosin, on the corticosterone response to restraint in rats. Neuropharmacology. 25:257–260.
- Gilliam-Davis S, Payne VS, Kasper SO, Tommasi EN, Robbins ME, Diz DI. 2007. Long-term AT1 receptor blockade improves metabolic function and provides renoprotection in Fischer-344 rats. Am J Physiol Heart Circ Physiol. 293:H1327–H1333.
- Gohlke P, Kox T, Jurgensen T, von KS, Rascher W, Unger T, Culman J. 2002. Peripherally applied candesartan inhibits central responses to angiotensin II in conscious rats. Naunyn Schmiedebergs Arch Pharmacol. 365:477–483.
- Hardie DG, Carling D. 1997. The AMP-activated protein kinase—fuel gauge of the mammalian cell?. Eur J Biochem. 246:259–273.
- Hardie DG, Hawley SA, Scott JW. 2006. AMP-activated protein kinase—development of the energy sensor concept. J.Physiol (Lond). 574:7–15.
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. 2003. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2:28.1–28.16.
- Jones ES, Black MJ, Widdop RE. 2004. Angiotensin AT2 receptor contributes to cardiovascular remodelling of aged rats during chronic AT1 receptor blockade. J Mol Cell Cardiol. 37:1023–1030.
- Kahn BB, Alquier T, Carling D, Hardie DG. 2005. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 1:15–25.
- Kola B. 2008. Role of AMP-activated protein kinase in the control of appetite. J Neuroendocrinol. 20:942–951.
- Kola B, Hubina E, Tucci SA, Kirkham TC, Garcia EA, Mitchell SE, Williams LM, Hawley SA, Hardie DG, Grossman AB, Korbonits M. 2005. Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. J Biol Chem. 280:25196–25201.
- Kola B, Grossman AB, Korbonits M. 2008. The role of AMP-activated protein kinase in obesity. Front Horm Res. 36:198–211.
- Leong DS, Terron JA, Falcon-Neri A, Armando I, Ito T, Johren O, Tonelli LH, Hoe KL, Saavedra JM. 2002. Restraint stress modulates brain, pituitary and adrenal expression of angiotensin II AT(1A), AT(1B) and AT(2) receptors. Neuroendocrinology. 75:227–240.
- Liu H, Kitazato KT, Uno M, Yagi K, Kanematsu Y, Tamura T, Tada Y, Kinouchi T, Nagahiro S. 2008. Protective mechanisms of the angiotensin II type 1 receptor blocker candesartan against cerebral ischemia: In-vivo and in-vitro studies. J Hypertens. 26:1435–1445.
- Li M, Zhao J, Hu Y, Lu H, Guo J. 2009. Oxygen free radicals regulate energy metabolism via AMPK pathway following cerebral ischemia. Neurol Res. 32:779–784.
- Machado LJ, Marubayashi U, Reis AM, Coimbra CC. 1998. The hyperglycemia induced by angiotensin II in rats is mediated by AT1 receptors. Braz J Med Biol Res. 31:1349–1352.
- McBride A, Hardie DG. 2009. AMP-activated protein kinase—a sensor of glycogen as well as AMP and ATP?. Acta Physiol (Oxf). 196:99–113.
- Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, Stuck BJ, Kahn BB. 2004. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 428:569–574.
- Muller-Fielitz H, Markert A, Wittmershaus C, Pahlke F, Johren O, Raasch W. 2011. Weight loss and hypophagia after high-dose AT1-blockade is only observed after high dosing and depends on regular leptin signalling but not blood pressure. Naunyn Schmiedebergs Arch Pharmacol. 383:373–384.
- Nishimura Y, Ito T, Hoe K, Saavedra JM. Chronic peripheral administration of the angiotensin II AT(1) receptor antagonist candesartan blocks brain AT(1) receptors. Brain Res. 2000a; 871:29–38.
- Nishimura Y, Ito T, Saavedra JM. Angiotensin II AT(1) blockade normalizes cerebrovascular autoregulation and reduces cerebral ischemia in spontaneously hypertensive rats. Stroke. 2000b; 31:2478–2486.
- Pacak K. 2000. Stressor-specific activation of the hypothalamic–pituitary–adrenocortical axis. Physiol Res. 49 Suppl 1: S11–S17.
- Palkovits M, Baffi JS, Pacak K. 1997. Stress-induced fos-like immunoreactivity in the pons and the medulla oblongata of rats. Stress. 1:155–168.
- Pavel J, Benicky J, Murakami Y, Sanchez-Lemus E, Saavedra JM. 2008. Peripherally administered angiotensin II AT1 receptor antagonists are anti-stress compounds in vivo. Ann NY Acad Sci. 1148:360–366.
- Pavlatou MG, Mastorakos G, Lekakis I, Liatis S, Vamvakou G, Zoumakis E, Papassotiriou I, Rabavilas AD, Katsilambros N, Chrousos GP. 2008. Chronic administration of an angiotensin II receptor antagonist resets the hypothalamic-pituitary-adrenal (HPA) axis and improves the affect of patients with diabetes mellitus type 2: Preliminary results. Stress. 11:62–72.
- Pelisch N, Hosomi N, Ueno M, Masugata H, Murao K, Hitomi H, Nakano D, Kobori H, Nishiyama A, Kohno M. 2010. Systemic candesartan reduces brain angiotensin II via downregulation of brain renin–angiotensin system. Hypertens Res. 33:161–164.
- Ramamurthy S, Ronnett GV. 2006. Developing a head for energy sensing: AMP-activated protein kinase as a multifunctional metabolic sensor in the brain. J Physiol (Lond). 574:85–93.
- Reis FM, Ribeiro-de-Oliveira AJr, Guerra RM, Reis AM, Coimbra CC. 1996. Blood glucose and prolactin in hyperprolactinemic rats exposed to restraint and surgical stress. Life Sci. 58:155–161.
- Ribeiro-de-Oliveira AJr, Guerra RM, Foscolo RB, Marubayashi U, Reis AM, Coimbra CC. 1999. Effects of chronic bromocriptine (CB-154) treatment on the plasma glucose and insulin secretion response to neurocytoglucopenia in rats. J Endocrinol. 162:237–242.
- Ribeiro-de-Oliveira A, Guerra RM, Foscolo RB, Marubayashi U, Reis AM, Coimbra CC. 2001. Bromocriptine-induced dissociation of hyperglycemia and prolactin response to restraint. Pharmacol Biochem Behav. 68:229–233.
- Ribeiro-Oliveira AJr, Nogueira AI, Pereira RM, Boas WW, dos Santos RA, Simoes E, Silva AC. 2008. The renin–angiotensin system and diabetes: An update. Vasc Health Risk Manag. 4:787–803.
- Ronnett GV, Ramamurthy S, Kleman AM, Landree LE, Aja S. 2009. AMPK in the brain: Its roles in energy balance and neuroprotection. J Neurochem. 109 Suppl 1: 17–23.
- Saavedra JM, Benicky J. 2007. Brain and peripheral angiotensin II play a major role in stress. Stress. 10:185–193.
- Unger T. 2003. Inhibiting angiotensin receptors in the brain: Possible therapeutic implications. Curr Med Res Opin. 19:449–451.
- Yamada F, Inoue S, Saitoh T, Tanaka K, Satoh S, Takamura Y. 1993. Glucoregulatory hormones in the immobilization stress-induced increase of plasma glucose in fasted and fed rats. Endocrinology. 132:2199–2205.
- Yamauchi T, Kadowaki T. 2008. Physiological and pathophysiological roles of adiponectin and adiponectin receptors in the integrated regulation of metabolic and cardiovascular diseases. Int J Obes (Lond). 32 Suppl 7: S13–S18.
- Yoshida D, Higashiura K, Shinshi Y, Satoh K, Hyakkoku M, Yoshida H, Miyazaki Y, Ura N, Shimamoto K. 2009. Effects of angiotensin II receptor blockade on glucose metabolism via AMP-activated protein kinase in insulin-resistant hypertensive rats. J Am Soc Hypertens. 3:3–8.
- Zhang J, Xie Z, Dong Y, Wang S. 2008. Liu C and Zou MH Identification of nitric oxide as an endogenous activator of the AMP-activated protein kinase in vascular endothelial cells. J Biol Chem. 283:27452–27461.
- Zorad S, Dou JT, Benicky J, Hutanu D, Tybitanclova K, Zhou J, Saavedra JM. 2006. Long-term angiotensin II AT1 receptor inhibition produces adipose tissue hypotrophy accompanied by increased expression of adiponectin and PPARgamma. Eur J Pharmacol. 552:112–122.