Abstract
We evaluated the host metabolic response to chronic varied stress during infection with the fungus Candida albicans. We used four groups of female Wistar rats: normal uninfected and unstressed, stressed, C. albicans infected and infected, and stressed. Infected rats reacted with rapid metabolic adjustments, evident as anorexia and body weight loss, partly mediated by glucocorticoids and TNF-α. Higher circulating levels of IL-6 and glucose (p < 0.05) revealed the progress and catabolic effect of the inflammatory response. Infected and stressed rats instead showed anorexia associated with infection and weight loss as the result of reduced food intake. This group exhibited a prompt reduction in circulating leptin on day 3 (p < 0.05), reduction in glucose levels and depletion of hepatic glycogen depots. We also evaluated the contribution of TNF-α, glucocorticoids, and food deprivation to liver damage. Lipid peroxidation in liver detected in the infected and infected-stressed groups was exacerbated by the glucocorticoid receptor antagonist RU 486, suggesting the modulatory activity of glucocorticoids, while hepatic fat accumulation and glycogen depletion decreased with anti-TNF-α treatment. Food deprivation exacerbated liver injury while the response to stress contributed to greater fungal colonization. Our findings emphasize the impact of metabolic alterations on tissue damage when the host immune activity is modulated by stress mediators.
Introduction
The capacity to store energy and the ability to elicit effective immune responses against pathogens are critical for the survival of multicellular organisms. Immune function requires adequate energy supply and the need to organize and distribute resources underlies the overlap between immune and metabolic pathways (Hotamisligil Citation2006; Hotamisligil and Erbay Citation2008). Hence, defects or surplus in energy reserves are associated with higher risk of infection, death, and poor wound healing. Infection produces negative energy balance with reduced food intake, weight loss, increased thermogenesis, and fever (Sarraf et al. Citation1997). Pro-inflammatory cytokines, such as IL-1, TNF-α, IL-6, or the adipose tissue-derived hormone leptin, released during infection, act in the brain inducing anorexia as well as in the periphery stimulating multiple metabolic changes (Asselah et al. Citation2006; Wallington et al. Citation2008). This complex network of mediators triggers an important negative feedback to cytokine production and toxicity (Gaillard et al. Citation2000). Moreover, insulin and glucose, which importantly contribute to the body's energy balance, can activate inflammatory pathways (Koteish and Diehl Citation2001; CitationLennie et al. 2001). Production of mediators such as the regulatory cytokine IL-10 also varies during metabolic disorders (Esposito et al. Citation2003).
The yeast Candida albicans is a member of the oral and gastrointestinal flora in healthy persons. While immunocompetent individuals control efficiently this fungus, dissemination of this opportunistic pathogen can occur in immunocompromised hosts. Previously, we described in rats the effect of daily stress exposure on the innate immune response during C. albicans infection (CitationRodríguez-Galán et al. 2001). Hyperactivity of the hypothalamus–pituitary–adrenal axis in our model was indicated by increments in circulating ACTH (CitationRodríguez-Galán et al. 2001) and corticosterone levels (Rodríguez-Galán et al. Citation2003). Other parameters relevant for the inflammatory response such as immune cell recruitment (CitationRodríguez-Galán et al. 2002), TNF-α production (Correa et al. Citation2004), or IL-1/IL-1ra balance (Rodríguez-Galán et al. Citation2003) were affected by stress hormones. After 3 days of stress treatment, macrophages exhibited functional deficiencies (CitationRodríguez-Galán et al. 2002) and reduced ability to kill the fungus (Rodríguez-Galán et al. Citation2003). At early stages of infection, the liver is important for controlling fungal spreading. In agreement, infected and stressed rats showed an intense fungal burden and severe liver damage with hepatocyte apoptosis (Renna et al. Citation2006), micro- and macro-vesicular steatosis (CitationRodríguez-Galán et al. 2001), marked lipid peroxidation, and higher levels of functional enzymes (Correa et al. Citation2004). Considering that liver metabolic cells (hepatocytes) are adjacent to immune cells (Kupffer cells), which allow continuous and dynamic interaction between metabolic and immune responses (Hotamisligil Citation2006; Hotamisligil and Erbay Citation2008; Wallington et al. Citation2008), we hypothesized that stress exposure could affect the metabolic adjustment and contribute to the worse outcome of the fungal infection in immunocompromised hosts. To evaluate this possibility, we compared the immune–metabolic response early during the fungal infection in normal and stressed rats, and we studied the role of TNF-α and glucocorticoids in that response.
Methods and materials
Animals
Outbred female Wistar rats (body weight, 150–200 g) were maintained at 22°C under 12 h light–dark cycles (lights on 07:00 h–19:00 h) with continuous access to food and water except when food was removed as part of a stress procedure. Rats were assigned among four groups: normal uninfected unstressed control; stressed; C. albicans-infected; infected; and stressed. Numbers of rats per group are given in and the Figure legends.
Table I. Effect of C. albicans infection and stress exposure on the weight of liver, spleen, and thymus.
Microorganism and infection
Pathogenic C. albicans strain N° 387 was from the stock culture collection of the Mycology Division, Department of Clinical Biochemistry, Faculty of Chemical Science, National University of Cordoba, Argentina. Yeast cells were grown on Sabouraud glucose agar slopes at 28°C, maintained by weekly subculture, and periodically checked for assimilation pattern and virulence (CitationRodríguez-Galán et al. 2001; Correa et al. Citation2004). We inoculated normal rats with 3 × 108 viable yeast cells by i.p. injection, and after 3 days liver homogenates were prepared as described previously (CitationRodríguez-Galán et al. 2001). Briefly, livers were removed under sterile conditions, weighed, and homogenized individually in Roswell Park Memorial Institute (RPMI) medium 1640 (Sigma, St Louis, Mo, USA), and plated onto Sabouraud agar to isolate the fungus. For each infection, yeast cells were harvested after 48 h of culture, centrifuged at 1000g, washed twice in sterile PBS-0.1% gentamicin, counted, and diluted to the desired concentration.
Stress procedure
Rats were exposed to different stressors between 14:00 h and 16:00 h except for food deprivation, which lasted 24 h. In our model, the stress paradigm involves daily different stressors during 3 days (Correa et al. Citation1998, Citation2004; CitationRodríguez-Galán et al. 2001, Citation2003) and includes: day 1, swim at 4°C for 5 min; day 2, restraint (in a tube 215 × 75 mm internal diameter made of 6 mm rigid opaque plastic, sealed at one end and containing a removable cap with a central 20 mm hole through which the animal may obtain fresh air) for 2 h; and day 3, food deprivation for 24 h. The Animal Experimentation Ethics Committee, Faculty of Chemical Science, National University of Cordoba, Argentina approved the protocols.
Experimental design
For experiments depicted in : infected stressed rats were injected i.p. with 3 × 108 yeasts on day 1 and stressed immediately and during the next 2 days (CitationRodríguez-Galán et al. 2001, Citation2003; Correa et al. Citation2004). Infected or stressed groups were either infected or stressed as above. Rats were weighed and food intake recorded each day. On day 4, rats were killed by cervical dislocation and spleen, thymus, and liver were removed, placed in individual petri dishes, weighed and processed for histological examination, and biochemical assays. For colony forming unit (CFU) determination, a 0.5-ml aliquot of liver homogenate was surface plated in duplicate on Sabouraud glucose agar. CFU were counted after 48 h of incubation at room temperature (CitationRodríguez-Galán et al. 2001). When required, blood was obtained by cardiac puncture on days 2–4 from representative rats of each group using light ether anesthesia. For experiments depicted in , rats were treated 6 h before infection with a single i.p. injection of 20 mg/kg in 400 μl of IgG2a anti-rat TNF-α monoclonal antibody (CENTOCOR Discovery Research, Horsham, PA, USA) as described (Scallon et al. Citation2004). For experiments shown in , the glucocorticoid antagonist RU 486 (20 mg/kg, in 100 μl; mifepristone, Sigma) was administered s.c. daily at the tail base 30 min before exposure to the corresponding stressor as described (Moore and Fewell Citation2006). For leptin replacement, rats received i.p. a single 200 μl injection of either rat recombinant leptin (1 μg/g initial body weight, Abcam, Cambridge, UK) or 0.9% saline (CitationLennie et al. 2001). For experiments described in , rats were infected as above and food deprived only on day 3 (food-deprived group) or infected and stressed like the infected+stressed group but crowded (4 rats/19.6 cm × 30.9 cm × 13.3 cm cage) for 24 h instead of food deprivation on day 3 (infected-stressed+crowded). Experiments were performed at least twice.
Figure 1 Changes in food intake and body weight during C. albicans infection in stressed rats. Wistar rats were assigned among four groups: normal uninfected and unstressed (N), stressed (S), C. albicans-infected (I), and infected-stressed (IS). IS rats were infected i.p. with 3 × 108 yeast cells on day 1 and stressed immediately after infection and during the next 2 days. Groups I or S were either infected or stressed as above. (A) Food intake was recorded every day. (B)–(D) Rats were weighed daily and body weight increment was expressed as body weight index (BWI): BW–BW initial/BW initial × 100. Data are mean+SD (Panel A); n = 13–15 per group. * vs. N p < 0.05; ** vs. N p < 0.01.
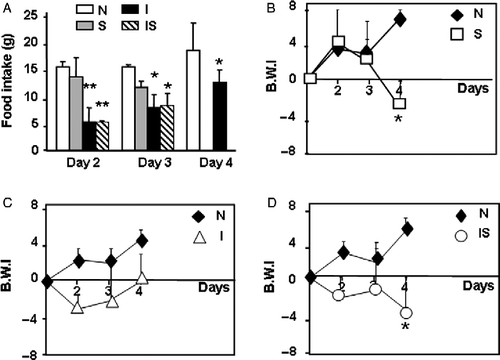
Figure 2 Serum cytokine concentrations during C. albicans infection. Wistar rats were assigned among four groups: normal uninfected and unstressed (N), stressed (S), C. albicans-infected (I), and infected-stressed (IS). IS rats were infected i.p. with 3 × 108 yeast cells on day 1 and stressed immediately after infection and during the next 2 days. Groups I or S were either infected or stressed as above. Representative rats from different groups were killed on days 2–4 and bled to assess serum IL-6 (A) and IL-10 (B) by ELISA. Data are mean+SD; number of samples per group for IL-6 measurement: 9–15; for IL-10 measurement: 5–9. On day 4, * vs. N p < 0.05; # vs. I and IS p < 0.01.
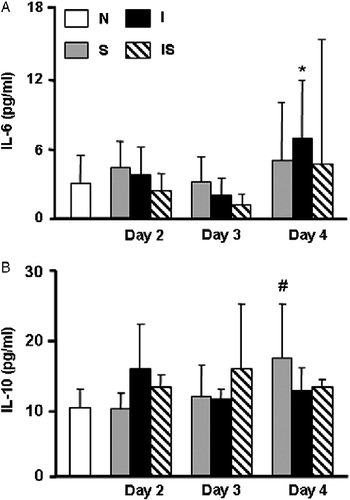
Figure 3 Serum concentrations of glucose and leptin during C. albicans infection. Wistar rats were assigned among four groups: normal uninfected and unstressed (N), stressed (S), C. albicans-infected (I), and infected-stressed (IS). IS rats were infected i.p. with 3 × 108 yeast cells on day 1 and stressed immediately after infection and during the next 2 days. Groups I or S were either infected or stressed as above. (A) On day 4, rats were bled to determine glucose levels; number of rats per group: 10–14. (B) Leptin was measured on samples from representative rats bled on days 2–4 of the experimental schedule. Number of rats per group: 7–9. Data are mean+SD. * vs. N p < 0.05; ** vs. N p < 0.01; # vs. I p < 0.05.
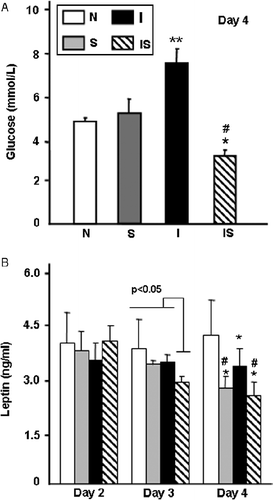
Figure 4 Involvement of TNF-α in metabolic balance. Rats were infected (I), infected-stressed (IS), treated with a single i.p. injection of 20 mg/kg IgG2a anti-rat TNF-α murine monoclonal antibody either 6 h before infection (IT) or 30 min before the stress procedure in the IS group (IST) as described in Methods and materials. (A) Rats were weighed daily and body weight increment was expressed as body weight index (BWI): BW-BW initial/BW initial × 100. (B) On day 4, four rats were bled to determine serum glucose concentrations. Data are mean+SD (panel B). Number of rats per group: 4–5. * vs. I p < 0.005; # vs. IS p < 0.02.
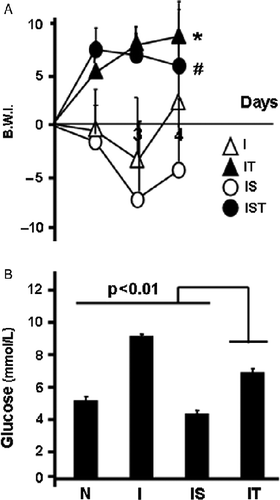
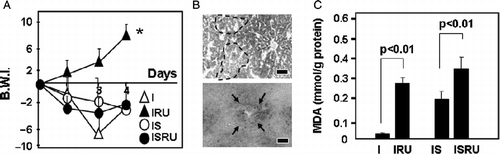
Figure 5 Involvement of glucocorticoids in metabolic balance and liver injury. Rats were infected (I), and infected-stressed (IS), injected s.c. with RU 486 daily in the I group (IRU) or 30 min before the stress procedure in the IS group (ISRU) as described in Methods and materials. (A) Rats were weighed daily and body weight increment was expressed as body weight index (BWI): BW-BW initial/BW initial × 100. Data are mean; number of rats per group: 6–7. (B) On day 4, livers were processed for histopathological studies. Microphotographs from representative sections from the ISRU group are shown. Upper: Alcian Blue–PAS (AB-PAS). Dotted lines mark boundaries of glycogen depletion areas. Lower: Sudan Black staining of the same rat in upper panel. Arrows point to lipid depots. Scale bars: 10 μm. (C) Lipid peroxidation, measured as MDA levels, was determined on day 4 in liver homogenates. Data are mean+SD. * vs. I p < 0.005.
IL-10 and IL-6 assays
Serum IL-10 and IL-6 concentrations were determined by solid-phase sandwich ELISA (Diagnostic BD Pharmingen, San Diego, CA, USA) according to the manufacturer's recommended protocols. The amount of each mediator was extrapolated from the standard curve, which was generated in 1:2 dilutions. The detection limit was 7.5 pg/ml for IL-10 and 1 pg/ml for IL-6. Results are expressed in pg/ml.
Leptin assay
Serum leptin concentrations were measured with an active murine leptin ELISA kit (DSL-10-24100, Diagnostic Systems Laboratories, Webster, Texas, USA), which detects both rat and mouse leptin. The minimum detection limit of the leptin assay was 0.04 ng ml. The intra-assay coefficient of variation for each assay was < 7%. Results are expressed in ng/ml.
Glucose assay
Serum glucose concentration was measured by an enzymatic method (CitationLott and Turner 1975) using commercial kits (Wiener Lab. Rosario, Argentina) according to the manufacturer's protocols. The minimum detection limit of the glucose assay was 0.002 mmol/l. The intra-assay coefficient of variation for each assay was < 2%. Results are expressed in mmol/l.
Histological studies
Livers were fixed with 10% formalin in PBS for at least 24 h, dehydrated in alcohol, cleared in xylol, and embedded in paraffin. Six micrometer sections were cut and stained with hematoxylin–eosin and Alcian Blue/Periodic acid-Schiff (PAS) stain. For specific lipid detection, Sudan Black staining was performed and livers were prepared as described previously (CitationRodríguez-Galán et al. 2001; Correa et al. Citation2004; Renna et al. Citation2006). Briefly, the fresh tissue was frozen using dry ice, cut with a microtome, and stained with Sudan Black. This technique was used to evaluate the distribution, extent, and morphology of lipid droplets and to classify the pathological injury using an arbitrary scale as follows: 0 (no damage), +(light), ++(moderate), and +++(intense). Pathological parameters were measured in at least two separate experiments with 4–6 rats per group.
Assay for hepatic lipid peroxidation
Liver homogenates, prepared as above (1 ml), were mixed with 2 ml of working solution containing 15% (w/v) trichloroacetic acid-0.375% (w/v) thiobarbituric acid-0.25 N HCl and heated for 15 min in boiling water. After cooling, the precipitate was removed by centrifugation at 1000g for 10 min. Absorbance was determined at 535 nm. Levels of malonaldehyde (MDA) were measured using 1,1,3,3-tetrametoxypropane as a standard. Results are expressed as mmol of MDA/g of protein (Wellen and Hotamisligil Citation2005).
Assay of hepatic enzymes
Serum alanine transferase (GPT) and aspartate amine transferase (GOT) activities were measured with commercial kits (Wiener Lab) according to the manufacturer's recommended protocols. The detection limit of each hepatic enzyme assay was 1.8 U/l. The intra-assay coefficient of variation for each assay was < 5%. Results are expressed in U/l.
Statistical analysis
Differences between group means were assessed using two-way ANOVA followed by Student–Newman–Keuls tests for multiple comparisons. Normality was evaluated by the Bartlett test. A p value < 0.05 was considered statistically significant. Data are shown as stated in Results and as mean ± SD, and in the Figures as mean+SD.
Results
Metabolic imbalance in infected, stressed, and infected-stressed rats
First, we evaluated the influence of stress, infection, or both on food intake and body weight in our experimental conditions. Food intake was not affected by stress alone (); no data were recorded for the stressed group on day 4 because rats were food deprived as part of the stress procedure. As expected, both groups of infected rats (infected and infected-stressed) exhibited significantly reduced food intake in the first 48 h of the infection (F(3,57) = 18.03; p < 0.001). The fall was still significant in the infected group on day 4 (p < 0.05); this parameter was not determined in the food-deprived infected-stressed group as explained above. Plots of body weight increments () showed three different profiles when compared with normal rats: weight loss on day 4 in the stressed food-deprived group (); in infected rats weight loss on days 2–3 that was reversed by day 4 (), and in the stressed-infected rats, a mixed pattern that includes the infection effect on days 2–3 and the food deprivation influence on day 4 ().
Second, we also measured serum concentrations of the inflammatory cytokine IL-6 and the regulatory cytokine IL-10 (). Differences were significant only on day 4, with increase in IL-6 concentration in the infected group (F(3,61) = 5.72; p = 0.002) and higher levels of IL-10 in rats only stressed (F(3,34) = 4.31; p < 0.01). In both groups with ongoing infection (infected and infected-stressed), increased liver (F(3,60) = 5.69; p = 0.013) and spleen (F(3,60) = 12.73; p < 0.01) weight was observed, whereas the thymus weight remained unchanged compared with controls ().
In spite of the changes found in body weight, we observed no differences in serum insulin concentrations between infected, stressed, and infected-stressed groups (data not shown). Serum glucose concentrations were modified with different treatments (). On day 4, normal and stressed groups showed similar glucose levels, while a significant increase was observed in the infected group (F(3,47) = 5.25; p < 0.001). In sharp contrast, infected-stressed rats exhibited reduction in serum glucose concentration.
Contribution of leptin to immune–metabolic imbalance in infected, stressed, and infected-stressed rats
shows that serum leptin concentration was reduced on day 4 in the infected, stressed, or infected-stressed groups compared with normal rats (F(11,97) = 7.3; p < 0.001); however, groups that were food deprived the day before (stressed and infected-stressed) exhibited the most pronounced reduction. Interestingly, leptin level was diminished already on day 3 in infected and stressed rats, and this effect was not related to the neuroendocrine effects of stress exposure, as leptin levels of the stressed group were comparable to normal rats at that time. We hypothesized that supply of exogenous leptin could attenuate the impairment of the infected-stressed group. However, administration of single dose of rat recombinant leptin on day 3 when deficiency started was unable to reverse changes described on day 4. Body weight increment was similar and no significant differences were found in glucose concentration in infected-stressed (3.11 ± 0.44 mmol/l) and infected-stressed+leptin (2.83 ± 0.88 mmol/l) groups (F(4,19) = 19.11; p < 0.001).
Contribution of TNF-α and glucocorticoids to metabolic imbalance
To assess the contribution of TNF-α and glucocorticoids, we used two strategies. First, we blocked any TNF-α released during the 3-day treatment with a monoclonal antibody. For comparison purposes, we included infected, infected and TNF-α blocked, infected-stressed (previously the addition of stress was found to reduce TNF-α release by 30%; Correa et al. Citation2004), and infected-stressed+TNF-α blocked rats. Anti TNF-α treatment efficiently overturned the weight loss observed in the infected groups (). Blocking TNF-α reduced glucose levels in infected rats, which reached levels intermediate between the infected and infected-stressed groups (F(3,9) = 12.29; p = 0.0003). Second, daily administration of the glucocorticoid receptor antagonist RU 486 to infected groups restored the weight loss experienced by infected rats (F(3,23) = 4.81; p < 0.009); however, no effect was observed in infected-stressed RU 486 treated rats (). Upon RU 486 treatment, no significant differences were observed in glucose values between infected (6.66 ± 1.22 mmol/l) and infected+RU 486 treated (5.33 ± 0.66 mmol/l) or between infected-stressed (3.11 ± 0.44 mmol/l) and infected-stressed+RU 486 (2.9 ± 1.05 mmol/l) groups.
Liver injury
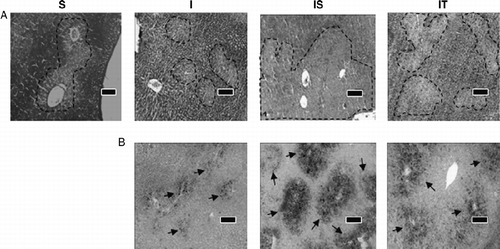
Representative liver sections from the infected unstressed group showed abundant glycogen staining with areas of mild depletion (). Hepatic glycogen stores were depleted in stressed rats whereas infected-stressed hosts exhibited almost complete exhaustion of glycogen stores. Rats treated with anti TNF-α exhibited an intermediate pattern, with more abundant glycogen stores than the infected-stressed group but less than stressed or infected rats. Moreover, the infected-stressed group exhibited pronounced perivascular micro- and macro-vesicular steatosis, in agreement with our previous findings (CitationRodríguez-Galán et al. 2001; Correa et al. Citation2004), in sharp contrast with the reduced steatosis of the infected group (). Remarkably, following the anti TNF-α treatment, the liver presented intermediate but more diffuse steatosis, suggesting the involvement of this pro-inflammatory cytokine in the abnormal lipid deposition observed in infected-stressed rats.
Figure 6 Histopathological changes in liver associated with metabolic adjustment. On day 4, livers from stressed (S), infected (I), infected-stressed (IS), and infected-anti TNF-α treated (IT) groups were processed for histopathological studies. Microphotographs from one representative liver section per group are shown. (A) AB-PAS. Dotted lines mark boundaries of glycogen depletion areas. (B) Sudan Black staining of the same rat liver in the upper panel. Arrows point to lipid depots. Scale bars: 10 μm.
The glucocorticoid antagonist produced moderate glycogen depletion and intermediate steatosis in infected-stressed rats (representative sections are shown in ; upper: PAS staining; lower: Sudan Black staining). Yet, RU-486 worsened lipid peroxidation (), with an approximately 10-fold rise in the infected group and a minor increase in the infected-stressed group (F(3,13) = 10.51; p = 0.002).
Contribution of food deprivation to alterations in infected and stressed rats
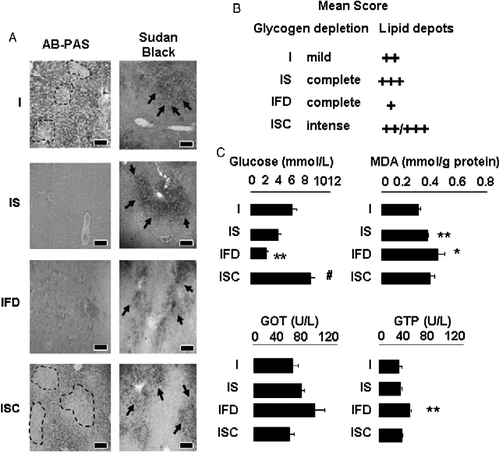
To test whether food deprivation could precipitate metabolic alterations observed in the infected-stressed group, we evaluated two additional groups: rats infected on day 1 and food deprived on day 3 (infected food deprived) and infected-stressed rats with 24 h crowding instead of food deprivation on day 3 (infected-stressed+crowded group). Representative liver sections stained to visualize glycogen stores and lipid deposition are shown in ; infected and infected-stressed groups are included for comparison. Food deprivation produced complete lack of glycogen in the infected food-deprived group with mild and diffuse steatosis. Interestingly, intense glycogen depletion was found in infected rats exposed to crowding (infected-stressed+crowded) with abundant micro- and macro-lipid droplets. Average score for glycogen depletion and lipid depots/group is shown in . Overall, these data indicate that glycogen depletion is highly affected by food deprivation whereas lipid accumulation is more dependent on stress hormones. shows that while fasting provoked blood glucose reduction (infected stressed and infected food-deprived groups), the alternative stressor (crowding) increased glycemic values (F(3,19) = 23.18; p < 0.0001). Interestingly, exacerbation of liver damage, as evaluated by transaminase levels (F(3,19) = 8.97; p = 0.01) and lipid peroxidation (F(3,16) = 11.06; p = 0.004) was found in fasted infected rats. Liver damage was similar or milder in the infected-stressed+crowded group compared with infected-stressed rats.
Figure 7 Role of fasting in liver injury. Rats were infected (I), infected, and food deprived (IFD) or/and infected-stressed (IS) or infected-stressed+crowded (ISC), as described in Methods and materials. (A) On day 4, livers were processed for histopathological studies. Microphotographs from representative liver sections per group stained with AB-PAS or Sudan Black are shown. Dotted lines mark boundaries of glycogen depletion areas; arrows point to lipid depots. Scale bars: 10 μm. (B) Average histological score for each group is shown. (C) On day 4, rats were bled to determine serum transaminase (GOT and GTP) and glucose levels; lipid peroxidation measured as MDA levels in liver homogenates was determined as in Figure 4. Data are mean+SD. number of rats per group: 4–5. * vs. I p < 0.05; ** vs. I p < 0.01; # vs. IS p < 0.02.
Influence of metabolic alterations on the ability to clear the fungus
Finally, we compared the liver fungal burden after 3 days of infection in the different immune–metabolic conditions studied here (F(6,65) = 8.41; p < 0.01; ). Even though food deprivation affected the metabolic balance and liver damage in the infected food-deprived group, the host immune competence remained similar to the infected group. In contrast, exposure to an alternative stress paradigm on day 3 (infected-stressed+crowded group) also conditioned the ability to clear efficiently the pathogen. Reduction in biological active TNF-α (infected-stressed and the infected anti TNF-α treated groups) as well as glucocorticoid antagonism (infected RU 486-treated group) promoted significantly higher CFU values. Together, metabolic alterations did not condition immune activity during pathogen challenge but contributed to worse tissue injury. Moreover, hyperactivity of the glucocorticoid response is pivotal in the immune failure of chronically stressed rats.
Figure 8 Fungal burden in immune competent and immune compromised hosts. Mean fungal loads (mean log CFU/g liver+SD) in liver homogenates (* vs. I p < 0.05) are shown. For comparative purposes, mean log CFU+SD of immune competent hosts (I) is depicted as gray-dotted area. Number of rats per group: 8–10. Abbreviations: I, C. albicans-infected; IRU, infected and RU 486 treated; IT, infected and anti TNF-α treated; IFD, infected and food deprived; IS, infected-stressed; ISRU, infected-stressed and RU 486 treated; ISC, infected-stressed+crowded.

Discussion
This study compared the immune–metabolic response of immune competent and immunocompromised hosts during fungal infection and describes the contribution of TNF-α, glucocorticoid, and food deprivation to liver damage. Immune defenses are energetically expensive to develop and operate (Koteish and Diehl Citation2001), and synchronized inflammatory and metabolic pathways guarantee redistribution of resources under different conditions. After infection, immune competent hosts (infected group) reacted with metabolic adjustments that included anorexia and body weight loss with recovery by day 4, as in other inflammatory conditions (Grunfeld et al. Citation1996). Conversely, infected-stressed hosts exhibited anorexia and sustained weight loss either as direct results of fasting or as a consequence of stress. A vast array of mediators contributes to weight loss (Morley et al. Citation2006): cytokine excess leads to decreased protein synthesis and proteolysis (Mitch and Goldberg Citation1996), and activates an ubiquitin-mediated proteolytic system involved in hypercatabolism (Acharyya et al. Citation2004). Stress mediators also induce enhancement of metabolic activity (Rybkin et al. Citation1997; Tamashiro et al. Citation2007) and reduction in food intake (Kramer et al. Citation1999). Rats lose weight after restraint stress ends, suggesting a persistent effect on food intake beyond the actual stress period (Harris et al. Citation1998). Hence, in the present study, restraint on day 2 and food deprivation on day 3 could have contributed to the profile detected in the infected-stressed group. Complete blockage of TNF-α, the most relevant cytokine in metabolic balance at the very beginning of infection (Morley et al. Citation2006), abolished weight loss in the infected and infected-stressed groups, in agreement with other studies employing neutralizing antibodies (Truyens et al.Citation1995), soluble TNF receptor (Granado et al. Citation2006), or TNF − / − mice (Volman et al. Citation2002). Although a differential effect of RU 486 on the weight loss observed in the infected and infected-stressed groups remains uncertain, several possibilities may explain our findings: first, high doses of RU 486 slow weight gain during treatment and may fail to block wasting (Pickering et al. Citation2003); second, an overproduction of TNF-α with RU 486 administration may produce increased sensitivity to TNF-α-related fasting (CitationLázár et al. 1992); and finally, epinephrine and norepinephrine could contribute by inhibiting anabolic hormones (Yamada et al. Citation1993; Nandi et al. Citation2002).
After food deprivation, hepatic glycogen stores are depleted and glucose metabolism shifts toward hepatic gluconeogenesis as the primary endogenous glucose source (Yamada et al. Citation1993). In agreement, the stressed uninfected group suffered depletion of hepatic glycogen depots and succeeded in maintaining glucose supply in the blood. However, infected-stressed and infected food-deprived rats showed depletion of glycogen stores and failure of hepatic gluconeogenesis to maintain glucose concentrations, as in septic (Ceppi et al. Citation1992) or endotoxic shock syndrome (Knowles et al. Citation1987). Changes observed in vivo are not exclusively due to regulatory hormones as inflammatory cytokines may have direct effects on hepatocytes, so reducing liver capacity to synthesize glycogen (Flores et al. Citation1990; Wallington et al. Citation2008). Indeed, acute treatment of animals with TNF-α produces severe hypoglycemia with depletion of liver glycogen (Chajek-Shaul et al. Citation1990). Accordingly, anti TNF-α treatment increased serum glucose levels and attenuated hepatic glycogen depletion in the infected group.
So long as infection progresses in immune competent hosts, increased release of IL-6, splenomegalia, hepatomegalia, and reduced leptinemia occurs, as described in patients with chronic inflammatory processes (Popa et al. Citation2005). Although soluble and transient inflammatory stimuli such as cytokines (Sarraf et al. Citation1997) or lipopolysaccharide produce acute increment in leptin levels (Grunfeld et al. Citation1996; Faggioni et al. Citation2000), injection of viable yeast cells leads to robust and sustained release of several mediators by the innate immune system. Here, the infected group showed reduction in both food intake and body weight 48 h after infection, and reduced instead of increased leptin levels might be expected. The early lack of leptin observed in the infected-stressed group is neither related to fasting (Ahima et al. Citation1996), as it was absent in rats that were only stressed, nor associated to inflammation, as the infected-stressed group showed reduced levels of TNF-α (CitationRodríguez-Galán et al. 2002; Correa et al. Citation2004) and IL-6. Perhaps leptin deficiency in the infected-stressed group represents a consistent marker of immune–metabolic impairment during the ongoing inflammatory response.
Hepatic lipid accumulation occurs when fatty acid input exceeds the amount that can be released or oxidized as an energy source (Asselah et al. Citation2006). While steatosis without concomitant inflammation is considered a benign condition, fatty degeneration increases liver sensitivity to hormones and cytokines (Koteish and Diehl Citation2001). In agreement, infected rats with simultaneous hyperactivity of the stress response (infected-stressed and infected-stressed+crowded) showed the most profuse pattern of steatosis. A double hit model has been proposed wherein steatosis makes the liver vulnerable to oxidative stress, lipid peroxidation, or cytokines such as TNF-α, TGF-β, IL-1β, IL-6, and IL-8 (Day and James Citation1998), leading to hepatic inflammation and fibrosis (Koteish and Diehl Citation2001). Supporting this concept of synergistic effects, both RU 486 and anti-TNF-α treatments decreased fat accumulation.
During fungal infection, liver injury occurs with the release of hepatic enzymes and lipid peroxidation that is exacerbated by stress input (CitationRodríguez-Galán et al. 2001; Correa et al. Citation2004). Whether an increase in glucocorticoid levels has a role in such lipid metabolism is unclear (Khovidhunkit et al. Citation2004). Lipid oxidation markers such as malondialdehyde–lysine and carboxymethyllysine are decreased by corticosterone treatment (Vendemiale et al. Citation2001). In agreement, the RU 486 blocking experiment in the present study induced the greatest peroxidation, with striking increase in malondialdehyde in the infected RU 486 treated group, illustrating the protective activity of endogenous glucocorticoids on liver damage. Differences between infected-stressed and infected-stressed+RU 486 treated rats could be minor because infected-stressed rats have already a significant increment in lipid peroxidation (Correa et al. Citation2004). Another possibility is the involvement of the peroxisomal pathway of fatty acid catabolism. Under normal physiological conditions, peroxisomal oxidation represents a minor pathway relative to the mitochondrial system. However, the peroxisomal pathway is engaged in rodents during periods of lipid overload due to metabolic disturbances (Kliewer et al. Citation2001; Vendemiale et al. Citation2001). The metabolic adjustment in infected-stressed and infected-food deprived rats could determine higher influx of lipids into the liver, with activation of this alternative pathway. Nevertheless, in food-deprived immunocompetent hosts with limited fat accumulation, exacerbated liver injury resulted, with elevated malondialdehyde production. Indeed, starvation weakens cellular detoxification systems in normal liver and exposes the hepatocyte to oxidative injury (Domenicali et al. Citation2001). In food-deprived rats, greater transaminase levels and oxidative damage in mitochondria are observed during ischemia–reperfusion injury (Adams et al. Citation2009). Moreover, feeding attenuates lipopolysaccharide-induced liver injury, while in fasted rats significant increase in serum cytokines and hepatic cyclooxygenase is observed (Miech Citation2005). Together, this previous evidence supports our findings of enhanced severity of liver damage in infected hosts exposed to an acute fast.
In terms of infection outcome, concurrence of marked metabolic imbalance with hyperactivity of the stress response led to higher fungal load (infected-stressed group), as described previously (CitationRodríguez-Galán et al. 2001, Citation2003). The heavier colonization in the RU 486 treated groups was also associated with severe liver injury, while blocking TNF-α (infected and anti-TNF-α treated) conditioned the heaviest fungal burden, as previously demonstrated (Netea et al. Citation1999). During infection, immune competent hosts elicit a robust immune–metabolic response that favors control of the pathogen. Deficiency (as in the infected anti-TNF-α treated, infected RU 486 treated, and infected-stressed RU 486 treated groups) or excess (as in the infected-stressed and infected-stressed+crowded groups) of key immune–endocrine mediators compromise the inflammatory response settlement and favor heavier yeast colonization. A single fasting event did not impair the immune activity but made the liver more susceptible to injury.
The present study demonstrates that differences in the outcome of fungal infection and liver damage strongly depend on the host metabolic and immune status. Understanding this complex response may help to explain the progression of systemic infections occurring under different metabolic conditions in immunocompromised hosts.
Acknowledgements
The murine monoclonal IgG2a anti-rat TNF-α was a kind gift of CENTOCOR Discovery Research (USA). MCRG, CP, CES, and SGC belong to the staff research of CONICET. We would like to thank Miss Paula Icely and Mr Luis Navarro for technical assistance.
This work was supported by grants from Agencia Nacional de Promoción Científica y Tecnológica, The International Society for Infectious Diseases, Ministerio de Ciencia y Técnica de la Provincia de Córdoba, Consejo Nacional de Investigaciones Científicas Técnicas Secretaría de Ciencia y Tecnología—UNC.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Acharyya S, Ladner KJ, Nelsen LL, Damrauer J, Reiser PJ, Swoap S, Guttridge DC. 2004. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J Clin Invest. 114:370–378.
- Adams SD, Delano BA, Helmer KS, Mercer DW. 2009. Fasting exacerbates and feeding diminishes LPS-induced liver injury in the rat. Dig Dis Sci. 54:767–773.
- Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, Flier JS. 1996. Role of leptin in the neuroendocrine response to fasting. Nature. 382:250–252.
- Asselah T, Rubbia-Brandt L, Marcellin P, Negro F. 2006. Steatosis in chronic hepatitis C: Why does it really matter?. Gut. 55:123–130.
- Ceppi ED, Knowles RG, Carpenter KM, Titheradge MA. 1992. Effect of treatment in vivo of rats with bacterial endotoxin on fructose 2,6-bisphosphate metabolism and l-pyruvate kinase activity and flux in isolated liver cells. Biochem J. 284:761–766.
- Chajek-Shaul T, Barash V, Weidenfeld J, Friedman G, Ziv E, Shohami E, Shiloni E. 1990. Lethal hypoglycemia and hypothermia induced by administration of low doses of tumor necrosis factor to adrenalectomized rats. Metabolism. 39:242–250.
- Correa SG, Rodriguez-Galán MC, Rivero VE, Riera CM. 1998. Chronic varied stress modulates experimental autoimmune encephalomyelitis in Wistar rats. Brain Behav Immun. 12:134–148.
- Correa SG, Rodríguez-Galán MC, Salido-Rentería B, Cano R, Cejas H, Sotomayor CE. 2004. High dissemination and hepatotoxicity in rats infected with Candida albicans after stress exposure: Potential sensitization to liver damage. Int Immunol. 16:1761–1768.
- Day CP, James OF. 1998. Steatohepatitis: A tale of two “hits”?. Gastroenterology. 114:842–845.
- Domenicali M, Caraceni P, Vendemiale G, Grattagliano I, Nardo B, Dall'Agata M, Santoni B, Trevisani F, Cavallari A, Altomare E, Bernardi M. 2001. Food deprivation exacerbates mitochondrial oxidative stress in rat liver exposed to ischemia–reperfusion injury. J Nutr. 131:105–110.
- Esposito K, Pontillo A, Giugliano F, Giugliano G, Marfella R, Nicoletti G, Giugliano D. 2003. Association of low interleukin-10 levels with the metabolic syndrome in obese women. J Clin Endocrinol Metab. 88:1055–1058.
- Faggioni R, Moser A, Feingold KR, Grunfeld C. 2000. Reduced leptin levels in starvation increase susceptibility to endotoxic shock. Am J Pathol. 156:1781–1787.
- Flores EA, Istfan N, Pomposelli JJ, Blackburn GL, Bistrian BR. 1990. Effect of interleukin-1 and tumor necrosis factor/cachectin on glucose turnover in the rat. Metabolism. 39:738–743.
- Gaillard RC, Spinedi E, Chautard T, Pralong FP. 2000. Cytokines, leptin, and the hypothalamo–pituitary–adrenal axis. Ann N Y Acad Sci. 917:647–657.
- Granado M, Martín AI, Priego T, López-Calderón A, Villanúa MA. 2006. Tumour necrosis factor blockade did not prevent the increase of muscular muscle RING finger-1 and muscle atrophy F-box in arthritic rats. J Endocrinol. 191:319–326.
- Grunfeld C, Zhao C, Fuller J, Pollack A, Moser A, Friedman J, Feingold KR. 1996. Endotoxin and cytokines induce expression of leptin, the ob gene product, in hamsters. J Clin Invest. 97:2152–2157.
- Harris RB, Zhou J, Youngblood BD, Rybkin II, GN Smagin, Ryan DH. 1998. Effect of repeated stress on body weight and body composition of rats fed low- and high-fat diets. Am J Physiol. 275:R1928–R1938.
- Hotamisligil GS. 2006. Inflammation and metabolic disorders. Nature. 444:860–867.
- Hotamisligil GS, Erbay E. 2008. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol. 8:923–934.
- Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, Feingold KR, Grunfeld C. 2004. Effects of infection and inflammation on lipid and lipoprotein metabolism: Mechanisms and consequences to the host. J Lipid Res. 45:1169–1196.
- Kliewer SA, Xu HE, Lambert MH, Willson TM. 2001. Peroxisome proliferator-activated receptors: From genes to physiology. Recent Prog Horm Res. 56:239–263.
- Knowles RG, McCabe JP, Beevers SJ, Pogson CI. 1987. The characteristics and site of inhibition of gluconeogenesis in rat liver cells by bacterial endotoxin. Stimulation of phosphofructokinase-1. Biochem. J. 242:721–728.
- Koteish A, Diehl AM. 2001. Animal models of steatosis. Semin Liver Dis. 21:89–104.
- Kramer M, Hiemke C, Fuchs E. 1999. Chronic psychosocial stress and antidepressant treatment in tree shrews: Time-dependent behavioral and endocrine effects. Neurosci Biobehav Rev. 23:937–947.
- Lennie TA, Wortman MD, Seeley RJ. 2001. Activity of body energy regulatory pathways in inflammation-induced anorexia. Physiol Behav. 73:517–523.
- Lott JA, Turner K. 1975. Evaluation of Trinder's glucose oxidase method for measuring glucose in serum and urine. Clin Chem. 21:1754–1760.
- Lázár G, Duda E, Lázár G. 1992. Effect of RU 38486 on TNF production and toxicity. FEBS Lett. 308:137–140.
- Miech RP. 2005. Pathophysiology of mifepristone-induced septic shock due to Clostridium sordellii. Ann Pharmacother. 39:438–483.
- Mitch WE, Goldberg AL. 1996. Mechanisms of muscle wasting. The role of the ubiquitin–proteasome pathway. N Engl J Med. 335:1897–1905.
- Moore SL, Fewell JE. 2006. Mifepristone (RU38486) influences the core temperature response of term pregnant rats to intraperitoneal lipopolysaccharide. Exp Physiol. 91:741–746.
- Morley JE, Thomas DR, Wilson MM. 2006. Cachexia: Pathophysiology and clinical relevance. Am J Clin Nutr. 83:735–743.
- Nandi J, Meguid MM, Inui A, Xu Y, Makarenko IG, Tada T, Chen C. 2002. Central mechanisms involved with catabolism. Curr Opin Clin Nutr Metab Care. 5:407–418.
- Netea MG, van Tits LJ, Curfs JH, Amiot F, Meis JF, van der Meer JW, Kullberg BJ. 1999. Increased susceptibility of TNF-alpha lymphotoxin-alpha double knockout mice to systemic candidiasis through impaired recruitment of neutrophils and phagocytosis of Candida albicans. J Immunol. 63:1498–1505.
- Pickering WP, Baker FE, Brown J, Butler HL, Govindji S, Parsons JM, Pawluczyk IZ, Walls J, Bevington A. 2003. Glucocorticoid antagonist RU38486 fails to block acid-induced muscle wasting in vivo or in vitro. Nephrol Dial Transplant. 8:1475–1484.
- Popa C, Netea MG, Radstake TR, van Riel PL, Barrera P, van der Meer JW. 2005. Markers of inflammation are negatively correlated with serum leptin in rheumatoid arthritis. Ann Rheum Dis. 64:1195–1198.
- Renna MS, Correa SG, Porporatto C, Figueredo CM, Aoki MP, Paraje MG, Sotomayor CE. 2006. Hepatocellular apoptosis during Candida albicans colonization: Involvement of TNF-alpha and infiltrating Fas-L positive lymphocytes. Int Immunol. 18:1719–1728.
- Rodríguez-Galán MC, Correa SG, Iribarren P, Sotomayor CE. 2002. Phenotypic and functional changes on phagocytic cells recruited at the site of Candida albicans infection after chronic varied stress exposure. Med Mycol. 40:485–492.
- Rodríguez-Galán MC, Correa SG, Cejas H, Sotomayor CE. 2001. Impaired activity of phagocytic cells in Candida albicans infection after exposure to chronic varied stress. Neuroimmunomodulation. 9:193–202.
- Rodríguez-Galán MC, Sotomayor C, Costamagna ME, Cabanillas AM, Rentería BS, Masini-Repiso AM, Correa S. 2003. Immunocompetence of macrophages in rats exposed to Candida albicans infection and stress. Am J Physiol Cell Physiol. 284:C111–C1118.
- Rybkin Zhou YII, Volaufova J, Smagin GN, Ryan DH, Harris RBS. 1997. Effect of restraint stress on food intake and body weight is determined by time of day. Am J Physiol Regul Integr Comp Physiol. 273:R1612–R1622.
- Sarraf P, Frederich RC, Turner EM, Ma G, Jaskowiak NT, Rivet DJ, Flier JS, Lowell BB, Fraker DL, Alexander HR. 1997. Multiple cytokines and acute inflammation raise mouse leptin levels: Potential role in inflammatory anorexia. J Exp Med. 185:171–175.
- Scallon B, Cai A, Radewonuk J, Naso M. 2004. Addition of an extra immunoglobulin domain to two anti-rodent TNF monoclonal antibodies substantially increased their potency. Mol Immunol. 41:73–80.
- Tamashiro KL, Hegeman MA, Nguyen MM, Melhorn SJ, Ma LY, Woods SC, Sakai RR. 2007. Dynamic body weight and body composition changes in response to subordination stress. Physiol Behav. 91:440–448.
- Truyens C, Torrico F, Angelo-Barrios A, Lucas R, Heremans H, De Baetselier P, Carlier Y. 1995. The cachexia associated with Trypanosoma cruzi acute infection in mice is attenuated by anti-TNF-alpha, but not by anti-IL-6 or anti-IFN-gamma antibodies. Parasite Immunol. 17:561–568.
- Vendemiale G, Grattagliano I, Caraceni P, Caraccio G, Domenicali M, Dall'Agata M, Trevisani F, Guerrieri F, Bernardi M, Altomare E. 2001. Mitochondrial oxidative injury and energy metabolism alteration in rat fatty liver: Effect of the nutritional status. Hepatology. 33:808–815.
- Volman TJ, Hendriks T, Verhofstad AA, Kullberg BJ, Goris RJ. 2002. Improved survival of TNF-deficient mice during the zymosan-induced multiple organ dysfunction syndrome. Shock. 17:468–472.
- Wallington J, Ning J, Titheradge MA. 2008. The control of hepatic glycogen metabolism in an in vitro model of sepsis. Mol Cell Biochem. 208:183–192.
- Wellen KE, Hotamisligil GS. 2005. Inflammation, stress, and diabetes. J Clin Invest. 115:1111–1119.
- Yamada F, Inoue S, Saitoh T, Tanaka K, Satoh S, Takamura Y. 1993. Glucoregulatory hormones in the immobilization stress-induced increase of plasma glucose in fasted and fed rats. Endocrinology. 132:2199–2205.