
Abstract
Perfluorooctanoate (PFOA) and perfluorooctanesulfonate (PFOS) are ubiquitous synthetic chemicals with no known effect on human cancer development. This article systematically and critically reviews the epidemiologic evidence regarding the association between PFOA and PFOS exposure and cancer risk in humans. Eighteen epidemiologic studies – eight of PFOA, four of PFOS, and six of both PFOA and PFOS – have estimated associations of exposure to these chemicals with cancer incidence or mortality, with studies equally divided between occupational and nonoccupational settings. Although some statistically significant positive associations have been reported, for example, with cancers of the prostate, kidney, testis, and thyroid, the majority of relative risk estimates for both PFOA and PFOS have been between 0.5 and 2.0 (with 95% confidence intervals including 1.0), inconsistently detected across studies, counterbalanced by negative associations, not indicative of a monotonic exposure-response relationship, and not coherent with toxicological evidence in animals, in which the primary target organs are the liver, testis (Leydig cells), and pancreas (acinar cells). Many positive associations with PFOA exposure were detected in community settings without occupational exposure and were not supported by results in exposed workers. Given that occupational exposure to PFOA and PFOS is one to two orders of magnitude higher than environmental exposure, the discrepant positive findings are likely due to chance, confounding, and/or bias. Taken together, the epidemiologic evidence does not support the hypothesis of a causal association between PFOA or PFOS exposure and cancer in humans.
Introduction
Perfluoroalkyl and polyfluoroalkyl substances (PFASs) have been used since the mid-twentieth century in a wide variety of polymer and surfactant applications (CitationBuck et al. 2011). Ammonium perfluorooctanoate (NH4+C7F15COO−) has been used as a processing aid in fluoropolymer manufacture and dispersion processing, and it rapidly dissociates in aqueous solution to the anion perfluorooctanoate (PFOA; C7F15COO−). Perfluorooctanesulfonyl-fluoride-based compounds, which can degrade or metabolize to perfluorooctanesulfonate (PFOS; C8F17SO3−), have been used in various surfactant and surface-protection products. For the sake of simplicity, this review refers to both PFOA and ammonium perfluorooctanoate by the acronym “PFOA” and to both PFOS and perfluorooctanesulfonyl fluoride by the acronym “PFOS.” PFOA, PFOS, and other PFASs are released to the environment through the industrial manufacture and use of these chemicals, use and disposal of consumer products that contain them, and abiotic or biotic degradation of precursors, which themselves can be environmentally released from industrial materials and consumer products (CitationBuck et al. 2011). PFOA and PFOS are widely and persistently detected in wildlife (CitationGiesy and Kannan 2001, CitationHoude et al. 2006) and nonoccupationally exposed humans (CitationButenhoff et al. 2006, CitationCalafat et al. 2007, CitationKannan et al. 2004). Consequently, 3M Company, a major international producer of PFOA and PFOS, voluntarily began phasing out the manufacture of these chemicals in May 2000, eventually eliminating the manufacture and use of PFOS in 2002 and PFOA in 2008 (Citation3M Company 2013). Following the initiation of the phase-out, significant declines in serum PFOS levels have been noted in the US general population (CitationKato et al. 2011, CitationOlsen et al. 2012). In 2006, the world's eight major fluoropolymer and telomer manufacturers signed on to the US Environmental Protection Agency's 2010/2015 PFOA Stewardship Program, which was designed to reduce emissions and product content of PFOA, higher homologues, and precursors by 95% no later than 2010, and to eliminate emission and production of these chemicals by 2015 (CitationU.S. EPA 2006).
Fourteen epidemiologic studies have evaluated the association between PFOA exposure and human cancer (CitationBarry et al. 2013, CitationBonefeld-Jorgensen et al. 2011, CitationConsonni et al. 2013, CitationEriksen et al. 2009, CitationGilliland and Mandel 1993, CitationHardell et al. 2014, CitationInnes et al. 2014, CitationLeonard et al. 2008, CitationLundin et al. 2009, CitationSteenland and Woskie 2012, CitationUbel et al. 1980, CitationVassiliadou et al. 2010, CitationVieira et al. 2013, CitationYeung et al. 2013) and ten have evaluated the association between PFOS exposure and human cancer (CitationAlexander and Olsen 2007, CitationAlexander et al. 2003, CitationBonefeld-Jorgensen et al. 2011, CitationEriksen et al. 2009, CitationGrice et al. 2007, CitationHardell et al. 2014, CitationInnes et al. 2014, CitationOlsen et al. 2004, CitationVassiliadou et al. 2010, CitationYeung et al. 2013), with some studies examining both exposures. These studies include investigations of workers with occupational exposure and community members predominantly without occupational exposure to PFOA and/or PFOS. The community studies, in turn, include investigations of persons exposed to PFOA as a result of industrial contamination of public water supply and several other studies of subjects without apparent unusual exposure to PFOA or PFOS.
Despite the publication of a relatively large number of studies in the past decade, no previous systematic review has summarized the epidemiologic evidence on the carcinogenicity of PFOA and PFOS. Although the production of both chemicals has largely ceased in North America and Europe, PFAS production has increased in China since 2000. It remains unclear whether human cancer risk is associated with past or recent occupational or environmental exposure to these compounds. To address this question in this review, we critically evaluate each epidemiologic study of PFOA and/or PFOS exposure in association with cancer risk or mortality and then weigh the totality of the evidence for and against a causal effect of these chemicals on cancer development in humans. Before undertaking this paper's main objective of reviewing the epidemiologic evidence on PFOA and PFOS in relation to human cancer risk, we begin with a brief review of the potentially relevant evidence for the carcinogenicity of these chemicals in laboratory animals and its potential relevance to human cancer risk.
Evidence for the carcinogenicity of PFOA and PFOS in laboratory animals
PFOA
The carcinogenicity potential of PFOA has been investigated in two long-term dietary studies. In the first, groups of 50 male and 50 female Sprague–Dawley (Crl:CD® BR) rats were fed diets containing 0, 30, or 300 ppm ammonium perfluorooctanoate for up to 2 years (CitationButenhoff et al. 2012a, CitationSibinski, 1987). Dose-related decreases in body weight gain were observed in both sexes, and the decreases were statistically significant in both treated groups. However, no mortality differences were observed between treated and control groups, and survival was actually increased somewhat in both treated groups relative to their respective controls. Histologic examination revealed increases in the frequency of various non-neoplastic lesions of the testis in males, the mammary gland in females, and the liver in both sexes. At the study's termination, testicular Leydig cell adenoma in the high-dose males and mammary fibroadenoma in both treated groups of females were statistically significantly increased compared with the incidence of these tumors in concurrent controls. However, the frequency of mammary fibroadenoma among the treated females was not elevated compared with that among 947 historical control female rats from the DuPont Haskell Laboratory, and a subsequent Pathology Working Group review of proliferative mammary gland lesions using the original study slides concluded that the incidence of mammary gland neoplasms was unaffected by treatment (CitationHardisty et al. 2010, CitationSykes 1987).
A second chronic feeding study was conducted using male Crl:CD® BR (CD) rats and a dietary PFOA concentration of either 0 or 300 ppm (CitationBiegel et al. 2001, CitationCook et al. 1992). The incidences of liver adenoma, Leydig cell adenoma, and pancreatic acinar cell adenoma/carcinoma were significantly increased in the treated group. Because the latter finding was not reported in the first carcinogenicity study (CitationButenhoff et al. 2012a, CitationSibinski, 1987), the histological slides from both PFOA studies were reviewed subsequently by independent pathologists, who concluded that PFOA did increase the incidence of proliferative acinar cell lesions in both studies at the highest dietary concentration of 300 ppm. Interstudy differences in these pancreatic lesions were characterized as quantitative rather than qualitative, with more and larger focal proliferative acinar cell lesions and a greater tendency for progression to adenoma in lesions from the second study compared with those from the first. The basis for these quantitative differences is not known, but is believed to be most likely attributable to differences in the diets used in the two different laboratories (CitationFrame and McConnell 2003).
Potential mechanisms of carcinogenicity were also investigated during this study using small groups of six to ten rats that were sacrificed at multiple interim time points (CitationBiegel et al. 2001, CitationCook et al. 1992). The liver and testes were evaluated for cell proliferation. Peroxisome proliferation was also assessed, and analyses of serum hormone levels (estradiol, testosterone, luteinizing hormone, follicle-stimulating hormone, and prolactin) were conducted. In rats exposed to PFOA, relative liver weights and hepatic β-oxidation activity were statistically significantly increased relative to controls at all of the sampling times. Absolute testis weights were also increased, but only at 24 months. No hepatic or Leydig cell proliferation was observed at any of the sampling times. In addition, serum testosterone, follicle-stimulating hormone, luteinizing hormone, and prolactin levels did not differ between PFOA-treated and control rats. However, serum estradiol concentrations were significantly increased in the treated rats at 1, 3, 6, 9, and 12 months.
PFOS
A 2-year feeding study of potassium PFOS (K+ PFOS) at concentrations up to 20 ppm in the diet using male and female Sprague–Dawley [Crl:CD® (SD)IGS BR] rats detected multiple non-neoplastic changes in the liver, including hepatocellular hypertrophy with proliferation of endoplasmic reticulum, vacuolation, and increased eosinophilic granulation of the cytoplasm in both males and females at the higher exposure concentrations (CitationButenhoff et al. 2012b). In addition, statistically significant increases in hepatocellular adenoma incidence were observed in both male and female rats from the 20-ppm dose groups that survived to the terminal sacrifice. While there were no treatment-related findings for thyroid tissue, the males in a 20-ppm “recovery” group (exposed to K+ PFOS for only the first 53 weeks of the study) exhibited a statistically significant increase in the incidence of thyroid follicular cell adenoma. This result was considered by the study authors to be a spurious finding in light of the absence of any response in the corresponding group that was exposed to 20 ppm K + PFOS for the full 2 years. Interestingly, among females, statistically significantly decreasing trends were detected in the incidences of mammary fibroadenoma and combined mammary adenoma and fibroadenoma with increasing K+ PFOS exposure.
Modes of carcinogenic action in rats and potential human relevance
The two chronic carcinogenicity studies of PFOA show that this compound induces benign liver adenomas, Leydig cell adenomas, and pancreatic acinar cell tumors in rats. PFOS also induces liver adenomas in rats. However, neither PFOA nor PFOS is genotoxic, and recent studies have indicated an important role for activation of the peroxisome proliferator-activated receptor alpha (PPARα) and, possibly as well, the constitutive androstane receptor (CAR) and the pregnane X receptor (PXR), in the production of benign liver tumors by both of these chemicals (CitationCorton et al. 2014, CitationElcombe et al. 2010, CitationElcombe et al. 2012, CitationElcombe et al. 2014, CitationKlaunig et al. 2003, CitationKlaunig et al. 2012). The combination of liver adenomas, Leydig cell adenomas, and pancreatic acinar cell tumors induced by PFOA is known as the “tumor triad” that has been associated with a number of compounds that activate PPARα in the liver (CitationKlaunig et al. 2003, CitationKlaunig et al. 2012).
A scientific workshop was held in September 2010 in Research Triangle Park, North Carolina, to conduct a comprehensive, systematic review and assessment of the potential human relevance of evidence regarding the nongenotoxic modes of liver tumorigenesis that are mediated by nuclear receptors, including PPARα, CAR, PXR, and the aryl hydrocarbon receptor (AhR). The workshop's panel deliberations and conclusions have recently been published in a series of comprehensive review papers (CitationAndersen et al. 2014, CitationBudinsky et al. 2014, CitationCorton et al. 2014, CitationElcombe et al. 2014).
For PPARα agonists, including PFOA and PFOS, the workshop panel identified the following sequence of key events in the mode of action for hepatic tumor induction in rodents: 1) PPARα activation in the liver; 2) alteration of cell growth pathways in the liver; 3) perturbation of hepatic cell growth and survival, leading to the formation of new preneoplastic liver cells and the induction of new focal liver lesions; 4) selective clonal expansion of preneoplastic foci; and 5) transformation and outgrowth of preneoplastic liver cells into adenomas (CitationCorton et al. 2014). The induction of testicular Leydig cell tumors and pancreatic acinar cell tumors in rats by PFOA is currently not as well understood as liver tumor induction, but the same first key step, namely, PPARα activation in the liver, is thought to be required before subsequent changes in the liver and other organs lead ultimately to testicular and pancreatic neoplasms (CitationKlaunig et al. 2003, CitationKlaunig et al. 2012).
For CAR agonists, again including PFOA and PFOS, the 2010 workshop panel identified a similar, but not identical, sequence of key events: 1) CAR activation in the liver; 2) altered hepatic gene expression specific to CAR activation; 3) increased hepatocellular proliferation; 4) selective clonal expansion of altered hepatic foci; and 5) transformation and outgrowth of neoplastic cells into hepatic adenomas and carcinomas (CitationElcombe et al. 2014). For PXR agonists, key events in a mode of carcinogenic action in rodents could not be definitively established due to data limitations, but PXR activation, increased cell proliferation, and clonal expansion of altered cells leading to altered foci were thought to be likely to be involved.
The mechanisms by which PFOA and PFOS induce liver tumors in rats appear not to be relevant to the potential carcinogenicity of these compounds in humans. For example, most of the key events involved in hepatocarcinogenesis by PPARα and CAR activators that are clearly demonstrated in rodents do not seem to occur in humans (CitationCorton et al. 2014, CitationElcombe et al. 2014, CitationKlaunig et al. 2003, CitationKlaunig et al. 2012). Only the first of the listed key events for the PPARα mode of action, namely, activation of this nuclear receptor, has been demonstrated clearly in humans, where PPARα is the critical target for numerous hypolipidemic drugs that are currently in widespread use (CitationCorton et al. 2014). Nevertheless, the 2010 workshop panel did not rule out the potential human relevance of the other key events (CitationCorton et al. 2014).
For the proposed CAR receptor mode of rodent liver tumor induction, similar uncertainties regarding its potential human relevance remain. Phenobarbital is a chemical that has been used as a sedative, hypnotic, and antieplileptic drug in humans for several decades (CitationIARC, 2001), but it is also the compound selected as the “model” CAR activator to focus discussions during the 2010 nuclear receptor workshop (CitationElcombe et al. 2014). In addition to inducing liver tumors in rodents, phenobarbital is a prototypical inducer of the 2B subfamily of hepatic cytochrome P450 enzymes (CYP2B) in rodent and human liver (CitationMartignoni et al. 2006, CitationPelkonen et al. 2008). However, phenobarbital has been shown not to increase cell proliferation in cultured human hepatocytes, and the development of altered hepatic foci in human liver has not been reported. Furthermore, despite the widespread use of phenobarbital as a drug in humans, a recent review of epidemiological studies of phenobarbital concluded that there was no evidence for a specific role of phenobarbital in human liver cancer risk (CitationLa Vecchia and Negri, 2014).
Finally, the marked interspecies variations in the toxicities and pharmacokinetics of PFOA and PFOS make it especially difficult to meaningfully extrapolate findings from laboratory animals to humans (CitationButenhoff et al. 2006, CitationKennedy et al. 2004). For example, while the clearance half-life of PFOA in serum or plasma in laboratory animals ranges from approximately 2 h in female rats to about 10 days in male rats (CitationHan et al. 2012), it is approximately 3.5 years in humans (CitationOlsen et al. 2007), while the clearance half-life of PFOS is 1–2 months in rodents and approximately 4.8 years in humans (CitationChang et al. 2012, CitationOlsen et al. 2007). These disparate half-lives highlight the substantial sex and species differences that exist in the bioaccumulation and biopersistence of these chemicals in the body. In such circumstances, internal serum concentrations are likely to provide far superior dose metrics for assessing the potential human relevance of PFOA and PFOS carcinogenicity in rats than do external exposure measures, such as drinking water concentrations or estimated intake rates.
The substantial differences in the clearance half-lives of PFOA and PFOS across species and sex have recently been attributed to related differences in organic ion transport proteins and their differential impacts on the active renal tubular reabsorption of these chemicals (CitationHan et al. 2012). The development of physiologically based pharmacokinetic models that incorporate this and other important renal tubular secretion and reabsorption pathways offers the promise of significantly improved quantitative prediction of both the pharmacokinetics and the potential carcinogenicity of PFOA and PFOS in humans (CitationAndersen et al. 2006, CitationHan et al. 2012, CitationLoccisano et al. 2012a, CitationLoccisano et al. 2012b, CitationLoccisano et al. 2013, CitationTan et al. 2008).
In summary, while laboratory studies have demonstrated clearly that PFOA and PFOS exposures induce tumors in rats and have also increased substantially our understanding of the processes by which these nongenotoxic compounds accomplish this effect, these animal findings may or may not be relevant to humans. In such circumstances, the human evidence is critically important in establishing whether or not exposures to these compounds pose any increased cancer risk to humans (CitationAdami et al. 2011).
Epidemiologic literature review methods
To identify all epidemiologic studies of PFOA and/or PFOS in relation to human cancer, two authors independently searched the peer-reviewed scientific literature for relevant articles. Searches were conducted in PubMed using keywords and keyword roots including PFOA, APFO, PFOS, PSOF, perfluorooctan*, perfluorinate*, fluorochemical*, perfluoroalkyl*, cancer, tumor, malignan*, neoplas*, mortality, cohort, and related terms. Titles and abstracts were initially assessed to identify potentially relevant articles for a full-text review. Bibliographies of retrieved papers were also examined to identify additional articles. All investigators agreed on the final list of articles included in this review.
Each study is described in the following paragraphs with respect to its design, study subjects, exposure assessment, outcome assessment, control for confounders, other potential sources of bias, the probability and magnitude of possible bias, observed results, and interpretation. Characteristics of each study of PFOA exposure are briefly summarized in , and their results [including results presented in online appendices for CitationBarry et al. (2013), CitationConsonni et al. (2013), CitationLundin et al. (2009), CitationVieira et al. (2013), and CitationYeung et al. (2013)] are summarized in . Characteristics of each study of PFOS exposure are summarized in , and their results are summarized in . Observed associations are evaluated with regard to whether they were likely to be causal or due to bias, taking into consideration the probable direction and magnitude of bias. However, individual associations must be interpreted in light of the results from other studies, especially to assess whether chance may explain inconsistent findings. Therefore, the weight of evidence regarding possible causal relationships of PFOA and PFOS exposure with human cancer risk is assessed in accordance with the Bradford Hill guidelines of strength of association, consistency, biological gradient, plausibility, and coherence with toxicological evidence (CitationHill, 1965). These guidelines are used to provide a convenient logical framework, albeit not strict criteria, for the evaluation of causality. The guideline of temporality is also discussed where relevant – for example, when exposure has been measured after disease onset. The other three Bradford Hill guidelines, namely, specificity, experiment, and analogy, are not systematically addressed here because they are less informative for the assessment of the possible causality of a hypothesis.
Table 1. Design of epidemiologic studies of perfluorooctanoic acid (PFOA) and cancer.
Table 2. Results of epidemiologic studies of perfluorooctanoic acid (PFOA) and cancer
Table 3. Design of epidemiologic studies of perfluorooctanesulfonate (PFOS) and cancer.
Table 4. Results of epidemiologic studies of perfluorooctanesulfonate (PFOS) and cancer.
Occupational studies of PFOA
Overview
Epidemiologic studies of cancer risk among workers occupationally exposed to PFOA include a set of retrospective cohort mortality studies at each of the two PFOA manufacturing facilities in Cottage Grove, Minnesota (CitationGilliland and Mandel, 1993, CitationLundin et al. 2009, CitationUbel et al. 1980), and Parkersburg, West Virginia (CitationLeonard et al. 2008, CitationSteenland and Woskie, 2012), as well as a pooled retrospective cohort mortality analysis of all European and US facilities producing polytetrafluoroethylene, for which polymerization involves the use of PFOA (CitationConsonni et al. 2013). Throughout this review, the terms “retrospective” and “prospective” are used to describe the timing of exposure assessment relative to outcome assessment, with “retrospective” referring to the collection of exposure information after the outcome has occurred. Details of these studies are provided in and .
Occupational studies of PFOA share several strengths and limitations in common. Nearly all studies had virtually complete enumeration of workers at each facility, nearly complete follow-up for vital status, a high rate of cause-of-death ascertainment for decedents, and a long duration of follow-up. All studies relied mainly or entirely on some form of job history as the best available information to estimate PFOA exposure, thereby resulting in some unknown degree of exposure misclassification. Although all potentially exposed workers were studied, the modest size of each cohort, as well as the relatively young age distribution of workers, restricted the number of observed cancer outcomes. Most studies ascertained only cancer mortality, not cancer incidence, which is a more sensitive indicator of cancer risk, especially for cancer types with high survival. Control for confounding was largely limited to age, sex, race, and calendar period, although few other major potential confounders have been identified for several of the cancer endpoints of interest. Considering these strengths and limitations, along with the substantially higher cumulative PFOA exposure among workers than among nonoccupationally exposed persons (CitationFrisbee et al. 2009, CitationKato et al. 2011, CitationOlsen et al. 2003, CitationOlsen et al. 2012) and the limited number of PFOA production facilities worldwide, the studies of the Cottage Grove and Parkersburg cohorts provide the most informative epidemiologic evidence on cancer risk following high average and cumulative exposure to PFOA.
Studies of the Cottage Grove, Minnesota, facility
The first study of health outcomes in PFOA production workers was published by CitationUbel et al. (1980), who reported qualitative results of a cross-sectional analysis and retrospective cohort mortality study of employees at the 3M facility in Cottage Grove (). This plant consists of several divisions, with PFOA production limited to the chemical division, which produced PFOA from 1947 to 2000. The chemical division also manufactured small amounts of fluorochemicals involving PFOS, but PFOA was the predominant fluorochemical product. Starting in 1976, voluntary medical surveillance examinations, which included measurement of total serum fluorine levels, were offered to fluorochemical workers. The authors reported that based on three annual health evaluations of approximately 300 employees per year beginning in late 1976 (∼90% of plant workers in each year, with ∼50% participating during all 3 years), “[n]o health problems related to exposure to fluorochemicals were encountered among those examined” (CitationUbel et al. 1980). They added that “a review of absenteeism and illness patterns in these employees does not suggest any work related problems.”
As described by CitationUbel et al. (1980), an independent research group conducted a retrospective cohort mortality study among 3,688 workers employed at the Cottage Grove facility for at least 6 months between 1948 and 1978, a period during which 180 deaths (177 with death certificates obtained) were identified. Among the male workers, analyses revealed “no disagreement between the observed mortality and that expected. This was true of all the various causes of death and also of various specific causes of death due to cancer” (CitationUbel et al. 1980). In analyses restricted to chemical division workers, there were also “no disagreements between observed and expected mortality for any cause of death.” Due to the brevity of the study description and the absence of quantitative results, the strengths and limitations of the study methods cannot be thoroughly evaluated. Although this study provides limited evidence regarding the association between PFOA and cancer risk, its findings suggest no notable increase in cancer mortality among fluorochemical workers at the Cottage Grove plant.
In an extended retrospective cohort mortality study of workers at the Cottage Grove plant, CitationGilliland and Mandel (1993) followed 3,537 workers (2,788 men and 749 women) employed for at least 6 months between January 1, 1947, and December 31, 1983, excluding six workers with incomplete employment records. Vital status was traced via the Social Security Administration for the period 1947–1982 and the National Death Index for the period 1979–1989, supplemented by additional tracing strategies, to obtain vital status information for 100% of the cohort. Death certificates were obtained primarily from state health departments for 99.5% of the 398 deaths (348 among men). Using job history records, exposure status was classified according to a binary variable that distinguished between exposed workers, defined as those employed for at least 1 month in the chemical division, and unexposed workers, defined as those who had never worked in the chemical division or worked there for less than 1 month. Months of employment in the chemical division was also used as an estimate of cumulative exposure to PFOA. Standardized mortality ratios (SMRs) were calculated by comparing the observed numbers of cause-specific deaths to the expected numbers of deaths in the Minnesota white male population and, because appropriate mortality rates were not available for Minnesota females, and national rates were widely used and more statistically stable, in the US white male and white female populations, standardized by age and calendar period. In addition, selected hazard ratios (HRs) among male employees in relation to months of employment in the chemical division were estimated using proportional hazards regression models controlling for age at first employment, year of first employment, and years of total employment at the plant.
After a mean follow-up of 24.6 years for women in the chemical division and 26.4 years for women in the nonchemical division, no significant difference was found between observed and expected deaths from total cancer among female employees overall (17 deaths observed; SMR = 0.71 [95% confidence interval = 0.42–1.14]), nor were any site-specific cancer SMRs among female workers significantly different from the null () (CitationGilliland and Mandel, 1993). The same was true in analyses stratified by time between first employment and death (10, 15, or 20 years) or duration of employment (5, 10, or 20 years). Among male employees, mean follow-up was 24.8 years in the chemical division and 26.0 years in the nonchemical division. Overall, total cancer mortality was not significantly different from that expected among Minnesota white males (103 deaths observed; SMR = 1.05 [0.86–1.27]Footnote1), and all site-specific cancer SMRs were likewise nonsignificantly different from 1.0. All SMRs were also statistically nonsignificant among male chemical division employees. Findings were similar when compared with expected deaths among US white males or when stratified by latency period or duration of employment. In proportional hazards models, duration of employment in the chemical division was not significantly associated with overall cancer mortality, but it was significantly positively associated with prostate cancer mortality (HR per 10-year increase in duration of chemical division employment = 3.3 [1.02–10.6]).
Strengths of this study include the availability of employment records for nearly all eligible workers, the complete follow-up for vital status and nearly complete retrieval of death certificates, and long duration of follow-up (CitationGilliland and Mandel, 1993). In addition, this study was strengthened by the use of an unexposed internal cohort of nonchemical division workers as a comparison group to minimize the healthy worker effect – that is, the tendency of all-cause and certain cause-specific (e.g., cardiovascular) mortality rates to be lower in occupational cohorts than in the general population, mainly because people in poor health often are not part of the active workforce, but are included in the general population. (In this study, standardized rate ratios directly comparing the two worker subgroups were statistically unstable and, therefore, not reported.) Exposure was crudely classified based on history of working in the chemical division, without any information on specific departments or jobs involved in PFOA production. In fact, only one of the four men who died from prostate cancer had worked directly in the PFOA production buildings (CitationOlsen et al. 1998). Subsequent studies have shown that duration of employment, on its own, is not a good predictor of measured blood PFOA levels in Cottage Grove workers (CitationOlsen et al. 2003). Chance must be considered as an explanation for the observed findings, especially given the many health outcomes evaluated. Nevertheless, due in part to the dearth of established risk factors for prostate cancer and, therefore, the shortage of known confounders, the finding of a positive association between duration of employment in the chemical division and prostate cancer mortality in this cohort is suggestive of a potential association with PFOA exposure.
Subsequently, CitationLundin et al. (2009) reported on the mortality experience of this cohort with a longer period of enrollment eligibility (1947 through 1997) and 13 additional years of follow-up through 2002, as well as a minimum employment requirement of 1 year, resulting in 3,993 employees with 807 deaths (99.6% with a known cause). An expert panel of veteran workers and plant industrial hygienists was engaged to review job titles and department codes by year to classify jobs by likelihood of PFOA exposure based on where perfluorochemicals were developed or produced. Through this process, jobs were classified as having definite exposure (i.e., exposure “on a regular basis with potential for high exposure” to PFOA), probable exposure (i.e., “possible, but likely lower or transient” exposure to PFOA), or no or minimal exposure (i.e., work primarily in the nonchemical division, with some opportunity for PFOA exposure due to work-site contamination). Using this scheme, cohort members were classified as ever having worked in a definite-exposure job (513 workers), ever having worked in a probable-exposure job but no definite-exposure jobs (1,688 workers), or having worked only in no-or-minimal-exposure jobs (1,792 workers). A 6-month minimum employment requirement was also used, such that high exposure entailed having worked in a definite-exposure job for at least 6 months; moderate exposure entailed having ever had a probable-exposure job but zero to less than 6 months of a definite-exposure job; and low exposure entailed having worked only in nonexposed jobs.
Alternatively, relative exposure weights were assigned to each exposure category, with weights based in part on serum PFOA concentrations collected in 2000 from 131 chemical division employees, to estimate relative cumulative PFOA exposure (CitationLundin et al. 2009). In the serum study, workers with definite-exposure jobs had median serum PFOA levels of 2,600–5,200 ng/mL (converted from ppm), and those with probable-exposure jobs had median serum PFOA levels of 300–1,500 ng/mL; no data were available for nonexposed jobs. Taking these serum values and the long serum half-life of PFOA in humans into consideration, jobs with no exposure were assigned a weight of 1, those with probable exposure were assigned a weight of 30, and those with definite exposure were assigned a weight of 100. Cumulative exposure for each worker was then calculated as a sum of the days of employment at each exposure level, multiplied by the exposure weighting factor, to estimate the equivalent of time spent employed in a job with definite exposure. In sensitivity analyses, alternative weighting schemes of 1, 10, and 50 and 1, 10, and 100 were used. SMRs standardized by age, sex, and calendar period were calculated based on Minnesota state mortality rates, and HRs compared with an internal referent group were estimated with time-dependent Cox proportional hazards regression models, which allowed for delayed entry into high-exposure categories. Models were adjusted for sex and year of birth, with additional evaluation of age at cohort entry, smoking status (abstracted from occupational medical records for 35.8% of the cohort, with missing data imputed using a multiple-imputation model), and wage type (hourly, salaried, or both; or dichotomized based on the predominant wage type) as potential confounders.
After a mean follow-up of 29.3 years for workers with ever definite exposure, 31.6 years for workers with ever probable exposure but no definite exposure, and 31.6 years for nonexposed workers, the SMRs for total cancer mortality were not different from expectation among definitely exposed (19 deaths observed; SMR = 0.87 [0.52–1.35]) and probably exposed workers (119 deaths observed; SMR = 0.94 [0.78–1.12]), and significantly lower than expected among nonexposed workers (108 deaths observed; SMR = 0.78 [0.64–0.95]) () (CitationLundin et al. 2009). No specific cause of cancer death was significantly elevated in any group of workers. Using the 6-month minimum employment criterion to define time-dependent low, moderate, and high exposure, prostate cancer mortality was significantly increased among workers with high versus low exposure (based on 2 deaths in the high-exposure group; HR = 6.6 [1.1–37.7]), as well as workers with either moderate or high exposure (12 deaths; HR = 3.2 [1.0–10.3]). Similar findings were observed using weighted exposure days, with significantly increased prostate cancer mortality among workers with the equivalent of at least 5 years of definite exposure, compared with those with less than 1 year of definite exposure (7 deaths with at least 5 years of exposure; HR = 3.7 [1.3–10.4]). However, risk was lowest among those with the equivalent of 1–4.9 years of definite exposure (1 death; HR = 0.4 [0.1–3.6]). Mortality from pancreatic or bladder cancer was not significantly associated with job exposure categories or cumulative exposure years, adjusting for sex and birth year. Results based on the alternative weighting schemes, using a 10-year exposure lag, controlling for additional covariates, or stratifying by wage type were not substantively changed.
Strengths of this study (CitationLundin et al. 2009) are similar to those of the earlier cohort mortality study (CitationGilliland and Mandel, 1993). This study was additionally strengthened by the expert classification of jobs based on potential for PFOA exposure, thereby reducing possible exposure misclassification. The finding that risk of prostate cancer mortality was greatest among workers with the highest levels of exposure to PFOA lends additional weight to the finding of a positive association between occupational PFOA exposure and prostate cancer mortality (which may or may not translate to an association with prostate cancer incidence). However, interpretation of these results is complicated by the fact that nonexposed workers, who comprised the reference group for internal comparisons, had significantly lower prostate cancer mortality than expected based on the Minnesota male population, suggesting the presence of confounding by unmeasured prostate cancer risk (or preventive) factors in the internal analyses, or chance. Uncontrolled confounding is unlikely to be the sole explanation for HRs of 6.6 and 3.2, but the 95% confidence intervals for these estimates included 1.1 and 1.0, respectively, providing an indication of the instability of the estimates, as well as a higher likelihood of being due to confounding or chance. At the same time, the lack of a significant excess of mortality from any other cancer site, including the liver, testis, and pancreas, provides no evidence to support a causal relationship between PFOA exposure and mortality from other malignancies.
Studies of the Parkersburg, West Virginia, facility
The DuPont polymer production facility in Parkersburg is the other US site where cancer mortality has been studied among workers occupationally exposed to PFOA. CitationLeonard et al. (2008) conducted a retrospective cohort mortality study of 6,027 employees (4,872 men and 1,155 women) who had ever worked at the Parkersburg facility anytime between plant start-up on 1 January 1948 (2 years before the start of PFOA use in 1950) and the end of follow-up on 31 December 2002 (). Ninety percent of cohort members were identified through the DuPont Epidemiology Registry, which has conducted standard mortality surveillance of all active and pensioned company employees in the United States since 1957, and the remaining cohort members were identified through plant work history records. Vital status was ascertained through the Social Security Administration and causes of death were determined through the National Death Index or the DuPont Epidemiology Registry. SMRs were calculated based on expected age-, sex-, and calendar-period-specific mortality rates in three reference groups: the US population, the West Virginia population, and 72,882 DuPont workers from other facilities in West Virginia, Ohio, Virginia, Kentucky, Indiana, Pennsylvania, Tennessee, and North Carolina (referred to as “DuPont Region 1”). Plant employees were not classified according to PFOA exposure status. However, according to a 2004 cross-sectional health survey conducted at the Parkersburg plant, 23% of active employees (including survey participants and nonparticipants) were currently assigned to PFOA areas of the plant (CitationSakr et al. 2007). Among the 1,025 survey participants (55% of all active employees), 25% were currently working in PFOA areas, an additional 26% had been assigned to PFOA areas in the past, and 16% had intermittent current PFOA exposure; thus, the majority of the cohort had occupational exposure to PFOA as of 2004.
With an average of 26 years of follow-up (19 years of employment) among men and 16 years of follow-up (10 years of employment) among women, total cancer mortality was significantly lower among Parkersburg workers (234 deaths observed) than among the US population (SMR = 0.74 [0.65–0.84]) and the West Virginia population (SMR = 0.69 [0.60–0.78]), and was no different from that in the DuPont reference worker group (SMR = 1.02 [0.89–1.16]) () (CitationLeonard et al. 2008). No SMRs for site-specific cancers were significantly elevated in comparison with the US or West Virginia population. The only statistically significant difference was observed for mortality from thyroid and other endocrine gland malignancies, which occurred at a significant excess in Parkersburg employees compared with the DuPont reference cohort (3 deaths observed; SMR = 6.29 [1.30–18.37]). Of note, the SMR for prostate cancer was significantly below unity among workers compared with the US population and marginally significantly reduced compared with the West Virginia population.
This study is strengthened by its long follow-up time, use of an unexposed but otherwise comparable regional group of workers to adjust for the healthy worker effect, and low loss to follow-up, although the authors acknowledged potential loss to follow-up of decedents prior to 1957, when the DuPont Epidemiology Registry began mortality surveillance (CitationLeonard et al. 2008). Limitations are similar to those of other cohort mortality studies, with the additional major limitation that workers were not classified according to their estimated PFOA exposure, but were instead considered as a single exposed group, precluding an analysis of exposure-response gradients. Also, the authors did not report whether the exclusion of short-term workers altered the findings. There is no reason to suspect that known thyroid cancer risk factors, such as a low-iodine diet, ionizing radiation exposure, and family history, differed substantially between Parkersburg and other DuPont workers. Therefore, confounding is unlikely to explain the association. However, because the observed excess was based on only three thyroid cancer deaths (which are not representative of incident thyroid cancer) and the authors did not state whether the decedents were employed in areas with high PFOA exposure, chance is a plausible explanation. Overall, the results of this study suggest no substantial increase in cancer mortality among polymer workers occupationally exposed to PFOA.
CitationSteenland and Woskie (2012) extended and augmented this study by continuing mortality follow-up through 2008 and, more importantly, by using a job-exposure matrix in combination with serum PFOA data from 1,308 workers in 1979–2004 (median = 580 ng/mL, converted from ppm; range = 160–2,880 ng/mL) to estimate serum PFOA levels over time in eight job category/job group combinations. These groups were as follows: 1) direct PFOA exposure in the Teflon production area (8% of jobs), 2) with a separate indicator for the chemical operator job group; 3) direct PFOA exposure in other copolymer production areas that used PFOA (10% of jobs); 4) intermittent direct non-PFOA-use jobs (1% of jobs), 5) with a separate indicator for working in a tetrafluoroethylene (TFE) monomer job group; 6) maintenance jobs with intermittent direct or plant background PFOA exposures (15% of jobs), 7) with a separate indicator for the Teflon/copolymer maintenance job group; and 8) non-Teflon/copolymer division jobs with no PFOA use (66% of jobs). Regression models were constructed to predict measured serum PFOA levels based on job category/job group combination and other variables, such as the cumulative number of prior years spent in potentially PFOA-exposed jobs, annual amount of PFOA product used at or emitted from the plant, and temporal process changes in direct-exposure jobs; these models were then used to estimate the cumulative serum PFOA level in each cohort member in each year. Modeled serum PFOA levels correlated well with measured levels by job category/job group overall and by decade (Spearman ρ = 0.8), although individual-level correlations were not reported (CitationWoskie et al. 2012).
For the analysis, estimated cumulative serum levels were expressed in terms of ppm-years (converted here to ng/mL-years) and were categorized into quartiles based on the cumulative serum levels of decedents, with separate cut points developed for analyses assuming no lag, a 10-year lag, and a 20-year lag period between exposure and death (CitationSteenland and Woskie, 2012). Of the 6,027 workers in the cohort analyzed by CitationLeonard et al. (2008), 226 (4%) had insufficient work history data to allow estimation of serum PFOA levels over time and 10 others were omitted due to missing dates of birth, resulting in 5,791 workers for analysis. The mean estimated cumulative exposure was 7,800 ng/mL-years (median = 4,300) and the mean annual serum concentration was 350 ng/mL (median = 230 ng/mL). SMRs were calculated based on reference mortality rates from the cohort of 86,698 DuPont Region 1 (Appalachian region) workers in 1955–2009, excluding the Parkersburg plant, and also from the US general population in 1940–2007, extrapolated to 2009.
After a mean of 30 years of follow-up, total cancer mortality did not differ significantly between Parkersburg plant workers in any quartile of estimated cumulative serum PFOA, compared with the DuPont reference cohort (e.g., SMR for highest quartile = 0.94 [0.76–1.16] based on 91 observed deaths), and total cancer mortality was significantly lower than expected in the general US population (SMR = 0.74 [0.66–0.83]). The only two cancer sites with noteworthy positive associations were kidney cancer, for which mortality was significantly increased in the highest quartile of estimated cumulative serum PFOA (8 deaths observed; SMR = 2.66 [1.15–5.24]), but not in the other three quartiles or in all workers combined compared with the DuPont reference cohort or the US population; and mesothelioma (based on data from 1999 onward), for which mortality was significantly increased in the highest quartile as well as in the overall worker cohort relative to the DuPont reference cohort and the general population. Results were comparable, albeit attenuated for mesothelioma, after a 10- or 20-year lag, and were similar for kidney cancer after the inclusion of contributing (in addition to underlying) causes of death. All 12 kidney cancer deaths observed in the cohort were previously reported by CitationLeonard et al. (2008), with no additional deaths from kidney cancer occurring between 2003 and 2008. According to the results reported by CitationLeonard et al. (2008) and CitationSteenland and Woskie (2012), 2.78 kidney cancer deaths were expected in the cohort during the extended follow-up period based on DuPont Region 1 mortality rates (SMR = 0, P = 0.10) and 3.11 were expected based on overall US mortality rates (SMR = 0, P = 0.08).
The strengths and limitations of this study are similar to those of the earlier study in this cohort (CitationLeonard et al. 2008), but this study was further strengthened by the use of a time-dependent job-exposure matrix bolstered by serum PFOA data from 1,308 workers over 25 years to estimate cumulative and annual serum PFOA in all workers based on job category/job group and other work-related factors (CitationSteenland and Woskie, 2012). This approach reduced potential exposure misclassification compared with the earlier approach of grouping all plant workers together, although model error remained (CitationWoskie et al. 2012). The authors largely dismissed the observed excess of mesothelioma mortality in the worker cohort as probably being due to confounding by occupational asbestos exposure. However, they appeared to give greater credence to the kidney cancer result, although they noted that TFE, which has been found to be a rodent kidney carcinogen (CitationNational Toxicology Program, 1997), was used at the Parkersburg plant and was highly correlated with PFOA (CitationSteenland and Woskie, 2012). The SMR for kidney cancer in a study of TFE-exposed workers was estimated at 1.44 (0.69–2.65) overall and 2.58 (0.95–5.62) for workers with medium exposure, but 0.81 (0.10–2.93) for workers with high exposure (CitationConsonni et al. 2013); however, most of this cohort was also occupationally exposed to PFOA. Thus, confounding by TFE exposure could have explained part of the observed association with PFOA, but the magnitude of the association between TFE and kidney cancer in humans has not been precisely estimated. The absence of any kidney cancer mortality in the more recent follow-up period also raises the possibility of chance as an explanation and emphasizes the need to evaluate the consistency of this finding across study settings.
Study of combined European and US facilities
CitationConsonni et al. (2013) conducted a retrospective cohort mortality study that combined 5,879 male workers (excluding 778 female workers with 16 deaths) at six of the seven TFE production sites in Europe and the United States (excluding a small plant in North Carolina that employed only 31 workers in TFE processes starting in 1979). Although TFE exposure was the main focus of this study, the authors separately analyzed associations with PFOA exposure, which was highly correlated with TFE exposure. The minimum employment tenure varied by facility; all employees at three plants in Italy, England, and New Jersey were included, employees for at least 6 months at the Parkersburg plant were included, and employees for at least 1 year at two plants in Germany and the Netherlands were included in the analysis. The period of follow-up was 1960–2008 at the Italian site, 1952–2008 at the English site, 1969–2007 at the New Jersey site, 1950–2002 at the Parkersburg site, 1965–2001 at the German site, and 1967–2002 at the Dutch site. Ascertainment of vital status was conducted through linkages to population registries or other statistical or health databases, and death certificates and/or cause-of-death codes were obtained for 98.8% of known decedents from company-wide, local, state, or national health departments or databases. Time-varying cumulative exposure to PFOA and TFE was estimated semiquantitatively by using a job-exposure matrix with annual PFOA and TFE values for each relevant job title at each production site. The presence or absence of asbestos or vinyl chloride monomer at each plant was also recorded. Expected numbers of cause-specific deaths were calculated based on national age- and calendar-period-specific mortality reference rates for males (white males in the United States), with regional or state mortality rates used in sensitivity analyses.
After an average of 25 years of follow-up, significantly fewer than expected deaths from cancer occurred among the 4,205 male workers ever occupationally exposed to PFOA (SMR = 0.79 [0.67–0.92]), and no site-specific cancer SMRs were significantly elevated () (CitationConsonni et al. 2013). When estimated cumulative exposure to PFOA was categorized according to tertiles among observed all-cause deaths in PFOA-exposed workers, no significant excess mortality from total cancer, leukemia, or esophageal, liver, pancreatic, lung, or kidney/other urinary organ cancer was detected in the highest tertile of cumulative exposure, nor was a significant exposure-response trend observed for any of these outcomes. When cumulative exposures to TFE and PFOA were cross-classified, no deaths from any cause were observed (0.8 expected) among workers with high cumulative PFOA exposure and low cumulative TFE exposure, and only three deaths from cancer were observed (6.0 expected) among those with medium cumulative PFOA exposure and low TFE exposure. Thus, associations with PFOA exposure independent of TFE exposure could not be estimated robustly. In general, results were similar when regional mortality rates were used as the reference.
Strengths of this multicenter study include its large size, uniform approach to exposure assessment across sites, long duration of follow-up, and high rates of vital status ascertainment and determination of causes of death (CitationConsonni et al. 2013). A limitation is the lack of a reference cohort of comparable workers. Cumulative PFOA and TFE exposure were estimated semiquantitatively in terms of arbitrary “unit-years,” thereby preventing direct comparisons with results from other cohorts. As noted earlier, due to the correlation between PFOA and TFE (Spearman ρ = 0.72), observed associations could not reliably be attributed to either exposure. The observed nonsignificant positive association with death from kidney and other urinary organ cancer may be attributable largely to the fact that 2,379 (40%) of the 5,879 cohort members were from the Parkersburg plant – by far the largest production site in the study – where a positive association between estimated cumulative serum PFOA and kidney cancer mortality was previously reported (CitationSteenland and Woskie, 2012). CitationConsonni et al. (2013) did not present results after the exclusion of Parkersburg workers, nor did they state how many of the 10 kidney/other urinary organ cancer deaths in the pooled cohort came from the Parkersburg site, where CitationSteenland and Woskie (2012) reported that 12 kidney cancer deaths occurred in the full study group. CitationConsonni et al. (2013) concluded that their results “could neither conclusively confirm nor refute the hypothesis that TFE poses a carcinogenic risk to human beings,” and the same interpretation holds for PFOA.
Community studies of PFOA
Overview
Studies of cancer risk among communities with nonoccupational exposure to PFOA are more variable in design than the occupational cohort studies. These include a cancer-registry-based case-control study (CitationVieira et al. 2013), a retrospective cohort study (CitationBarry et al. 2013), a prospective case-cohort study (CitationEriksen et al. 2009), two retrospective case-control studies (CitationBonefeld-Jorgensen et al. 2011, CitationHardell et al. 2014), and three cross-sectional studies (CitationInnes et al. 2014, CitationVassiliadou et al. 2010, CitationYeung et al. 2013). Three of these studies were conducted in the Mid-Ohio River Valley near the DuPont plant in Parkersburg, West Virginia (CitationBarry et al. 2013, CitationInnes et al. 2014, CitationVieira et al. 2013), while the rest were conducted in Europe and Australia. Details of these studies are provided in and .
Studies of the Mid-Ohio Valley community
The C8 Health Project, a cross-sectional survey and serum study of 69,030 residents of the Mid-Ohio Valley in 2005–2006, was conducted as part of the settlement from a class-action lawsuit against DuPont, with the purpose of investigating the potential human health effects of PFOA exposure from contaminated drinking water (CitationFrisbee et al. 2009). Related research conducted by members of the C8 Science Panel, which was appointed by attorneys for the community and for DuPont to assess health outcomes in relation to community PFOA exposure, includes a cancer-registry-based case-control study by CitationVieira et al. (2013). This study was based in 13 Ohio and West Virginia counties encompassing six contaminated water districts: Little Hocking (where median serum PFOA concentrations were estimated in 1995 at 125 μg/L), Lubeck (65.8 μg/L), Tupper Plains (23.9 μg/L), Belpre (18.7 μg/L), Pomeroy (10.7 μg/L), and Mason (5.3 μg/L). Estimated PFOA exposure was compared between cases, who were adults diagnosed with each of 18 different cancers in 1996–2005, and controls, who were adults diagnosed with all other cancers (excluding kidney, pancreas, testis, and liver, as well as cancer sites with fewer than 100 cases in Ohio or that had not previously been evaluated in toxicological or epidemiologic studies of PFOA) in the same region and time period. All subjects were identified from the state-wide cancer registries in Ohio and West Virginia. The study dataset included 7,869 Ohio patients and 17,238 West Virginia patients with the following malignancies: bladder, brain, female breast, cervix, colon/rectum, kidney, leukemia, liver, lung, melanoma of the skin, multiple myeloma, non-Hodgkin lymphoma, ovary, pancreas, prostate, testis, thyroid, and uterus.
In analyses including cancer patients from both states, PFOA exposure was estimated based on residential water district at the time of diagnosis. In analyses restricted to Ohio, where 92% of cancer patients were geocoded to their street address at diagnosis and the remaining 8% were geocoded at the ZIP-code level, PFOA exposure was estimated based on a model that estimated individual serum PFOA levels using linked environmental, exposure, and pharmacokinetic models (CitationShin et al. 2011a, CitationShin et al. 2011b). In these analyses, because only the residential address at diagnosis was known, annual serum PFOA levels were estimated from 1951 to the date of diagnosis based on the assumption that patients had lived at that address for 10 years (or, in sensitivity analyses, for their lifetime), with an assumed exposure lag period of zero or 10 years. Estimated annual serum PFOA levels were summed over the assumed years of exposure to calculate a cumulative exposure estimate, and both the annual and cumulative measures were categorized based on the distribution among exposed cases. Logistic regression models were adjusted for age, sex, diagnosis year, smoking status (available for 90% of subjects), and insurance provider (available for 93% of subjects), as well as race in Ohio.
Site-specific cancer odds ratios (ORs) were significantly elevated for lung cancer (OR = 1.2 [1.1–1.3]) and non- Hodgkin lymphoma (OR = 1.2 [1.0–1.5]) when comparing all six contaminated water districts to uncontaminated water districts in the same counties (CitationVieira et al. 2013). In the Little Hocking water district, where estimated median serum PFOA levels were highest in 1995 (CitationShin et al. 2011b), the OR was significantly elevated for testicular cancer (OR = 5.1 [1.6–15.6]). However, no clear exposure-response patterns emerged across the remaining water districts, and ORs for testicular cancer were below 1.0 or not estimated (due to zero cases; i.e., OR = 0) in the other five water districts. In the analysis of estimated annual serum PFOA levels in Ohio cancer patients, assuming 10-year residency and lag period, the OR for kidney cancer was significantly elevated among cases with “very high” or “high” levels, compared with unexposed (OR = 2.0 [1.0–3.9] and OR = 2.0 [1.3–3.2], respectively), while the OR for non-Hodgkin lymphoma was significantly elevated among cases with “very high” or “medium” but not “high” levels. The association of kidney cancer with “very high” annual serum PFOA was detected only among women (OR = 3.5 [1.4–8.3]) and not among men (OR = 1.0 [0.3–3.4]). Borderline positive associations between “very high” serum PFOA and female breast, ovarian, prostate, and testicular cancers were counterbalanced by nonsignificant inverse associations with the same cancers in other exposure categories, including “high,” “medium,” and “low” versus none. Results were comparable when using estimated cumulative serum PFOA, assuming no exposure lag period, assuming lifetime residency, including kidney, liver, pancreas, and testis cancers in the control group, imputing missing data for smoking and health insurance, or stratifying by sex (except for kidney cancer).
Although this study (CitationVieira et al. 2013) benefits from its population-based setting, its large overall size, and its quantitative estimation of PFOA exposure based on a model that fairly accurately predicted serum PFOA levels in 2005–2006 (Spearman ρ = 0.67) (CitationShin et al. 2011b), several shortcomings must be noted. First, exposure to PFOA was assessed ecologically according to water district of residence at diagnosis, that is, at the group level rather than at the individual level. Water usage varies among individuals, and PFOA exposure was not uniform across water districts. Due to the ecological design, it is impossible to determine whether the individuals who were most highly exposed were those who developed a given cancer type; thus, associations observed at the water-district level cannot be assumed to hold for individual persons. The degree of potential ecological bias cannot be estimated. Second, PFOA exposure was estimated rather than measured, and while the exposure model was used to estimate serum PFOA levels since 1951, it was validated against levels measured only in 2005–2006. Third, information on residential history was not available, and the assumption of a minimum 10-year duration of residency was not validated among cancer patients. Therefore, estimation of PFOA exposure based on current address at the time of cancer diagnosis may not have captured long-term exposure at other previous addresses. Such misclassification could have been differential if residential relocation patterns differed by cancer type. Finally, limitations that affected other studies described earlier, including small numbers of rare cancers, minimal control for confounding, and multiple hypothesis testing, also applied to this study. In light of the highly imprecise OR estimates and the absence of any positive exposure-response trends, the results of this study provide only a hint of a possible association between PFOA exposure and elevated risk of various cancers.
In another study from the C8 Science Panel, CitationBarry et al. (2013) conducted a retrospective cohort study based on repeated interviews in 2008–2011 of participants in the 2005–2006 cross-sectional C8 Health Project, combined with additional subjects in the retrospective cohort mortality study of Parkersburg plant workers. The C8 Health Project enrolled 69,030 people (an estimated 80% of eligible subjects; 81% of those aged 20 years and older) who lived, worked, or attended school for at least 1 year in one of the six contaminated water districts near the Parkersburg plant between 1950 and 3 December 2004 (CitationFrisbee et al. 2009). Of the participants aged 20 years and older, 74% consented to further contact by the C8 Science Panel, and 82% of these (61% of the original cohort; an estimated 49% of eligible subjects) participated in one or two follow-up surveys in 2008–2011. From the cohort mortality study of Parkersburg plant workers employed for at least 1 day between 1948 and 2002 (CitationSteenland and Woskie, 2012), 4,391 (73%) of 6,026 workers were interviewed. After the exclusion of 0.07% of community members and 15% of workers who lacked retrospective PFOA exposure estimates, the analytic cohort consisted of 32,254 adults, including 3,713 workers (1,890 of whom were also enrolled in the cross-sectional C8 Health Project) and 28,541 community members with no evidence of having worked at the Parkersburg plant.
Using the same serum PFOA model as used by CitationVieira et al. (2013) (CitationShin et al. 2011a, CitationShin et al. 2011b), CitationBarry et al. (2013) estimated each participant's annual serum PFOA concentration from 1952 or birth through 2011 based on PFOA emission and dispersion data, individual residential history and water consumption, and a model for PFOA absorption, distribution, metabolism, and excretion. For plant workers, occupational PFOA exposure was added based on the job-exposure matrix used by CitationSteenland and Woskie (2012). The estimated median annual PFOA serum level was 19.4 (range = 2.8–9,217) ng/mL in community members and 174.4 (range = 5.2–3,683) ng/mL in workers. Lifetime cancer history was self-reported on the questionnaire, and the authors sought to validate reported diagnoses through medical chart review (if consent was granted) or through linkage to the Ohio and West Virginia state cancer registries. The analysis was restricted to validated primary cancers, which comprised 69% of community-reported diagnoses and 75% of worker-reported diagnoses. Estimated cumulative serum PFOA, calculated as the sum of all annual estimates up to a given age, was modeled on the logarithmic scale with respect to cancer diagnosis using Cox proportional hazards regression models with age as the time scale, adjusting for time-varying smoking, time-varying alcohol consumption, sex, education, and 5-year birth period, and assuming a 10-year lag.
With an average of 32 years of follow-up after age 20 for community residents and 38 years for workers, assuming a 10-year lag, marginally nonsignificant positive associations were detected between a one-unit increase in estimated log cumulative serum PFOA and the risk of testicular cancer (HR = 1.28 [0.95–1.73]) and kidney cancer (HR = 1.09 [0.97–1.21]), while borderline or significant inverse associations were detected with breast and oral cancers () (CitationBarry et al. 2013). The positive associations were slightly stronger but similar with no lag assumption and were reportedly similar with a 20-year lag. When estimated cumulative serum PFOA was categorized by quartile among the thyroid, kidney, and testicular cancer cases, significant positive exposure-response trends were detected for testicular cancer with a 10-year lag (P = 0.02 for a linear trend test across quartiles using exposure category midpoints; P = 0.10 for a linear trend test using continuous log estimated cumulative serum PFOA) or with no lag (P = 0.04 and 0.05, respectively). For kidney cancer and thyroid cancer, no significant exposure-response trend was detected with either lag. Results were comparable when estimated person-time prior to living or working on one of the six contaminated water districts was excluded. After stratification between the community and worker cohorts, associations with continuous log cumulative serum PFOA were similar in both cohorts for testicular and thyroid cancers with a 10-year lag, but positive associations with no lag were detected only in the community cohort. For kidney cancer, a borderline significant positive association with unlagged continuous cumulative serum PFOA was observed only in the community cohort. When exposure was categorized into quartiles, positive exposure-response trends were observed for thyroid cancer only in the worker cohort and for testicular and kidney cancers only in the community cohort.
Advantages of this study include the estimation of annual and cumulative serum PFOA based on a detailed model that incorporated serum PFOA measurements from 45,276 participants in the C8 Health Project along with additional relevant data; and the validation of self-reported cancer history (CitationBarry et al. 2013). However, several important limitations deserve comment. Because only positive cancer histories were validated, the extent of underascertainment of cancer cases is unknown. If participants living in water districts known to have higher PFOA levels were more likely to report a positive history, then differential outcome misclassification could have positively biased the results (toward higher HRs). Similarly, selection bias could have distorted the findings if participation in the C8 Health Project or follow-up questionnaires was directly or indirectly related to both PFOA exposure status and cancer history. Such selection bias is plausible, given that factors that predict serum PFOA levels, such as demographic, socioeconomic, and behavioral factors (CitationCalafat et al. 2007, CitationJain 2013, CitationJain 2014, CitationKato et al. 2011), can influence both cancer risk and the decision to participate in a health research study. For example, higher educational level has been shown to be associated with higher serum PFOA levels (CitationCalafat et al. 2007), a greater likelihood of study participation (CitationLissner et al. 2003), and elevated risk of testicular (CitationRichardson et al. 2012) and thyroid cancers (CitationLi et al. 2013), thereby potentially resulting in positive bias. The study was also limited by probable exposure misclassification (of an unknown degree, given that the serum PFOA model was validated only against 2005–2006 serum PFOA measurements), small numbers of site-specific cancers, and multiple hypothesis testing. Consequently, chance and various sources of bias are plausible explanations for the observed associations.
CitationInnes et al. (2014) conducted a cross-sectional study of 208 prevalent colorectal cancer cases and 47,151 adults without cancer who participated in the C8 Health Project baseline survey and blood sampling in 2005–2006. Participants who had ever been diagnosed with colon and/or rectal cancer were identified from the self-reported health survey, with validation of positive reports based on medical records, and controls were other adult participants who had not been diagnosed with cancer and had complete data (excluding 0.6% of participants with missing data on PFOA and PFOS and 3.2% with missing data on other covariates of interest). PFOA, PFOS, and eight other PFASs were measured in serum collected at the time of the health survey. Associations between serum PFAS levels (as quartiles or continuous variables) and colorectal cancer diagnosis were estimated with adjustment for age only; age, race, sex, socioeconomic status, marital status, smoking, alcohol consumption, vegetarian diet, and exercise; or all of these covariates plus serum lipid profiles, C-reactive protein, estradiol, uric acid, and gastrointestinal symptoms. Additional adjustment for other PFASs, anemia, osteoarthritis, rheumatoid arthritis, or fibromyalgia did not affect the results.
Higher serum levels of PFOA were significantly associated with a lower prevalence of colorectal cancer in age-adjusted and multivariate-adjusted models () (CitationInnes et al. 2014). For example, in the model adjusted for all covariates listed in the preceding paragraph, the OR of colorectal cancer associated with the highest quartile (≥ 71.3 ng/mL) versus lowest quartile (0.25–13.4 ng/mL) of serum PFOA was 0.64 (0.44–0.94), with a statistically significant inverse trend (P = 0.002), although continuous PFOA was not significantly associated (OR per ng/mL = 1.00 [1.00–1.00], P = 0.46). The significant inverse association of serum PFOA with colorectal cancer diagnosis was detected among men but not among women, in nonobese but not in obese adults, and for cases diagnosed in 2000 or later but not for those diagnosed earlier; however, the association did not vary significantly by age or colorectal cancer treatment method. Restricting the analysis to participants who had lived at the same residence since 1990–1995 or before and to cases diagnosed within the previous 6 years strengthened the inverse association (adjusted OR for highest versus lowest quartile = 0.4 [0.2–0.5]), as did restricting the analysis to adults with serum PFOA ≤ 20 ng/mL (age-adjusted OR = 0.4 [0.2–0.7], P-trend = 0.009). Results were unchanged after restriction to primary colon cancer cases, to those not undergoing current treatment, or to those who had not received chemotherapy, or after the inclusion of all 281 self-reported colorectal cancer cases.
This study benefits from its direct measurement of serum PFOA with a broad exposure range, the availability of data to adjust for numerous potential confounders, and the validation of self-reported cancer diagnoses using medical records (CitationInnes et al. 2014). Colorectal cancer may have been underascertained, with possible differences in cancer reporting based on the place of residence and PFOA exposure status, although it seems unlikely that residents of water districts with higher PFOA levels would be less likely to report a positive colorectal cancer history than those in low-PFOA districts. Likewise, selection bias based on study participation or survey completion is possible, but again it is improbable that residents of high-PFOA water districts with a history of colorectal cancer would be less motivated to participate. A key limitation is that serum PFOA levels measured in 2005–2006 may be etiologically irrelevant to contemporaneously or previously diagnosed colorectal cancer, and it is possible (although not studied) that disease could produce lower levels. Overall, bias is not a probable explanation for the observed inverse exposure-response trends, but the postdiagnostic measurement of serum PFOA prevents a causal interpretation.
Studies of other groups
A prospective case-cohort study was conducted in Denmark by CitationEriksen et al. (2009). From a population-based cohort of 57,053 Danish-born men and women aged 50–65 years without a history of cancer as of enrollment in 1 December 1993, through 31 May 1997, the authors identified all incident cases of cancer of the prostate (N = 713), bladder (N = 332), pancreas (N = 128), and liver (N = 67) by linkage with the Danish Cancer Registry and Danish Pathology Data Bank with follow-up through 1 July 2006. A subcohort of 680 men and 92 women was randomly selected as a reference group. Plasma PFOA levels were measured in specimens collected at cohort entry, with a mean coefficient of variation of 5.9% (and 1.8% for PFOS, which was also measured in plasma). Analyses were conducted according to the unweighted case-cohort approach by Cox proportional hazards regression, stratified by sex, with age as the time scale, and with plasma PFOA categorized into quartiles based on the distribution among patients with each cancer type. Models for prostate cancer were adjusted for education, body mass index, dietary fat intake, and fruit and vegetable intake. Models for bladder cancer were adjusted for smoking status, smoking intensity, smoking duration, education, and occupation associated with risk for bladder cancer. Models for pancreatic cancer were adjusted for smoking status, smoking intensity, smoking duration, dietary fat intake, and fruit and vegetable intake. Models for liver cancer were adjusted for smoking status, education, alcohol intake, and occupation associated with risk for liver cancer.
No significant association was detected between plasma PFOA and risk of any of the four cancer outcomes, whether plasma PFOA was analyzed in quartiles or as a continuous variable () (CitationEriksen et al. 2009). For example, compared with the lowest quartile of plasma PFOA, the relative risk (RR, estimated as incidence rate ratio) of prostate cancer for the highest quartile of plasma PFOA was 1.18 (0.84–1.65), that for bladder cancer was 0.81 (0.53–1.24), that for pancreatic cancer was 1.55 (0.85–2.80), and that for liver cancer was 0.60 (0.26–1.37). The median plasma concentration of PFOA among the groups ranged from about 5 to 7 ng/mL. Results were similar after stratification by sex.
Noteworthy strengths of this study are the prospective design, with plasma PFOA measured prior to onset, and the direct assessment of plasma PFOA concentration in all subjects, without misclassification resulting from exposure estimation (CitationEriksen et al. 2009). Additional strengths include the control for several confounders, the selection of an appropriate comparison group, the presumably complete ascertainment of cases by linkage to high-quality Danish disease registries, and the larger numbers of incident cases than most other studies of PFOA [with the exception of CitationVieira et al. (2013)]. Plasma PFOA was measured only once per individual at a variable point in time (median = 7 years, range = 0–12 years) prior to cancer diagnosis. It is unclear whether a single measurement is adequate to estimate relative differences in long-term PFOA exposure, although the long biological half-life of PFOA in humans (CitationOlsen et al. 2007) suggests that this is possible. Median plasma PFOA concentrations in these subjects were relatively low – considerably lower than the measured median PFOA serum levels of 24.2 (range = 0.25–4,752) ng/mL among community members and 112.7 (range = 0.25–22,412) ng/mL among plant workers in the study by CitationBarry et al. (2013), for example – such that associations with higher levels of PFOA exposure could not be estimated. Thus, although this study convincingly shows no association between nonoccupational plasma PFOA levels and risk of prostate, bladder, pancreatic, or liver cancer, it was not designed to address the question of a potential association with high-level PFOA exposure.
CitationBonefeld-Jorgensen et al. (2011) conducted a case-control study of breast cancer in Greenland Inuit women among whom serum levels of several persistent organic pollutants, metals, and PFASs, including PFOA, were compared. Thirty-one incident breast cancer cases, representing approximately 80% of eligible cases in Greenland, were diagnosed and enrolled in 2000–2003 from a single hospital in Nuuk where all breast cancer cases in Greenland are registered. One hundred and fifteen controls, frequency-matched to the cases on age and region of residence, were selected from two cross-sectional serological studies. One of these studies enrolled 153 randomly selected Greenland women living in Nuuk, with a 95% participation rate (CitationCote et al. 2006), and the other enrolled 50 Inuit women randomly selected from each of the five districts in Greenland in 1999–2005, with a participation rate of 90% in Nuuk and nearly 100% in the other districts (CitationDeutch et al. 2007). Serum PFASs, including PFOA and PFOS, were measured in specimens collected from cases at diagnosis and from controls at study enrollment. Associations with breast cancer risk were estimated using logistic regression adjusting for age, body mass index, number of full-term pregnancies, breastfeeding history, menopausal status, and serum cotinine level.
The median serum PFOA level among the 31 cases was 2.5 (range = 0.2–7.2) ng/mL and that among the 98 controls with available data was 1.6 (range = 0.2–7.6) ng/mL (CitationBonefeld-Jorgensen et al. 2011). In addition, the median serum level of the sum of perfluorocarboxylated acids (including PFOA as well as perfluoroheptanoic acid, perfluorononanoic acid, perfluorodecanoic acid, perfluoroundecanoic acid, perfluorododecanoic acid, and perfluorotridecanoic acid) was 8.0 (range = 0.3–21.4) ng/mL among cases and that among controls was 5.2 (range = 1.0–28.1) ng/mL. Estimated ORs showed no significant association between a 1-ng/mL increase in serum PFOA and risk of breast cancer, whether in unadjusted models including all 31 cases and 98 controls with PFOA data, unadjusted models including the 7 cases and 61 controls with data on PFOA and all adjustment covariates, or fully adjusted models including the 7 cases and 61 controls with complete data (). Likewise, no significant association was detected with a 1-ng/mL increase in total serum perfluorocarboxylated acids.
A strength of this study is its high case ascertainment and control participation rates, such that the subjects were probably representative of the general Greenland Inuit population, even though the study was not strictly population-based because the case and control ascertainment periods and geographic regions were somewhat different (CitationBonefeld-Jorgensen et al. 2011). Another strength is the direct measurement, rather than estimation, of PFOA exposure in all participants. Due to the retrospective design, serum PFOA concentration was measured after breast cancer diagnosis in cases, although whether the disease affects serum levels has not been studied. Nevertheless, levels measured only once at the time of disease diagnosis may not be etiologically relevant. Also, the authors examined associations only with continuous serum PFOA exposure, without consideration of nonlinear exposure-response associations. Due to the small number of cases with full covariate data, it is unlikely that the models could adjust sufficiently for confounding by measured risk factors such as age, body mass index, and reproductive history, which were crudely classified. Diet, alcohol consumption, and other unmeasured breast cancer risk factors may also have acted as confounders. Such confounding could have resulted in positive bias, since serum levels of PFASs have been positively associated with alcohol consumption, and inversely associated with parity and breastfeeding (CitationJain, 2013, CitationJain, 2014). Nevertheless, the results of this study suggest no association between nonoccupational serum levels of PFOA or total perfluorocarboxylated acids and breast cancer risk in Greenland Inuit women.
Another case-control study was conducted in Sweden by CitationHardell et al. (2014), who measured whole-blood levels of six perfluorinated carboxylates and two perfluorinated sulfonates, including PFOA and PFOS, in 201 incident cases of prostate cancer and 186 population controls. Prostate cancer patients were admitted consecutively to University Hospital in Örebro for radiotherapy or chemotherapy in 2007–2011, with a participation rate of 79%. Controls were individually matched to eligible cases on age and county of residence and selected from the Swedish population registry, with a participation rate of 54% after excluding those with prior cancer. PFASs were measured in whole blood collected during the same time period for cases and controls. ORs for risk of prostate cancer, including subgroups defined by Gleason score, prostate-specific antigen (PSA) level, and first-degree family history of prostate cancer, were estimated using unconditional logistic regression adjusting for age, body mass index, and year of blood draw.
The median level of PFOA in whole blood was 2.0 (range = 0.320–15) ng/mL among cases and 1.9 (range = 0.345–8.4) ng/mL among controls (CitationHardell et al. 2014). The median among controls was used as the cutoff point for analyses of higher versus lower PFOA levels, with the 75th percentile used for alternative analyses. No significant association was detected between elevated blood PFOA and risk of prostate cancer overall, nor were any significant associations detected after subdividing cases by Gleason score (2–6 versus 7–10) or PSA level (≤ 10 versus ≥ 11 ng/mL) (). When cases and controls were classified according to both their first-degree family history of prostate cancer and their blood PFOA level, significantly higher prostate cancer risk was detected among those with both a positive family history and elevated blood PFOA, relative to those with neither (OR = 2.6 [1.2–6.0]).
In an unconventional design, the controls in this study were selected from a population-based registry, whereas the cases were hospital-based (CitationHardell et al. 2014). Thus, it is unclear if the controls were representative of the source population for the cases and whether the cases were representative of all incident prostate cancers in the study base. Moreover, the low participation rate among controls increases the likelihood of selection bias based on demographic, socioeconomic, and behavioral factors, such as level of education, which can influence serum PFOA concentration and the decision to participate, as well as prostate cancer risk, thereby resulting in overestimated ORs. The benefit of directly measuring PFOA exposure is offset by the fact that blood PFOA levels were measured only once after prostate cancer diagnosis in cases, making their etiologic relevance unclear. Because blood PFOA concentration was dichotomized, exposure-response trends could not be analyzed. The fact that a first-degree family history of prostate cancer – an established risk factor – was not significantly associated with prostate cancer risk among those with lower blood PFOA suggests error in the classification of this variable. The positive association in those with a family history could also be due to chance, given that many subgroup analyses were conducted. The primary results of this study indicate no association between nonoccupational exposure to PFOA and risk of prostate cancer.
Two cross-sectional studies provide limited information on the relationship between PFOA exposure and human cancer risk. In 2009, CitationVassiliadou et al. (2010) measured serum levels of PFOA and PFOS in 40 cancer patients (with unspecified cancer sites) hospitalized at a specialized cancer treatment center in Athens, Greece; 56 healthy employees at an Athens research center who were undergoing an annual health check-up; and 86 ambulatory patients and healthy individuals undergoing a medical check-up at a hospital in Argolida, located in a semiurban and rural area of Greece. The median serum PFOA concentration was 2.27 (range = 1.29–6.89) ng/mL in 17 male cancer patients and 1.85 (range = 0.75–3.26) ng/mL in 23 female cancer patients, 3.14 (range = 1.68–10.21) ng/mL in 27 Athens male controls and 1.70 (range = 0.57–6.57) ng/mL in 29 Athens female controls, and 1.81 (range = 0.48–5.60) ng/mL in 27 Argolida male controls and 1.71 (range = 0.55–6.29) ng/mL in 59 Argolida female controls (). The authors reported a statistically significant difference (P < 0.05 based on one-way analysis of variance) in unadjusted mean serum PFOA values, with the highest levels detected among Athens controls (2.95 ng/mL), followed by cancer patients (2.31 ng/mL) and Argolida controls (1.97 ng/mL). Serum PFOA levels were measured at a single time point after diagnosis in cases, cancer sites were not specified, the controls were selected from different source populations than the cases, no information was provided on participation rates, no confounders were controlled for, and the study size was limited. Therefore, this study does not offer convincing data on the association between serum PFOA levels and cancer risk, although the results are consistent with no such association.
Similarly, CitationYeung et al. (2013) cross-sectionally measured levels of 12 PFASs, including PFOA and PFOS, in the serum and liver tissue of several patient groups in Melbourne, Australia. These groups comprised patients with hepatocellular carcinoma (HCC) but presumably without evidence of hepatitis C virus (HCV) infection (24 serum specimens, 12 liver specimens); patients with both HCC and HCV infection (13 serum specimens, 14 liver specimens); patients with liver cirrhosis and HCV infection (38 serum specimens, 38 liver specimens); patients with amyloidosis or acute liver failure (4 serum specimens, 2 liver specimens); healthy donors without any known liver disease (25 serum specimens); and patients with colorectal cancer metastasis to the liver, with tissue taken well clear of the tumor margin (9 liver specimens). PFOA levels in serum and liver tissue were poorly correlated in HCV-negative HCC cases (Spearman ρ = − 0.227), HCV-positive HCC cases (ρ = 0.189), and HCV cases (ρ = 0.298). CitationYeung et al. (2013) reported the distribution of serum and liver tissue PFOA concentrations in each patient group, but did not perform a statistical comparison across groups. However, the median serum PFOA level was comparable between HCV-negative HCC cases (2.48 ng/mL) and healthy donors (2.34 ng/mL), and it was lower among HCV-negative HCC cases than among patients with HCV-positive liver cirrhosis (3.55 ng/mL), whose levels were similar to those in patients with HCV-positive HCC (3.43 ng/mL) (). In liver tissue, median PFOA levels were lower in HCV-negative HCC cases (0.495 ng/g) than in normal liver tissue from colorectal metastasis patients (0.506 ng/mL) and comparable between HCV-positive patients with HCC (0.454 ng/g) and with cirrhosis (0.416 ng/g). In paired serum and liver specimens, the ratio of PFOA concentration in liver to that in serum did not differ significantly across groups (P > 0.05). The shortcomings of this study, including the single post-diagnosis measurement of PFOA in serum and liver tissue, the potentially different source populations for the various patient groups, the lack of information to evaluate possible selection bias due to nonparticipation, the absence of control for confounding, and the modest study size, combine to limit the utility of the results for addressing the association between PFOA exposure and liver cancer risk, although the findings are compatible with no such association.
Summary of epidemiologic evidence on PFOA and cancer in humans
In this section, we evaluate the weight of evidence for or against the hypothesis of a causal effect of PFOA on human cancer based on the collective epidemiologic evidence to date. Here, the community-based case-control studies (CitationBonefeld-Jorgensen et al. 2011, CitationHardell et al. 2014) and cross-sectional studies (CitationVassiliadou et al. 2010, CitationYeung et al. 2013), which yielded generally statistically null results, are not considered because their methodological limitations render them largely uninformative for addressing the hypothesis of interest. The cross-sectional study of colorectal cancer in the C8 Health Project (CitationInnes et al. 2014) is included because of its relevance to communities exposed to higher environmental levels of PFOA.
Strength of association
A strong RR for the association between a suspected risk factor and a disease adds credibility to a causal interpretation of the association (CitationHill, 1965), since a strong association is less likely than a weak one to be explained by bias, confounding, or chance. As shown in , the majority of RR estimates were between 0.5 and 2.0, with 95% confidence intervals including 1.0. The rare exceptions were typically based on fewer than five exposed cases or deaths, making the estimates unstable.
A few stronger associations were detected based on at least five exposed subjects, but none was consistent across studies. Given the high potential for uncontrolled confounding in all of these studies, and selection bias in several studies, the observed associations cannot reasonably be attributed to PFOA exposure. In fact, the elevated SMRs for mesothelioma in the Parkersburg plant were attributed by the authors to occupational asbestos exposure (CitationSteenland and Woskie, 2012), thereby illustrating the potential for observed associations to be explained by known uncontrolled strong confounders.
Exposure misclassification in these studies may not be nondifferential between cancer cases and noncases and independent of other errors. Exposure misclassification is especially likely to be differential in cross-sectional and case-control studies, where exposure status is classified after or simultaneously with disease status, but differential misclassification may also occur in cohort studies, resulting in an unpredictable direction of bias on RR estimates. For example, in a cohort study using a job-exposure matrix to classify exposure, differential error might occur if job title were associated with both the degree of exposure misclassification and the probability of developing or being ascertained with cancer via socioeconomic status (i.e., apart from its role as a surrogate for exposure level). Moreover, even in the presence of nondifferential exposure misclassification, reported associations are not necessarily underestimated. Additional conditions must be satisfied for the bias to be toward the null, and even when all such conditions are met, a given estimate may by chance be biased away from the null (CitationJurek et al. 2005, CitationJurek et al. 2008). Thus, it cannot be assumed that more accurate classification of PFOA exposure would necessarily have led to stronger associations in these studies.
Consistency of association
The repeated observation of an association across multiple study settings can lend support to a causal hypothesis (CitationHill, 1965). Across the retrospective cohort studies of occupational PFOA exposure and cancer mortality, overall cancer mortality was consistently close to or below unity in comparison with general populations or other worker cohorts. There was a consistent lack of a significant positive association for most specific cancer sites. Although some positive RRs were reported for some sites, the estimates were imprecise (and mostly statistically nonsignificant) mainly due to small study sizes.
An issue relevant to many of the results reviewed here is that of multiple comparisons. Many of the positive findings were reported in studies that tested associations with numerous outcomes, without any adjustment for multiple testing. Thus, several erroneous rejections of the null hypothesis (i.e., false-positive results) would be expected from these studies. Several unreplicated positive associations were reported by CitationVieira et al. (2013), who reported more than 400 tests of association. For example, CitationVieira et al. (2013) found a small (20–30%) excess risk of lung cancer in all PFOA-contaminated water districts combined, the Mason water district, and the Tuppers water district, compared with uncontaminated water districts. However, lung cancer SMRs were nearly all around or below 1.0 in cohort studies of PFOA-exposed workers or community members (CitationBarry et al. 2013, CitationConsonni et al. 2013, CitationGilliland and Mandel, 1993, CitationLeonard et al. 2008, CitationLundin et al. 2009, CitationSteenland and Woskie, 2012). CitationVieira et al. (2013) reported a doubling of ovarian cancer risk in the highest category of estimated annual or cumulative serum PFOA level in communities around the Parkersburg facility, and a 70% increase in uterine cancer risk in the second-highest category of estimated annual serum PFOA. However, CitationBarry et al. (2013) found no excess of ovarian or uterine cancer risk after using the same exposure model to estimate cumulative serum PFOA level in residents of the same region. CitationVieira et al. (2013) found a 40% increase in melanoma of the skin among residents of the contaminated Belpre water district, but both CitationLeonard et al. (2008) and CitationBarry et al. (2013) reported no association of melanoma with PFOA exposure. Modest (50–100%) excesses of brain/central nervous system cancer and non-Hodgkin lymphoma in specific categories of estimated annual or cumulative serum PFOA (CitationVieira et al. 2013) also were not supported by the results of other studies (CitationBarry et al. 2013, CitationConsonni et al. 2013, CitationLeonard et al. 2008, CitationLundin et al. 2009, CitationSteenland and Woskie, 2012).
CitationGilliland and Mandel (1993) reported a positive association between duration of employment in the chemical division of the Cottage Grove facility and prostate cancer mortality. In the same cohort with longer follow-up, CitationLundin et al. (2009) also found a significant and substantial excess of prostate cancer mortality among the most highly exposed workers at the plant. Additionally, CitationVieira et al. (2013) found a statistically nonsignificant 50% excess of prostate cancer in the highest category of estimated annual or cumulative serum PFOA in residents around the Parkersburg facility. However, these positive findings were counterbalanced by firmly null (CitationBarry et al. 2013, CitationEriksen et al. 2009, CitationSteenland and Woskie, 2012) or even inverse (CitationConsonni et al. 2013, CitationLeonard et al. 2008) results for prostate cancer in other studies of occupationally or nonoccupationally exposed subjects.
Some evidence of a positive association between estimated serum PFOA level and testicular cancer risk was found among residents in Ohio and West Virginia communities with a PFOA-contaminated public water supply. CitationBarry et al. (2013) reported a 30% increase in testicular cancer risk (50–70% among community members) per unit increase of logged cumulative serum PFOA and substantial, albeit statistically unreliable and nonsignificant, increases in testicular cancer risk in the highest quartile of estimated cumulative serum PFOA. Using the same exposure model, CitationVieira et al. (2013) observed a five-fold excess of testicular cancer risk in the most highly contaminated water district and a nearly three-fold excess of testicular cancer risk in the highest category of estimated annual or cumulative serum PFOA exposure. CitationGilliland and Mandel (1993) found an elevated, statistically nonsignificant SMR for testicular cancer among chemical division workers at Cottage Grove, but this estimate was based on only one testicular cancer death. Otherwise, no apparent associations with testicular cancer mortality were reported in other studies (CitationConsonni et al. 2013, CitationLeonard et al. 2008, CitationLundin et al. 2009, CitationSteenland and Woskie, 2012). Thus, the associations reported in the Parkersburg community were not detected consistently in other study groups, including Parkersburg workers. However, cohort mortality studies are not well suited for assessing the risk of testicular cancer due to the high survival from this disease.
A positive association between PFOA exposure and kidney cancer mortality was detected in both studies of workers at the Parkersburg facility, with a 30–80% excess among Parkersburg workers compared with other regional DuPont workers, and a nearly threefold excess in the highest quartile of estimated cumulative serum PFOA level (CitationLeonard et al. 2008, CitationSteenland and Woskie, 2012). This excess was based completely on the 12 kidney cancer deaths reported by CitationLeonard et al. (2008) during the first follow-up period (1948–2002), as none were observed during the extended follow-up period (2003–2008), representing a nonsignificant deficit. In a pooled analysis that included the Parkersburg cohort, CitationConsonni et al. (2013) found a nonsignificant twofold excess of kidney cancer mortality in the highest category of cumulative APFO exposure, but this statistically unstable estimate was based on only four deaths (none with low TFE exposure). In the cancer-registry-based study of Ohio residents near Parkersburg, kidney cancer risk was also doubled in the two highest categories of estimated annual serum PFOA exposure (CitationVieira et al. 2013). However, results for community members in the cohort study of regional residents near Parkersburg were variable, with a doubling of kidney cancer risk among those in the third and fourth quartiles of estimated cumulative serum PFOA with no lag period, but no such excess after a 10-year lag (CitationBarry et al. 2013). In the same study, Parkersburg plant workers had a three- to four-fold excess of kidney cancer in the third quartile of estimated cumulative serum PFOA, but not in the highest quartile. By contrast, CitationLundin et al. (2009) detected a nonsignificant deficit of kidney cancer mortality (SMR = 0.53) among Cottage Grove workers with ever probable/never definite PFOA exposure and no kidney cancer deaths among those with ever definite exposure.
CitationSteenland and Woskie (2012) observed a more than two-fold excess of bladder cancer mortality among workers in the second-lowest category of estimated cumulative PFOA exposure at the Parkersburg plant. This finding was not supported by the results of other studies, all of which reported convincingly null RR estimates (CitationBarry et al. 2013, CitationConsonni et al. 2013, CitationEriksen et al. 2009, CitationGilliland and Mandel, 1993, CitationLeonard et al. 2008, CitationLundin et al. 2009, CitationVieira et al. 2013).
CitationLeonard et al. (2008) reported a six-fold excess of thyroid and other endocrine gland cancer based on three deaths among workers at the Parkersburg facility, compared with other regional DuPont workers. Surprisingly, results for thyroid cancer were not reported in the update of this cohort (CitationSteenland and Woskie, 2012). Using the same exposure model, CitationBarry et al. (2013) found a borderline significant twofold excess of thyroid cancer among workers at the Parkersburg plant with no lag, but not after a 10-year lag or in community members. Analyses by quartile of estimated cumulative serum PFOA level yielded high HR estimates for thyroid cancer in workers, but these were statistically unreliable and nonsignificant. An increasing trend was detected with occupational but not community PFOA exposure. In the same geographic region, CitationVieira et al. (2013) found no association between residential water district or estimated annual serum PFOA level and thyroid cancer risk. CitationLundin et al. (2009) reported a single thyroid cancer death in a worker with no occupational PFOA exposure.
The 40% reduction in colorectal cancer diagnosis associated with the highest quartile of serum PFOA in community members around the Parkersburg plant (CitationInnes et al. 2014) was mirrored in a significantly lower rate of colon cancer mortality in the pooled analysis of TFE workers (CitationConsonni et al. 2013) and nonsignificant inverse associations among Parkersburg plant workers (CitationLeonard et al. 2008) and residents of the most highly PFOA-contaminated water districts around the plant (CitationVieira et al. 2013). Other reported associations with colon, rectal, or colorectal cancer, however, were close to the null (CitationBarry et al. 2013, CitationGilliland and Mandel, 1993, CitationLundin et al. 2009).
Overall, there was no consistent finding across all or even most studies. Perhaps the only positive association that showed some consistency across multiple studies is that with kidney cancer. However, it should be recognized that all of the studies that observed a positive association between estimated PFOA exposure and kidney cancer risk or mortality were based at the Parkersburg plant or in the community surrounding the Parkersburg plant [or, in the case of CitationConsonni et al. (2013), in a study cohort that comprised largely Parkersburg workers] (CitationBarry et al. 2013, CitationConsonni et al. 2013, CitationLeonard et al. 2008, CitationSteenland and Woskie, 2012, CitationVieira et al. 2013). The three occupational study groups overlapped substantially (CitationConsonni et al. 2013, CitationLeonard et al. 2008, CitationSteenland and Woskie, 2012), as did the two community study groups (CitationBarry et al. 2013, CitationVieira et al. 2013), in which the same exposure estimation model was applied. Thus, the results of these studies do not constitute independent replications. The only study that reported on kidney cancer outside of the Parkersburg region (CitationLundin et al. 2009) found that kidney cancer mortality was nonsignificantly lower than expected among workers who were probably directly exposed to PFOA, with no kidney cancer deaths among definitely exposed workers. These findings call into question the consistency and generalizability of the observed kidney cancer association.
Exposure-response gradient
The observation of a monotonic exposure-response relationship, where disease frequency increases unidirectionally, albeit not necessarily linearly, in concert with increasing exposure level, can strengthen the evidence in favor of a causal association (CitationHill, 1965). Among the studies that examined cancer risk across increasing levels of PFOA exposure, few monotonic exposure-response gradients were detected. In the cohort of Cottage Grove facility workers, CitationGilliland and Mandel (1993) found a positive relationship between increasing duration of employment in the chemical division and prostate cancer mortality. In the updated analysis of this cohort, this finding was echoed in a positive trend toward increasing prostate cancer mortality with higher estimated occupational exposure to PFOA when categorized by job classification (CitationLundin et al. 2009), although the apparent gradient may have been an artifact of the lower-than-expected prostate cancer mortality in the nonexposed group. Moreover, the association between estimated cumulative PFOA exposure and prostate cancer mortality was not monotonic, as risk was lower in the middle than in the lowest category. Other studies that examined the exposure-response relationship between PFOA exposure and prostate cancer risk or mortality did not detect an apparent pattern (CitationBarry et al. 2013, CitationEriksen et al. 2009, CitationSteenland and Woskie, 2012, CitationVieira et al. 2013). In fact, the Parkersburg worker cohort had a monotonically decreasing trend in prostate cancer mortality with increasing exposure (CitationSteenland and Woskie, 2012).
Among workers at the Parkersburg facility, CitationSteenland and Woskie (2012) reported positive exposure-response relationships between estimated annual and/or cumulative serum PFOA level and mortality from mesothelioma and kidney cancer, but not other malignancies. When CitationBarry et al. (2013) categorized estimated cumulative serum PFOA concentration into quartiles, they observed a positive trend in the HR for kidney cancer among community members, but not among workers, for whom the lowest HR was detected in the highest exposure quartile. CitationVieira et al. (2013) found that the OR for kidney cancer increased with higher estimated serum PFOA levels in Ohio residents, but not with higher water-district-level average serum PFOA concentration in the residents of both Ohio and West Virginia. CitationConsonni et al. (2013) did not detect a robust monotonic trend between estimated cumulative PFOA exposure and kidney cancer mortality in pooled TFE workers, and CitationLundin et al. (2009) observed no exposure-response trend between job-level PFOA exposure and kidney cancer mortality in the Cottage Grove cohort.
For liver cancer, the suggestion of a positive trend with estimated cumulative PFOA exposure among pooled TFE workers (CitationConsonni et al. 2013) was contradicted by the lack of an apparent exposure-response trend in Cottage Grove workers (CitationLundin et al. 2009), Parkersburg workers (CitationSteenland and Woskie, 2012), the Parkersburg regional community (CitationBarry et al. 2013, CitationVieira et al. 2013), and Danish community members (CitationEriksen et al. 2009).
CitationBarry et al. (2013) detected positive exposure-response gradients between estimated cumulative serum PFOA level and risk of testicular cancer in community members with and without a 10-year lag, but not in Parkersburg workers. CitationVieira et al. (2013) reported increased testicular cancer risk only in the most highly contaminated water district and the highest category of annual or cumulative serum PFOA level, with nonsignificant deficits of testicular cancer in all other categories and no apparent monotonic trend. The apparent increasing trend in ovarian cancer risk with higher estimated serum PFOA exposure among Ohio residents near Parkersburg (CitationVieira et al. 2013) was counterbalanced by the lack of any trend in the cohort study based in the same region (CitationBarry et al. 2013). A positive trend toward higher thyroid cancer risk with increasing estimated cumulative serum PFOA was detected among occupationally exposed workers at the Parkersburg plant, but not among community members (CitationBarry et al. 2013, CitationVieira et al. 2013) – although such a pattern could be consistent with a monotonic trend only above a certain threshold level of serum PFOA. Also in the community around the Parkersburg plant, CitationInnes et al. (2014) detected a statistically significant inverse trend between serum PFOA and colorectal cancer prevalence, but others did not observe such a trend (CitationBarry et al. 2013, CitationLundin et al. 2009, CitationVieira et al. 2013).
When considering exposure-response gradients, it is important to recognize that the magnitude of probable exposure to PFOA differs substantially among occupational and community groups. As shown in , median serum PFOA levels among directly exposed fluorochemical workers at the Parkersburg plant in 1979–2004 (CitationWoskie et al. 2012), the Cottage Grove plant in 1993–1997 (CitationOlsen et al. 2000), the Decatur, Alabama, plant in 1998 (where levels were reported as the geometric mean, which is generally close to the median in studies that reported both) (CitationOlsen et al. 2003), and the Cottage Grove, Decatur, and Antwerp, Belgium, plants in 2000 (CitationOlsen and Zobel, 2007) ranged from approximately 1,000 to 2,880 ng/mL (1–2.88 ppm). By contrast, median serum PFOA levels were approximately 15–30% as high among intermittently directly exposed workers and 5–10% as high among indirectly (background) exposed workers in Parkersburg (CitationWoskie et al. 2012), and geometric mean levels were 5% as high among background-exposed film division workers in Decatur (CitationOlsen et al. 2003). Median serum PFOA concentrations among residents of the six PFOA-contaminated public water districts in Ohio and West Virginia near the Parkersburg plant in 2005–2006 were generally between 20 and 40 ng/mL, depending on age group and sex (CitationFrisbee et al. 2009), a level comparable to the background exposure level at the Decatur plant. Median serum PFOA levels were an order of magnitude lower among participants in the US population-based National Health and Nutrition Examination Survey (NHANES) in 1999–2008 (CitationKato et al. 2011) and among American Red Cross adult volunteer blood donors in 2000–2010 (CitationOlsen et al. 2012), with declining levels over time.
Figure 1. Median (or geometric mean) serum levels of perfluorooctanoic acid (PFOA) measured in directly, intermittently, and indirectly (background) exposed workers (Parkersburg, West Virginia; Cottage Grove, Minnesota; Decatur, Alabama; and Antwerp, Belgium) and in community members in Parkersburg and elsewhere in the United States. PTFE: polytetrafluoroethylene.
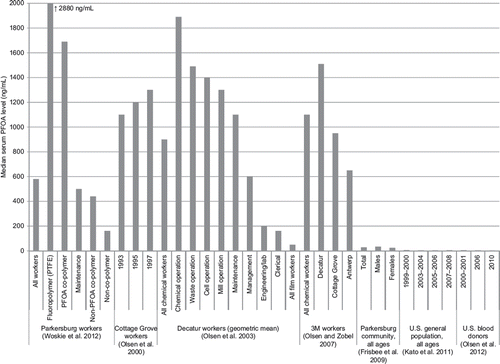
Thus, average exposure to PFOA differed by up to two orders of magnitude between directly exposed workers and nonoccupationally exposed community members, and by another order of magnitude between directly exposed workers and indirectly exposed workers or residents near the Parkersburg plant (). However, many of the positive associations with cancer outcomes were observed with environmental rather than occupational exposures to PFOA (CitationBarry et al. 2013, CitationVieira et al. 2013). This pattern might be explained by greater statistical power in the community-based studies, or by chance, confounding, and/or bias. In light of the fact that most SMR and RR point estimates in occupational studies were close to unity, insufficient statistical power cannot be the only reason for the generally null findings. Instead, chance, confounding, and bias (with an unknown degree and direction of impact) are more plausible explanations for the apparently stronger associations in less-exposed study groups.
Plausibility and coherence with toxicological evidence
Although animal toxicology data on PFOA are not readily translated to humans, a causal interpretation of an observed association may be better justified if it is coherent with laboratory evidence (CitationHill, 1965). Such evidence can also support the biological plausibility of a causal hypothesis (CitationHill, 1965). A priori, based on the results of experimental animal studies, the organs of greatest concern with respect to a potential carcinogenic effect of PFOA are the liver, testis (Leydig cells), and pancreas (acinar cells). However, no convincing associations with malignancies affecting any of these organs have been observed in epidemiologic studies of humans. Only testicular cancer has been associated with PFOA exposure in any of these studies (CitationBarry et al. 2013, CitationVieira et al. 2013), with ambiguous exposure-response trends. On the other hand, given the relatively poor site concordance between animals and humans for many known human carcinogens, the lack of associations between PFOA exposure and liver, testicular, and pancreatic cancers among humans does not constitute evidence against human carcinogenicity of PFOA; rather, it provides no evidence to support such an effect.
Of note, nearly all testicular cancers in humans are of germ-cell origin, with Leydig cell tumors constituting only an estimated 1–3% of testicular malignancies (CitationSarma et al. 2006). Therefore, it is questionable whether a positive association between PFOA exposure and testicular cancer risk in humans, even if well established, could accurately be described as being coherent with the finding of excess Leydig cell adenomas in rats fed with PFOA. Likewise, pancreatic acinar cell carcinomas account for only approximately 1% of pancreatic exocrine tumors in humans (CitationKlimstra et al. 1992), and mammary fibroadenomas [which were not significantly increased in rats fed with PFOA (CitationHardisty et al. 2010)] are not precursors of breast cancer or indicators of increased breast cancer risk in humans (CitationFitzgibbons et al. 1998).
TFE – which was used to manufacture fluoropolymers in the Parkersburg plant (CitationSteenland and Woskie, 2012) and five European plants (CitationConsonni et al. 2013), but not the Cottage Grove plant – is a kidney, liver, hematopoietic, and possibly testicular carcinogen in rodents. Specifically, 2-year whole-body inhalation exposure resulted in significant increases in renal tubule adenoma, renal tubule adenoma and carcinoma combined, hepatocellular adenoma, HCC, liver hemangiosarcoma, and mononuclear cell leukemia, as well as slight increases in testicular interstitial cell adenoma, in F344/N rats (CitationNational Toxicology Program, 1997). In B6C3F1 mice, the same exposure resulted in significant increases in liver hemangioma, liver hemangiosarcoma, hepatocellular adenoma, HCC, and histiocytic sarcoma of the liver, lung, spleen, lymph nodes, bone marrow, and kidney (CitationNational Toxicology Program, 1997). Thus, although epidemiologic data on TFE are inconclusive, animal toxicology data are coherent with the hypothesis that TFE, which was highly correlated with PFOA at the Parkersburg facility and at the six combined US and European facilities in the pooled analysis (CitationConsonni et al. 2013, CitationSteenland and Woskie, 2012), was responsible for the apparent positive association between PFOA exposure and kidney cancer mortality in these study groups. As stated by CitationConsonni et al. (2013), toxicological evidence in animals suggests that TFE could also have contributed to the modest, statistically nonsignificant excesses of liver cancer, testicular cancer, and leukemia mortality observed in the pooled TFE cohorts, as well as in some comparisons in the Parkersburg cohort (CitationLeonard et al. 2008, CitationSteenland and Woskie, 2012). Given that the Cottage Grove facility manufactured PFOA but did not use it for polymer production, TFE probably was not used in Cottage Grove, and its absence could plausibly explain the lack of excess kidney cancer mortality in that worker cohort (CitationLundin et al. 2009).
Occupational studies of PFOS
Overview
To date, all epidemiologic studies of cancer risk in association with occupational exposure to PFOS have been conducted at a 3M facility in Decatur, Alabama, that manufactured PFOS-based fluorochemicals in its chemical division between 1961 and 2002 (CitationAlexander and Olsen, 2007, CitationAlexander et al. 2003, CitationGrice et al. 2007, CitationOlsen et al. 2004). Details of the four studies conducted at this facility are provided in and . Worldwide, PFOS-based fluorochemicals were produced mostly at the Decatur facility and one other facility in Belgium (CitationPrevedouros et al. 2006), where cancer risk or mortality has not been studied. Of note, PFOA is a residual by-product of PFOS production (CitationSigurdson et al. 2003); therefore, chemical workers were potentially occupationally exposed to PFOA, as well as to other fluorochemicals and nonfluorochemicals. A 1998 biomonitoring study of randomly selected employees at the Decatur plant (with 80% participation) found that geometric mean serum levels of PFOS, PFOA (of which levels were slightly lower than PFOS levels), perfluorohexanesulfonate, N-ethyl perfluorooctanesulfonamidoacetate, N-methyl perfluorooctanesulfonamidoacetate, perfluorooctanesulfonamide, and perfluorooctanesulfonamidoacetate were approximately one order of magnitude higher in 126 chemical division workers than in 60 film division workers (CitationOlsen et al. 2003). Because the Decatur plant primarily manufactured PFOS-based chemicals, this plant has been studied only with respect to occupational PFOS exposure.
Like the studies of the Cottage Grove and Parkersburg facilities, studies of the Decatur facility are strengthened by complete cohort enumeration and availability of job records, but limited by potential exposure misclassification, modest numbers of subjects, and possible confounding. Despite these limitations, the studies of occupational PFOS exposure again provide the best available epidemiologic evidence on the association between high average and cumulative PFOS exposure and cancer risk in humans.
Studies of the Decatur, Alabama, facility
In a retrospective cohort mortality study, CitationAlexander et al. (2003) identified 2,083 workers employed in the chemical division and/or film division (located approximately 300 yards from the chemical division) at the Decatur plant for at least 365 days between 1961 and the end of 1997. Follow-up for vital status continued through 1998 by linkage to the National Death Index, Social Security Administration data, and/or Social Security Death Index. Cause of death was obtained and coded from death certificates for 96% of the decedents. The 1998 serum study mentioned earlier found that the geometric mean serum PFOS level was 941 ng/mL for chemical plant workers and 136 ng/mL for film plant workers, most of whom had no direct occupational exposure to fluorochemicals (CitationOlsen et al. 2003). [By comparison, contemporaneous geometric mean serum PFOS levels in the general population were approximately 30–35 ng/mL (CitationKato et al. 2011, CitationOlsen et al. 2012).] Among chemical plant workers, the highest geometric mean serum PFOS levels were measured in cell operators (2,000 ng/mL), followed by waste operators (1,500 ng/mL), chemical operators (1,500 ng/mL), maintenance workers (1,300 ng/mL), supervisors/managers (900 ng/mL), mill operators (600 ng/mL), engineers/lab workers (400 ng/mL), and administrative assistants (400 ng/mL). Based on these results, and the knowledge that production processes were constant over time, a company industrial hygienist and epidemiologist created a simple job-exposure matrix with three exposure categories: no workplace exposure to PFOS-based fluorochemicals (including film division jobs; 39% of the study cohort), low potential exposure (including engineers, quality control technicians, administrative assistants, managers, and environmental, health, and safety workers; 14% of the study cohort), and high potential exposure (including cell operators, chemical operators, maintenance workers, mill operators, waste operators, and crew supervisors; 47% of the study cohort).
After a median follow-up of 25.9 years, 145 deaths had occurred, including 65 among workers ever employed in a high-exposure job, 27 among workers ever employed in a low-exposure job but never a high-exposure job, and 53 among workers employed only in a no- or minimal-exposure job (CitationAlexander et al. 2003). The median duration of employment was 16.7 years in the high-exposure group, 10.4 years in the low-exposure group, and 9.9 years in the no-exposure group. The total cancer mortality rate in the overall cohort was significantly lower than expected based on Alabama rates (SMR = 0.72 [0.51–0.98]) (). No statistically significant SMRs were detected for site-specific cancers, but a borderline significant excess of bladder and other urinary organ cancer mortality was detected in the overall cohort. This excess was statistically significant when the analysis was limited to high-exposure workers, among whom all three deaths from bladder cancer occurred (SMR = 12.77 [2.63–37.35]). When the analysis was restricted to workers employed for at least 1 year in a high-exposure job, including all three workers who died from bladder cancer, the SMR was even higher (SMR = 16.12 [3.32–47.14]). The three subjects who died from bladder cancer had worked mostly in maintenance or in the plant incinerator or wastewater treatment plant. Results were similar when SMRs were calculated using the 23-county regional population as the reference group. No significant excess of overall or site-specific cancer mortality was detected in the low-exposure and no-exposure groups.
Strengths and limitations of this study (CitationAlexander et al. 2003) are similar to those of the occupational cohort studies of PFOA described earlier. Although chance may explain the three deaths from bladder cancer, the very high SMRs and the fact that all three deaths occurred among long-term high-exposure workers provide cause for further inquiry. The authors reported that a review of known or potential bladder carcinogens yielded a list of five compounds currently or formerly used at the Decatur facility. Four of these (4,4- methylene-dianiline, orthotoluidine, benzidine salts, and butyl benzyl phthalate) had not been used since the 1960s and 1970s, during which time they were not widely used, but had limited information on exposure monitoring and use. The other compound, melamine, was currently in use in a nonfluorochemical product line, with low anticipated exposures based on a qualitative exposure assessment that found short exposure task durations. Given that four of these compounds were phased out by the 1970s, an analysis of date of first employment of the workers who died from bladder cancer might have clarified the potential for confounding. In the absence of such information, these alternative causes cannot be ruled out as plausible explanations for the observed excess of bladder and other urinary tract mortality in this cohort.
To further investigate the association between PFOS exposure and bladder cancer in workers at the Decatur facility, CitationAlexander and Olsen (2007) sought to identify additional bladder cancer cases in the same cohort of workers employed for at least 365 days by the end of 1997. In 2002, following informational meetings with current employees and retirees, a self-administered questionnaire was mailed to all living members of the cohort to report a past diagnosis of bladder cancer and smoking history; nonrespondents were also contacted by telephone. Overall, the response rate to the questionnaire was 74% (1,400 of 1,895) with 24% refusing to participate and 2% lacking a valid contact address or phone number; response rates were 75.8% in the no-exposure group, 81.4% in the group with low exposure only or high exposure for less than 1 year, and 67.2% in the group with high exposure for at least 1 year. Participants who reported bladder cancer were recontacted to seek permission for physician contact to verify the diagnosis. Deaths from bladder cancer were also ascertained from death certificates that were obtained for 185 (98%) of the 188 decedents.
Potential exposure to PFOS was classified as no/minimal exposure, low exposure, or high exposure using the same approach as described by CitationAlexander et al. (2003). To estimate cumulative exposure, the exposure categories were assigned weights of 1, 3, and 10, respectively, and multiplied by the number of years spent in each job (CitationAlexander and Olsen, 2007). In the cohort of questionnaire respondents and workers who had died by the end of follow-up in 2002, standardized incidence ratios (SIRs) were estimated in comparison with age, sex, and calendar-year-specific reference cancer incidence rates from the US Surveillance, Epidemiology, and End Results cancer registries for 1970 (the revised start of study follow-up) through 1999, with referent rates for 1999 applied to 2000–2002. In addition, associations with categories of time-dependent estimated cumulative PFOS exposure within the cohort were estimated using Poisson rate ratios adjusted for age and sex. In a sensitivity analysis to evaluate potential selection bias due to nonparticipation, the number of expected bladder cancer cases among nonrespondents in each exposure category was estimated using incidence rates, assuming a twofold excess of bladder cancer among nonrespondents.
Five bladder cases were identified from death certificates and six others were reported on the questionnaire, including two that were confirmed and four that lacked consent for validation (CitationAlexander and Olsen, 2007). Two had never worked in PFOS-exposed areas, while six of the nine who had ever worked in a low- or high-exposure job had worked for at least 1 year in these jobs. Only three subjects with bladder cancer had worked in a high-exposure job for at least 1 year. SIRs were estimated separately for workers who never had an exposed job, ever had a low-exposure job, ever had a high-exposure job, ever had a low- or high-exposure job, had a low- or high-exposure job for at least 1 year, or had a high-exposure job for at least 1 year (). The SIR was highest in the group ever employed in a low-exposure job (7 cases observed; SIR = 2.26 [0.91–4.67]) and not substantially or significantly elevated in the group employed for at least 1 year in a high-exposure job (3 cases observed; SIR = 1.12 [0.23–3.27]). In an analysis by cumulative exposure, the SIR did not follow a monotonic exposure-response trend with increasing years of employment in the equivalent of high-exposure jobs. Likewise, a monotonic exposure-response trend was not detected across categories of estimated cumulative PFOS exposure. In the sensitivity analysis, assuming that the equivalent of nearly two additional bladder cancer cases was expected among nonrespondents, estimated SIRs remained statistically nonsignificant with no evidence of a positive exposure-response trend.
Although this study is strengthened by its setting in a well-defined occupational cohort and its use of serum PFOS data as the basis for exposure classification, it has several limitations (CitationAlexander and Olsen, 2007). Perhaps the most important consideration is whether bias resulted from underascertainment of bladder cancer due to nonparticipation in the study survey, underreporting of past bladder cancer among study participants, and/or failure to report bladder cancer as the underlying cause of death on death certificates. The authors performed a sensitivity analysis that accounted for case underascertainment among the 495 nonparticipants and found little change in the results. Minimal bias might also be expected from case underascertainment among the 183 decedents without reported bladder cancer. It is worth noting that in the study by CitationBarry et al. (2013), 96.5% of 115 self-reported bladder cancer cases were validated, and 100% of 50 self-reported bladder cancer cases in a US study of radiologic technologists were validated (CitationSigurdson et al. 2003). Thus, a self-reported diagnosis of bladder cancer appears to have high specificity, but the sensitivity remains unknown. The questionnaire participation rate was significantly higher among workers with any PFOS exposure than those with no exposure, suggesting potential bias toward greater underreporting in the nonexposed, which would have resulted in higher RRs for exposed versus nonexposed. However, among exposed workers, the participation rate was greater among those with low exposure than those with high exposure for at least 1 year, a difference that could have accounted for the higher RR in low-exposure than in high-exposure groups. Another limitation is the inadequate adjustment for confounding, especially by bladder cancer risk factors such as smoking, which was more common among workers with higher cumulative PFOS exposure than those with lower PFOS exposure, and therefore may have been a positive confounder in the RR analyses. These results do not conclusively rule out a positive association between PFOS and bladder cancer risk, but they also do not confirm the excess risk of bladder cancer mortality previously reported among highly exposed workers at the Decatur plant (CitationAlexander et al. 2003).
As part of a “qualitative screening evaluation of the health experience” of the Decatur facility workforce, CitationOlsen et al. (2004) analyzed health claims data for 652 chemical division employees and 659 film division employees (96% of eligible employees) from 1993 through 1998. The cohort comprised all full-time and inactive workers (including those on short- or long-term disability) employed at the Decatur site for at least 1 year as of 1 January 1993, with continued follow-up through 1998. The distribution of workers by work status was comparable between the chemical and film divisions. Health claims were grouped into “episodes of care,” which were defined as sets of one or more claims data records that were categorized into discrete disease entities by a computerized algorithm based on diagnosis codes, revenue, procedure codes, and drug codes, taking into account all inpatient and outpatient visits, procedures, ancillary services, and prescription drugs used in the diagnosis, treatment, and management of more than 400 diseases or conditions. Potential exposure to PFOS was classified based on job records, with each worker assigned a job title that best described his or her usual job activity. One set of analyses compared all 652 chemical division workers with all 659 film division workers. To reduce exposure misclassification, a second set of analyses compared 211 workers who had high-exposure jobs in the chemical division for at least 10 years prior to the study with 345 workers who had similar but unexposed task-like jobs in the film division for at least 10 years prior to the study. For each division, the observed number of health claims was compared with an expected value based on all other 3M manufacturing workers in the United States (approximately 20,000 workers), using indirect standardization to adjust for age and sex. An RR estimate was then calculated based on the ratio of the two SIRs, referred to by the authors as the “risk ratio episodes of care” and simplified here to “RR.”
On average, chemical division employees underwent 2.7 episodes of care per person per year, whereas film division employees underwent 3.0 episodes of care per person per year (CitationOlsen et al. 2004). Among long-term workers, chemical division employees underwent an average of 3.1 episodes of care per person per year versus 3.3 in the film division. RRs were higher in the chemical division than in the film division for all malignant neoplasms, but estimates were imprecise (). The only significant difference observed was for malignant melanoma of the skin, for which there were five episodes of care in chemical workers (versus 2.2 expected) and none in film workers (versus 2.6 expected), for an RR of 12 ([1.0–> 100]; calculated by considering 0.5 deaths as observed episodes of care among film workers). A marginally significant excess of prostate cancer was also observed among chemical workers, who had five episodes of care (versus 3.1 expected), compared with film workers, who had one episode of care (versus 4.7 expected; RR = 7.7 [0.9–> 100]). Of note, one film worker [who was not one of the three decedents identified by CitationAlexander et al. (2003)] and no chemical workers underwent an episode of care for bladder cancer. No significant findings were observed in the analysis restricted to long-term workers. A statistically significant excess of episodes of care for benign colonic polyps was observed among chemical workers (RR = 2.4 [1.3–4.5]).
This study benefited from the ability to compare workers in a single facility with stark differences in potential PFOS exposure, with further reduction of exposure misclassification in the analysis restricted to long-term workers (CitationOlsen et al. 2004). Nevertheless, due to the limitations of using health claims data to define outcomes, the authors appropriately cautioned that the analysis “should only be considered a screening study for diseases and conditions and not a definitive measure of risk” (CitationOlsen et al. 2004). Medical history prior to study entry was not taken into account, and episodes of care could not be interpreted as indicators of incident rather than prevalent or recurrent disease, some of which may have preceded employment at the Decatur facility. The authors also noted that episodes of care are not equivalent to definitive diagnoses. An additional limitation is the relatively short follow-up period, as a longer study period might have enabled classification of diseases that were likely to be newly diagnosed. The slightly higher average number of episodes of care per person among film workers than among chemical workers suggests that systematic differences in care-seeking patterns could have resulted in underestimated RRs. However, the authors noted that “in 1997 there was heightened awareness for colon cancer screening among chemical plant employees,” which may have explained at least part of the increased frequency of episodes of care for benign colonic polyps and colorectal cancer among chemical plant employees. Thus, care-seeking patterns may have varied by health outcome, with different directions and magnitudes of bias. Overall, the results of this study must be considered as hypothesis-generating and only minimally informative regarding a potential causal association between PFOS exposure and cancer risk.
Using the same methods as CitationAlexander and Olsen (2007), CitationGrice et al. (2007) conducted a case-control study of self-reported outcomes other than bladder cancer among current, retired, and former workers employed for at least 1 year at the Decatur facility. As described earlier, 1,400 (74%) of 1,895 living active, retired, and former employees who had worked for at least 1 year at the Decatur facility completed a questionnaire on selected diseases and health conditions. Permission was sought to obtain medical records for validation of self-reported diagnoses of prostate cancer, colon cancer, breast cancer, and melanoma. Most self-reported prostate cancers (22 of 29) and about half of the colon cancers (12 of 22) were confirmed with medical records. Other than one self-reported prostate cancer that was reported by the physician not to be cancer, the remaining self-reported prostate and colon cancers were unvalidated due to a lack of patient consent for medical records release or physician inability to retrieve the records. Of 39 self-reported melanomas, medical records were obtained for 22, and only 8 of these were confirmed as melanoma, whereas 12 were nonmelanoma skin cancers and 2 were noncancerous lesions. Given the high validation rate for prostate and colon cancers and the low validation rate for melanoma, self-reported diagnoses were analyzed for the first two outcomes, but only confirmed diagnoses were analyzed for melanoma. Cancers reported on decedents’ death certificates were also included in the analysis under the assumption that these reports were valid. Exposure classification was based on the same approach as used by CitationAlexander et al. (2003) and CitationAlexander and Olsen (2007).
No significant association or apparent monotonic exposure-response trend was detected between categories of potential workplace PFOS exposure (never, ever low- or high-exposure, low or high exposure for at least 1 year, or high exposure for more than 1 year) and risk of validated melanoma, self-reported prostate cancer, or self-reported colon cancer () (CitationGrice et al. 2007). Comparable results were obtained when only validated prostate and colon cancers and self-reported melanomas were evaluated. No significant associations were found with estimated cumulative PFOS exposure calculated using relative weights assigned to each exposure category. Four cases of breast cancer, no cases of liver cancer, and no cases of thyroid cancer were self-reported; these cancers were not analyzed as outcomes.
As in the study by CitationAlexander and Olsen (2007), underascertainment of cancer diagnoses among survey nonparticipants is unlikely to have substantially affected the results of CitationGrice et al. (2007). Thus, even though workers with at least 1 year of high exposure had the lowest participation rate (and those with low exposure or less than 1 year of high exposure had the highest participation rate), thereby potentially obscuring exposure- response trends, the magnitude of bias was probably small. The high positive predictive value of self-reported prostate, colon, and breast cancers are in line with the findings of CitationBarry et al. (2013), who reported that 88.9% of 515 self-reported prostate cancers, 88.7% of 311 self-reported colorectal cancers, and 95.6% of 608 self-reported breast cancers were confirmed by medical records or cancer registry documentation; confirmation rates were even higher (96.2%, 92.9%, and 96.8%, respectively) for patients with retrievable records. Like CitationGrice et al. (2007), CitationBarry et al. (2013) found a low positive predictive value for self-reported melanoma (47.2% confirmed of all self-reported cases; 59.1% confirmed of those with records). However, the negative predictive value of self-reported data on these cancers is unknown. Overall, these results do not demonstrate an association between PFOS exposure and risk of prostate cancer, colon cancer, or melanoma.
Community studies of PFOS
Overview
All six studies of cancer risk in relation to nonoccupational exposure to PFOS were described earlier in the section on community studies of PFOA (CitationBonefeld-Jorgensen et al. 2011, CitationEriksen et al. 2009, CitationHardell et al. 2014, CitationInnes et al. 2014, CitationVassiliadou et al. 2010, CitationYeung et al. 2013). Therefore, the study methods, strengths, and limitations are not described again in this section.
In the cross-sectional analysis of serum PFASs in colorectal cancer cases and controls from the Mid-Ohio Valley, CitationInnes et al. (2014) found a significant inverse association between serum PFOS and colorectal cancer prevalence in nearly all reported statistical models. For example, in the fully adjusted model, the OR for the highest quartile (≥ 29.2 ng/mL) versus the lowest quartile (0.25–13.5 ng/mL) of serum PFOS was 0.24 (0.16–0.37), with a highly significant inverse trend (P-trend < 0.00001) and a significant decrement in colorectal cancer prevalence per 1-ng/mL increase in continuous serum PFOS (OR = 0.96 [0.95–0.97]; P-trend < 0.00001). The significant inverse association was detected after stratification by sex, body mass index, age, or colorectal cancer treatment method, but it was more pronounced in cases diagnosed in 2000 and later than in those diagnosed earlier. The inverse association also persisted after restriction to participants who had lived at the same address since 1990–1995 or before and to cases diagnosed in 2000 or 2005–6 or later, restriction to participants with serum PFOS ≤ 20 ng/mL, exclusion of primary rectal cancer cases, those undergoing current treatment, or those who had received chemotherapy, or inclusion of all self-reported cases. These findings point to a strong inverse association between serum PFOS around or after the time of colorectal cancer diagnosis, but the timing of serum collection after cancer diagnosis precludes an interpretation of a protective effect.
In the Danish case-cohort study, CitationEriksen et al. (2009) reported median plasma PFOS concentrations of 36.8 (5th to 95th percentiles = 18.2–62.5) ng/mL in prostate cancer cases, 32.3 (15.2–58.0) ng/mL in bladder cancer cases, 32.7 (15.2–56.4) ng/mL in pancreatic cancer cases, 31.0 (15.8–62.9) ng/mL in liver cancer cases, and 34.3 (16.2–61.8) ng/mL in the noncancer subcohort. Plasma PFOS and PFOA concentrations were highly correlated (Spearman ρ = 0.70). No statistically significant associations were detected between plasma PFOS, whether categorized in quartiles or expressed as a continuous variable, and risk of bladder, pancreatic, or liver cancer, with RRs at or below the null for the highest quartile of plasma PFOS for all three malignancies (). Positive associations were detected between the second, third, and fourth quartiles of plasma PFOS and risk of prostate cancer (RR for highest quartile = 1.38 [0.99–1.93]). However, no apparent exposure-response trend was detected (RR for a 10-ng/mL increase in plasma PFOS = 1.05 [0.97–1.14]), suggesting that the positive associations were attributable to the lower risk of prostate cancer in the bottom quartile, which, in turn, might be due to chance or a threshold effect. Overall, these findings indicate no association between low-level nonoccupational exposure to PFOS and short- to intermediate-term risk of bladder, pancreatic, or liver cancer, whereas the potential evidence of a threshold association with risk of prostate cancer requires confirmation in other studies.
CitationBonefeld-Jorgensen et al. (2011) observed a median serum PFOS level of 45.6 (range = 11.6–124) ng/mL among 31 breast cancer patients and 21.9 (range = 1.5–172) ng/mL among 98 controls in Greenland. In both unadjusted models (OR per 1-ng/mL increase in serum PFOS = 1.01 [1.003–1.02] including all subjects; OR = 1.01 [0.99–1.03] including subjects with covariate data) and an adjusted model (OR = 1.03 [1.001–1.07]), a borderline significant positive association was detected with breast cancer risk (). The same was true for the sum of perfluorosulfonated acids, which included PFOS along with perfluorohexane sulfonate and perfluorooctane sulfonamide (unadjusted OR = 1.013 [1.002–1.023] for all subjects; unadjusted OR = 1.01 [0.99–1.02] for subjects with covariate data; adjusted OR = 1.03 [1.00–1.05]). These findings provide weak evidence of an association, potentially explained by bias or chance, between nonoccupational PFOS exposure and breast cancer risk in Greenland Inuit women.
In the Swedish case-control study of prostate cancer, CitationHardell et al. (2014) reported that the median concentration of PFOS in whole blood was 9.0 (range = 1.4–69) ng/mL among cases and 8.3 (range = 1.7–49) ng/mL among controls. Elevated blood PFOS above the median among controls was not associated with risk of prostate cancer overall, nor was it significantly associated with risk of low-grade or high-grade prostate cancer, or risk of prostate cancer with PSA ≤ 10 or ≥ 11 ng/mL (). When cases and controls were cross-classified according to their first-degree family history of prostate cancer and blood PFOS concentration, a significantly increased risk was detected among those with both (OR = 2.7 [1.04–6.8]), relative to those with neither. Again, however, family history unexpectedly was not significantly associated with increased risk among those with lower blood PFOS levels, raising concerns about chance and bias as explanations for the results. Overall, the findings suggest no association between nonoccupational PFOS exposure and risk of prostate cancer.
CitationVassiliadou et al. (2010) found no apparent difference in median serum PFOS concentrations among cancer patients (median = 11.33 ng/mL, range = 4.98–26.38 ng/mL in 17 males; median = 8.00 ng/mL, range = 2.12–25.70 ng/mL in 23 females), Athens controls (median = 13.69 ng/mL, range = 6.97–30.36 ng/mL in males; median = 7.03 ng/mL, range = 2.27–16.63 ng/mL in females), and Argolida controls (median = 10.47 ng/mL, range = 3.46–40.36 ng/mL in males; median = 8.47 ng/mL, range = 2.63–26.36 ng/mL in females) (). A one-way analysis of variance comparing means across the three subject groups yielded a statistically nonsignificant P-value (> 0.05). These findings provide little evidence either for or against a causal role of PFOS in cancer development.
CitationYeung et al. (2013) reported different patterns of tissue-specific correlation for PFOS than for PFOA, which was not correlated between serum and liver tissue. PFOS levels were correlated between paired serum and liver tissue samples in HCV-positive cirrhosis patients (ρ = 0.699) and in HCV-positive HCC cases (ρ = 0.503), but not correlated in HCV-negative HCC cases (ρ = − 0.064) (). PFOA and PFOS levels were correlated with each other in control serum (Spearman ρ = 0.708) and in control liver tissue (ρ = 0.850). Again, the authors did not statistically compare median serum PFOS levels across patient groups. However, median serum PFOS levels were highest in HCV-positive cirrhosis patients (13.7 ng/mL, range = 1.12–126 ng/mL), followed by HCV-negative HCC patients (11.5 ng/mL, range = 4.36–48.4 ng/mL) and HCV-positive HCC patients (11.4 ng/mL, range = 4.04–26.4 ng/mL), and lowest in healthy controls (7.29 ng/mL, range = 1.43–34.9 ng/mL) (). By contrast, median liver tissue serum PFOS levels were highest in control liver tissue (5.03 ng/g, range = 1.03–10.8 ng/g), followed by HCV-negative HCC (4.96 ng/g, range = 1.92–13.7), HCV-positive HCC (4.12 ng/g, range = 2.28–42.5), and lastly HCV-positive cirrhosis (2.35 ng/g, range = 0.375–12.5). The ratio of liver PFOS to serum PFOS in paired specimens did not differ significantly among patient groups (P > 0.05). Again, this study provides only weak evidence against an association between nonoccupational PFOS exposure and liver cancer risk.
Summary of epidemiologic evidence on PFOS and cancer in humans
As before, in this section, we use the main Bradford Hill guidelines (CitationHill, 1965) as a framework to consider the weight of evidence for or against the hypothesis of a causal effect of PFOS on human cancer risk, excluding lower-quality studies (CitationBonefeld-Jorgensen et al. 2011, CitationHardell et al. 2014, CitationVassiliadou et al. 2010, CitationYeung et al. 2013) from consideration.
Strength of association
As shown in , most estimated associations between PFOS exposure and cancer have been in the range of 0.5 to 2.0. Except for the striking inverse association between serum PFOS and colorectal cancer prevalence (CitationInnes et al. 2014), RR estimates falling outside this range were typically based on five or fewer cases, with correspondingly imprecise 95% CIs consistent with no association. Confounding, bias, and chance could readily explain such observed associations.
Consistency of association
Only two retrospective cohort studies of PFOS exposure have evaluated more than four cancer outcomes (CitationAlexander et al. 2003, CitationOlsen et al. 2004). Consequently, few opportunities are available for independent replication of observed associations with site-specific cancer mortality, incidence, or prevalence. In particular, only CitationAlexander et al. (2003) evaluated associations between PFOS exposure and cancers of the digestive organs, esophagus, lung/bronchus/trachea, urinary organs, and lymphatic and hematopoietic system. Only CitationOlsen et al. (2004) reported associations between PFOS exposure and cancers of the rectum and thyroid, and only CitationEriksen et al. (2009) reported associations with pancreatic cancer. Therefore, the consistency of these associations, all of which were statistically null or unreliable, could not be assessed.
Otherwise, no associations, including null findings, were consistently detected across studies. A statistically nonsignificant elevated risk of episodes of care for colon cancer was detected in chemical division workers, especially in long-term, high-exposure workers, at the Decatur plant (CitationOlsen et al. 2004), but no association was found between occupational PFOS exposure and colon cancer mortality or self-reported colon cancer at the same plant (CitationAlexander et al. 2003, CitationGrice et al. 2007), whereas an inverse association was observed in Mid-Ohio Valley residents (CitationInnes et al. 2014). A nonsignificant excess of liver cancer mortality was reported in Decatur chemical division workers, but no association was found between estimated PFOS exposure and episodes of care for liver cancer (CitationOlsen et al. 2004) or incident liver cancer (CitationEriksen et al. 2009). A nonsignificant excess of episodes of care for respiratory system cancer was observed in chemical versus film division workers in Decatur (CitationOlsen et al. 2004), but this was contradicted by a nonsignificant deficit of respiratory cancer mortality in the same facility (CitationAlexander et al. 2003). A nonsignificant excess of prostate cancer episodes of care was reported in chemical versus film division workers at the Decatur facility (CitationOlsen et al. 2004), and a weak, statistically nonsignificant association with plasma PFOS concentration was found for incident prostate cancer in Denmark (CitationEriksen et al. 2009), but no association with occupational PFOS exposure was found in relation to self-reported prostate cancer in Decatur workers (CitationGrice et al. 2007). While a substantial and statistically significant excess of mortality from bladder and other urinary organ cancer was originally detected among highly exposed workers at the Decatur plant (CitationAlexander et al. 2003), later studies of this worker group found no apparent excess of episodes of care for bladder cancer among chemical division workers (CitationOlsen et al. 2004) and no apparent association between estimated cumulative occupational PFOS exposure and self-reported bladder cancer (CitationAlexander and Olsen, 2007), nor was an association between plasma PFOS level and incident bladder cancer observed in Denmark (CitationEriksen et al. 2009). Finally, high but statistically unstable RRs for malignant melanoma episodes of care among chemical division workers at Decatur (CitationOlsen et al. 2004) were countered by nonsignificantly elevated SMRs and null ORs for melanoma in the same workplace (CitationAlexander et al. 2003, CitationGrice et al. 2007).
Given that all four occupational studies of PFOS exposure and cancer were conducted at the Decatur facility (CitationAlexander and Olsen, 2007, CitationAlexander et al. 2003, CitationGrice et al. 2007, CitationOlsen et al. 2004), one might have expected to find consistent associations in these workers, despite the major differences in outcome ascertainment and classification across the studies. The fact that findings were inconsistent among these studies, as well as across the community-based studies of PFOS and cancer, underscores the tenuousness of reported associations with estimated PFOS exposure in any given study and their collective failure to support any conclusion that the relationship is causal.
Exposure-response gradient
Most studies evaluated associations with different levels of potential PFOS exposure, thereby enabling at least rudimentary exposure-response analyses. With the exception of the highly statistically significant inverse association between serum PFOS and colorectal cancer prevalence in the C8 Health Study Project (CitationInnes et al. 2014), no other monotonic exposure-response trends were convincingly established. CitationAlexander et al. (2003) detected a positive trend toward increasing SMRs for bladder and other urinary tract cancer with increasing job-based PFOS exposure, especially long-term high-level exposure. By contrast, no such trend was detected in relation to similar exposure categories or estimated cumulative occupational PFOS exposure in a follow-up study of self-reported and fatal bladder cancer (CitationAlexander and Olsen, 2007), and the observed trend between serum PFOS exposure and bladder cancer risk in Denmark was nonsignificantly inverse (CitationEriksen et al. 2009).
Using episodes of care to define cancer outcomes, CitationOlsen et al. (2004) reported stronger colon and rectal cancer RR estimates for long-term, high-exposure chemical division workers than for all chemical division workers combined (versus comparable film division workers), and CitationGrice et al. (2007) also found a modest positive trend between job-based PFOS exposure and self-reported colon cancer in the same cohort of Decatur plant workers. However, these trends were not corroborated by findings for colon cancer mortality at the Decatur plant (CitationAlexander et al. 2003) and were directly contradicted by the inverse trend detected in the community around the Parkersburg plant (CitationInnes et al. 2014).
The small, nonsignificant increase in prostate cancer risk associated with higher quartiles of plasma PFOS in Denmark did not follow a monotonic pattern, nor was any association detected between continuous measures of PFOS in plasma and prostate cancer risk in that study (CitationEriksen et al. 2009).
As with PFOA, biomonitoring studies of serum PFOS levels show major differences among occupational and community groups (). The geometric mean level was 941 ng/mL (0.941 ppm) among fluorochemical workers at the Decatur plant in 1998 (CitationOlsen et al. 2003) and the median was 1,000 ng/mL at the same plant in 2000 (CitationOlsen and Zobel, 2007). At the Antwerp and Cottage Grove plants, the median levels were 550 and 450 ng/mL, respectively (CitationOlsen and Zobel, 2007), while the geometric mean level among background-exposed film division workers at the Decatur plant was 136 ng/mL (CitationOlsen et al. 2003). By contrast, median serum PFOS levels were up to two orders of magnitude lower in Ohio and West Virginia residents near the Parkersburg plant (approximately 20 ng/mL in 2005–2006), where industrial use of PFOS did not occur (CitationFrisbee et al. 2009). Median serum PFOS levels were comparable in US general population participants in NHANES (30.2 ng/mL in 1999–2000 and 13.6 ng/mL in 2007–2008) (CitationKato et al. 2011), and in American Red Cross adult volunteer blood donors (35.8 ng/mL in 2000–2001 and 8.6 ng/mL in 2010) (CitationOlsen et al. 2012). Again, these differences must be considered when contemplating the plausibility of observed positive associations in community, but not in occupational, settings.
Figure 2. Median (or geometric mean) serum levels of perfluorooctanesulfonic acid (PFOS) measured in directly and indirectly (background) exposed workers (Decatur, Alabama; Cottage Grove, Minnesota; and Antwerp, Belgium) and in community members in Parkersburg, West Virginia, and elsewhere in the United States.
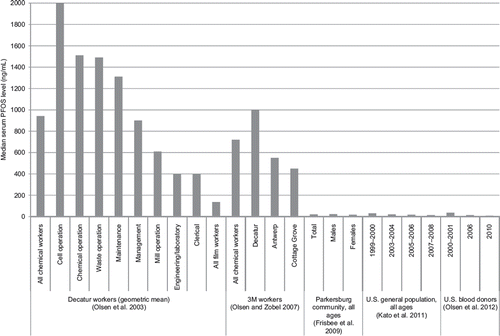
Plausibility and coherence with toxicological evidence
Toxicological studies in animals clearly pinpoint the liver as the main target organ for a potential carcinogenic effect of PFOS. Although CitationAlexander et al. (2003) reported elevated SMRs for liver cancer among workers with low or high potential PFOS exposure, these estimates were based on only one death each and, therefore, highly unstable. CitationOlsen et al. (2004) reported no episodes of care for liver cancer among chemical division workers, compared with one such episode among film division workers. The inverse RR estimates for liver cancer in association with higher quartiles of plasma PFOS concentration reported by CitationEriksen et al. (2009) in Denmark also are not consistent with a hepatocarcinogenic effect of PFOS in humans, at least at relatively low concentrations.
The 2-year rat feeding study of PFOS detected a potentially spurious increase in thyroid follicular cell adenoma among male rats fed with PFOS for 1 year and followed for a 2nd year, but not among those fed with PFOS for the full 2 years (CitationSeacat et al. 2002). Only CitationOlsen et al. (2004) reported on thyroid cancer as an outcome, with one episode of care (versus 1.0 expected) in a short-term and/or low-exposure chemical division worker and none among long-term, high-exposure chemical division workers or film division workers. Thus, although concordance of sites of carcinogenesis across species is not a requirement for establishing human cancer hazards, a comparison of results from animal and human studies offers little to no support for a causal relationship between PFOS exposure and human cancer.
Conclusions
The epidemiologic studies on PFOA or PFOS and risk of cancer in humans include six studies of PFOA in occupationally exposed workers (CitationConsonni et al. 2013, CitationGilliland and Mandel, 1993, CitationLeonard et al. 2008, CitationLundin et al. 2009, CitationSteenland and Woskie, 2012, CitationUbel et al. 1980), two studies of PFOA in environmentally exposed communities (CitationBarry et al. 2013, CitationVieira et al. 2013), four studies of PFOS in occupationally exposed workers (CitationAlexander and Olsen, 2007, CitationAlexander et al. 2003, CitationGrice et al. 2007, CitationOlsen et al. 2004), and six studies of both PFOA and PFOS in environmentally exposed communities (CitationBonefeld-Jorgensen et al. 2011, CitationEriksen et al. 2009, CitationHardell et al. 2014, CitationInnes et al. 2014, CitationVassiliadou et al. 2010, CitationYeung et al. 2013). The vast majority of reported associations with cancer mortality, incidence, or prevalence have been consistent with the null hypothesis of no effect. The few observed positive associations have not met the Bradford Hill guidelines, that is, they are weak, inconsistent, offset by negative associations, not in keeping with a positive exposure-response gradient, and not coherent with the toxicological findings of liver, testicular Leydig cell, and pancreatic acinar cell tumors in animals exposed to PFOA and liver tumors in those exposed to PFOS. Moreover, confounding, bias, and chance (especially in light of multiple comparisons) cannot be ruled out as explanations for the reported positive associations, many of which were observed in studies of environmentally exposed communities, but not in occupational settings where exposure to PFOA and PFOS was one to two orders of magnitude higher. Toxicological and mechanistic data in animals do not conflict with the epidemiologic data in humans and may even be interpreted as offering evidence against a carcinogenic effect of PFOA and PFOS in humans, given that the mechanisms by which these chemicals induce tumors in rodents may not be involved in human carcinogenesis.
The Health Council of the Netherlands (HCN) recently reviewed the scientific evidence on the carcinogenicity and genotoxicity of PFOA from human, laboratory animal, and mechanistic studies, and concluded that the available data on PFOA and its salts are “insufficient to evaluate the carcinogenic properties (category 3)” (CitationHCN, 2013). Regarding the epidemiologic evidence in particular, HCN concluded: “The reported results of a relatively substantial number of human longitudinal studies have such a high degree of inconsistency that the Committee classifies the human data as inadequate for firm conclusion about whether or not a cancer risk exists from exposure to PFOA in these studies.” HCN also concluded that “Overall … there is no cancer type that is consistently elevated in these studies.”
This classification is consistent with our conclusion that the existing epidemiologic evidence does not support the hypothesis of a causal association between PFOA or PFOS exposure and cancer in humans. However, further research on this topic is warranted. Quantitative exposure assessment in previously unstudied occupational settings – for example, at industrial facilities in Asia that continue to produce or use PFOA and/or PFOS (CitationLim et al. 2011) – could provide the basis for future cohort studies once sufficient follow-up time has accrued. More readily, continued follow-up of existing cohorts and linkage to cancer registries to ascertain cancer incidence might provide additional insight into whether these compounds affect cancer risk in humans.
Declaration of interest
The work of all authors on this manuscript was supported by the 3M Company. 3M was not involved in the preparation or approval of the manuscript. The findings and conclusions in this manuscript are those of the authors and do not necessarily represent the views of 3M.
JSM and ETC are consultants to 3M, PB consulted to industry on PFOA issues, and TBS serves pro bono as chairman of the North Carolina Science Advisory Board for Toxic Air Pollutants, which, upon special request from the North Carolina Division of Water Quality, reviewed and recently recommended a two-fold reduction of the Interim Maximum Allowable Level of PFOA in North Carolina groundwater. HOA and PC declare no other potential conflicts of interest in the past 5 years.
Notes
1All confidence intervals reported hereafter are 95% and two-sided, and are indicated by parentheses or brackets as appropriate.
References
- 3M Company. (2013). 3M's Phase Out and New Technologies. Available at: http://www.solutions.3m.com/wps/portal/3M/en_US/PFOS/PFOA/Information/phase-out-technologies/. Last accessed: 09/25/2013.
- Adami HO, Berry SC, Breckenridge CB, Smith LL, Swenberg JA, Trichopoulos D, et al. (2011). Toxicology and epidemiology: improving the science with a framework for combining toxicological and epidemiological evidence to establish causal inference. Toxicol Sci, 122, 223–34.
- Alexander BH, Olsen GW, Burris JM, Mandel JH, Mandel JS. (2003). Mortality of employees of a perfluorooctanesulphonyl fluoride manufacturing facility. Occup Environ Med, 60, 722–9.
- Alexander BH, Olsen GW. (2007). Bladder cancer in perfluorooctanesulfonyl fluoride manufacturing workers. Ann Epidemiol, 17, 471–8.
- Andersen ME, Clewell HJ III, Tan YM, Butenhoff JL, Olsen GW. (2006). Pharmacokinetic modeling of saturable, renal resorption of perfluoroalkylacids in monkeys–probing the determinants of long plasma half-lives. Toxicology, 227, 156–64.
- Andersen ME, Preston RJ, Maier A, Willis AM, Patterson J. (2014). Dose-response approaches for nuclear receptor-mediated modes of action for liver carcinogenicity: results of a workshop. Crit Rev Toxicol, 44, 50–63.
- Barry V, Winquist A, Steenland K. (2013). Perfluorooctanoic Acid (PFOA) exposures and incident cancers among adults living near a chemical plant. Environ Health Perspect, 121, 1313–18.
- Biegel LB, Hurtt ME, Frame SR, O’Connor JC, Cook JC. (2001). Mechanisms of extrahepatic tumor induction by peroxisome proliferators in male CD rats. Toxicol Sci, 60, 44–55.
- Bonefeld-Jorgensen EC, Long M, Bossi R, Ayotte P, Asmund G, Kruger T, et al. (2011). Perfluorinated compounds are related to breast cancer risk in Greenlandic Inuit: a case control study. Environ Health, 10, 88.
- Buck RC, Franklin J, Berger U, Conder JM, Cousins IT, de Voogt P, et al. (2011). Perfluoroalkyl and polyfluoroalkyl substances in the environment: terminology, classification, and origins. Integr Environ Assess Manag, 7, 513–41.
- Budinsky RA, Schrenk D, Simon T, Van den Berg M, Reichard JF, Silkworth JB, et al. (2014). Mode of action and dose-response framework analysis for receptor-mediated toxicity: the aryl hydrocarbon receptor as a case study. Crit Rev Toxicol, 44, 83–119.
- Butenhoff JL, Chang SC, Olsen GW, Thomford PJ. (2012b). Chronic dietary toxicity and carcinogenicity study with potassium perfluorooctanesulfonate in Sprague Dawley rats. Toxicology, 293, 1–15.
- Butenhoff JL, Kennedy GL Jr, Chang SC, Olsen GW. (2012a). Chronic dietary toxicity and carcinogenicity study with ammonium perfluorooctanoate in Sprague-Dawley rats. Toxicology, 298, 1–13.
- Butenhoff JL, Olsen GW, Pfahles-Hutchens A. (2006). The applicability of biomonitoring data for perfluorooctanesulfonate to the environmental public health continuum. Environ Health Perspect, 114, 1776–82.
- Calafat AM, Kuklenyik Z, Reidy JA, Caudill SP, Tully JS, Needham LL. (2007). Serum concentrations of 11 polyfluoroalkyl compounds in the U.S. population: data from the National Health and Nutrition Examination Survey (NHANES). Environ Sci Technol, 41, 2237–42.
- Chang SC, Noker PE, Gorman GS, Gibson SJ, Hart JA, Ehresman DJ, Butenhoff JL. (2012). Comparative pharmacokinetics of perfluorooctanesulfonate (PFOS) in rats, mice, and monkeys. Reprod Toxicol, 33, 428–40.
- Consonni D, Straif K, Symons JM, Tomenson JA, van Amelsvoort LG, Sleeuwenhoek A, et al. (2013). Cancer risk among tetrafluoroethylene synthesis and polymerization workers. Am J Epidemiol, 178, 350–8.
- Cook JC, Murray SM, Frame SR, Hurtt ME. (1992). Induction of Leydig cell adenomas by ammonium perfluorooctanoate: a possible endocrine-related mechanism. Toxicol Appl Pharmacol, 113, 209–17.
- Corton JC, Cunningham ML, Hummer BT, Lau C, Meek B, Peters JM, et al. (2014). Mode of action framework analysis for receptor-mediated toxicity: the peroxisome proliferator-activated receptor alpha (PPARalpha) as a case study. Crit Rev Toxicol, 44, 1–49.
- Cote S, Ayotte P, Dodin S, Blanchet C, Mulvad G, Petersen HS, et al. (2006). Plasma organochlorine concentrations and bone ultrasound measurements: a cross-sectional study in peri-and postmenopausal Inuit women from Greenland. Environ Health, 5, 33.
- Deutch B, Pedersen HS, Asmund G, Hansen JC. (2007). Contaminants, diet, plasma fatty acids and smoking in Greenland 1999–2005. Sci Total Environ, 372, 486–96.
- Elcombe CR, Elcombe BM, Foster JR, Chang SC, Ehresman DJ, Butenhoff JL. (2012). Hepatocellular hypertrophy and cell proliferation in Sprague-Dawley rats from dietary exposure to potassium perfluorooctanesulfonate results from increased expression of xenosensor nuclear receptors PPARalpha and CAR/PXR. Toxicology, 293, 16–29.
- Elcombe CR, Elcombe BM, Foster JR, Farrar DG, Jung R, Chang SC, et al. (2010). Hepatocellular hypertrophy and cell proliferation in Sprague-Dawley rats following dietary exposure to ammonium perfluorooctanoate occurs through increased activation of the xenosensor nuclear receptors PPARalpha and CAR/PXR. Arch Toxicol, 84, 787–98.
- Elcombe CR, Peffer RC, Wolf DC, Bailey J, Bars R, Bell D, et al. (2014). Mode of action and human relevance analysis for nuclear receptor-mediated liver toxicity: a case study with phenobarbital as a model constitutive androstane receptor (CAR) activator. Crit Rev Toxicol, 44, 64–82.
- Eriksen KT, Sorensen M, McLaughlin JK, Lipworth L, Tjonneland A, Overvad K, et al. (2009). Perfluorooctanoate and perfluorooctanesulfonate plasma levels and risk of cancer in the general Danish population. J Natl Cancer Inst, 101, 605–9.
- Fitzgibbons PL, Henson DE, Hutter RV. (1998). Benign breast changes and the risk for subsequent breast cancer: an update of the 1985 consensus statement. Cancer Committee of the College of American Pathologists. Arch Pathol Lab Med, 122, 1053–5.
- Frame SR, McConnell EE. (2003). Review of proliferative lesions of the exocrine pancreas in two chronic studies in rates with ammonium perfluorooctanoate. DuPont-13788 dated October 16, 2003.
- Frisbee SJ, Brooks AP Jr, Maher A, Flensborg P, Arnold S, Fletcher T, et al. (2009). The C8 health project: design, methods, and participants. Environ Health Perspect, 117, 1873–82.
- Giesy JP, Kannan K. (2001). Global distribution of perfluorooctane sulfonate in wildlife. Environ Sci Technol, 35, 1339–42.
- Gilliland FD, Mandel JS. (1993). Mortality among employees of a perfluorooctanoic acid production plant. J Occup Med, 35, 950–4.
- Grice MM, Alexander BH, Hoffbeck R, Kampa DM. (2007). Self-reported medical conditions in perfluorooctanesulfonyl fluoride manufacturing workers. J Occup Environ Med, 49, 722–9.
- Han X, Nabb DL, Russell MH, Kennedy GL, Rickard RW. (2012). Renal elimination of perfluorocarboxylates (PFCAs). Chem Res Toxicol, 25, 35–46.
- Hardell E, Karrman A, van Bavel B, Bao J, Carlberg M, Hardell L. (2014). Case-control study on perfluorinated alkyl acids (PFAAs) and the risk of prostate cancer. Environ Int, 63, 35–9.
- Hardisty JF, Willson GA, Brown WR, McConnell EE, Frame SR, Gaylor DW, et al. (2010). Pathology Working Group review and evaluation of proliferative lesions of mammary gland tissues in female rats fed ammonium perfluorooctanoate (APFO) in the diet for 2 years. Drug Chem Toxicol, 33, 131–7.
- HCN. (2013). Perfluorooctanoic Acid and Its Salts – Evaluation of the Carcinogenicity and Genotoxicity. The Hague: Health Council of the Netherlands (HCN).
- Hill AB. (1965). The environment and disease: association or causation?Proc R Soc Med, 58, 295–300.
- Houde M, Martin JW, Letcher RJ, Solomon KR, Muir DC. (2006). Biological monitoring of polyfluoroalkyl substances: a review. Environ Sci Technol, 40, 3463–73.
- IARC. (2001). IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Volume 79. Some Thyrotropic Agents. Lyon, France: International Association for Research on Cancer (IARC) Press.
- Innes KE, Wimsatt JH, Frisbee S, Ducatman AM. (2014). Inverse association of colorectal cancer prevalence to serum levels of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) in a large Appalachian population. BMC Cancer, 14, 45.
- Jain RB. (2013). Effect of pregnancy on the levels of selected perfluoroalkyl compounds for females aged 17–39 years: data from National Health and Nutrition Examination Survey 2003–2008. J Toxicol Environ Health A, 76, 409–21.
- Jain RB. (2014). Contribution of diet and other factors to the levels of selected polyfluorinated compounds: data from NHANES 2003–2008. Int J Hyg Environ Health, 217, 52–61.
- Jurek AM, Greenland S, Maldonado G, Church TR. (2005). Proper interpretation of non-differential misclassification effects: expectations vs. observations. Int J Epidemiol, 34, 680–7.
- Jurek AM, Greenland S, Maldonado G. (2008). How far from non-differential does exposure or disease misclassification have to be to bias measures of association away from the null?Int J Epidemiol, 37, 382–5.
- Kannan K, Corsolini S, Falandysz J, Fillmann G, Kumar KS, Loganathan BG, et al. (2004). Perfluorooctanesulfonate and related fluorochemicals in human blood from several countries. Environ Sci Technol, 38, 4489–95.
- Kato K, Wong LY, Jia LT, Kuklenyik Z, Calafat AM. (2011). Trends in exposure to polyfluoroalkyl chemicals in the U.S. Population: 1999–2008. Environ Sci Technol, 45, 8037–45.
- Kennedy GL Jr, Butenhoff JL, Olsen GW, O’Connor JC, Seacat AM, Perkins RG, et al. (2004). The toxicology of perfluorooctanoate. Crit Rev Toxicol, 34, 351–84.
- Klaunig JE, Babich MA, Baetcke KP, Cook JC, Corton JC, David RM, et al. (2003). PPARalpha agonist-induced rodent tumors: modes of action and human relevance. Crit Rev Toxicol, 33, 655–780.
- Klaunig JE, Hocevar BA, Kamendulis LM. (2012). Mode of action analysis of perfluorooctanoic acid (PFOA) tumorigenicity and human relevance. Reprod Toxicol, 33, 410–8.
- Klimstra DS, Heffess CS, Oertel JE, Rosai J. (1992). Acinar cell carcinoma of the pancreas. A clinicopathologic study of 28 cases. Am J Surg Pathol, 16, 815–37.
- La Vecchia C, Negri E. (2014). A review of epidemiological data on epilepsy, phenobarbital, and risk of liver cancer. Eur J Cancer Prev, 23, 1–7.
- Leonard RC, Kreckmann KH, Sakr CJ, Symons JM. (2008). Retrospective cohort mortality study of workers in a polymer production plant including a reference population of regional workers. Ann Epidemiol, 18, 15–22.
- Li N, Du XL, Reitzel LR, Xu L, Sturgis EM. (2013). Impact of enhanced detection on the increase in thyroid cancer incidence in the United States: review of incidence trends by socioeconomic status within the surveillance, epidemiology, and end results registry, 1980–2008. Thyroid, 23, 103–10.
- Lim TC, Wang B, Huang J, Deng S, Yu G. (2011). Emission inventory for PFOS in China: review of past methodologies and suggestions. ScientificWorldJournal, 11, 1963–80.
- Lissner L, Skoog I, Andersson K, Beckman N, Sundh V, Waern M, et al. (2003). Participation bias in longitudinal studies: experience from the population study of women in Gothenburg, Sweden. Scand J Prim Health Care, 21, 242–7.
- Loccisano AE, Campbell JL Jr, Butenhoff JL, Andersen ME, Clewell HJ III. (2012a). Comparison and evaluation of pharmacokinetics of PFOA and PFOS in the adult rat using a physiologically based pharmacokinetic model. Reprod Toxicol, 33, 452–67.
- Loccisano AE, Campbell JL Jr, Butenhoff JL, Andersen ME, Clewell HJ III. (2012b). Evaluation of placental and lactational pharmacokinetics of PFOA and PFOS in the pregnant, lactating, fetal and neonatal rat using a physiologically based pharmacokinetic model. Reprod Toxicol, 33, 468–90.
- Loccisano AE, Longnecker MP, Campbell JL Jr, Andersen ME, Clewell HJ III. (2013). Development of PBPK models for PFOA and PFOS for human pregnancy and lactation life stages. J Toxicol Environ Health A, 76, 25–57.
- Lundin JI, Alexander BH, Olsen GW, Church TR. (2009). Ammonium perfluorooctanoate production and occupational mortality. Epidemiology, 20, 921–8.
- Martignoni M, Groothuis GM, de Kanter R. (2006). Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol, 2, 875–94.
- National Toxicology Program. (1997). NTP toxicology and carcinogenesis studies of tetrafluoroethylene (CAS No. 116-14-3) in F344 rats and B6C3F1 mice (inhalation studies). Natl Toxicol Program Tech Rep Ser, 450, 1–321.
- Olsen GW, Burlew MM, Marshall JC, Burris JM, Mandel JH. (2004). Analysis of episodes of care in a perfluorooctanesulfonyl fluoride production facility. J Occup Environ Med, 46, 837–46.
- Olsen GW, Burris JM, Burlew MM, Mandel JH. (2000). Plasma cholecystokinin and hepatic enzymes, cholesterol and lipoproteins in ammonium perfluorooctanoate production workers. Drug Chem Toxicol, 23, 603–20.
- Olsen GW, Burris JM, Ehresman DJ, Froehlich JW, Seacat AM, Butenhoff JL, Zobel LR. (2007). Half-life of serum elimination of perfluorooctanesulfonate, perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers. Environ Health Perspect, 115, 1298–305.
- Olsen GW, Gilliland FD, Burlew MM, Burris JM, Mandel JS, Mandel JH. (1998). An epidemiologic investigation of reproductive hormones in men with occupational exposure to perfluorooctanoic acid. J Occup Environ Med, 40, 614–22.
- Olsen GW, Lange CC, Ellefson ME, Mair DC, Church TR, Goldberg CL, et al. (2012). Temporal trends of perfluoroalkyl concentrations in American Red Cross adult blood donors, 2000–2010. Environ Sci Technol, 46, 6330–8.
- Olsen GW, Logan PW, Hansen KJ, Simpson CA, Burris JM, Burlew MM, et al. (2003). An occupational exposure assessment of a perfluorooctanesulfonyl fluoride production site: biomonitoring. AIHA J (Fairfax, Va), 64, 651–9.
- Olsen GW, Zobel LR. (2007). Assessment of lipid, hepatic, and thyroid parameters with serum perfluorooctanoate (PFOA) concentrations in fluorochemical production workers. Int Arch Occup Environ Health, 81, 231–46.
- Pelkonen O, Turpeinen M, Hakkola J, Honkakoski P, Hukkanen J, Raunio H. (2008). Inhibition and induction of human cytochrome P450 enzymes: current status. Arch Toxicol, 82, 667–715.
- Prevedouros K, Cousins IT, Buck RC, Korzeniowski SH. (2006). Sources, fate and transport of perfluorocarboxylates. Environ Sci Technol, 40, 32–44.
- Richardson LC, Neri AJ, Tai E, Glenn JD. (2012). Testicular cancer: a narrative review of the role of socioeconomic position from risk to survivorship. Urol Oncol, 30, 95–101.
- Sakr CJ, Kreckmann KH, Green JW, Gillies PJ, Reynolds JL, Leonard RC. (2007). Cross-sectional study of lipids and liver enzymes related to a serum biomarker of exposure (ammonium perfluorooctanoate or APFO) as part of a general health survey in a cohort of occupationally exposed workers. J Occup Environ Med, 49, 1086–96.
- Sarma AV, McLaughlin JC, Schottenfeld D. (2006). Chapter 60: Testicular cancer. In: Schottenfeld D, Fraumeni JF Jr, Eds. Cancer Epidemiology and Prevention, 3rd edition. New York: Oxford University Press 1151–65..
- Seacat AM, Thomford PJ, Butenhoff JL. (2002). Terminal observations in Sprague-Dawley rats after lifetime dietary exposure to potassium perfluorooctanesulfonate [Abstract]. Toxicologist, 66, 185.
- Shin HM, Vieira VM, Ryan PB, Detwiler R, Sanders B, Steenland K, Bartell SM. (2011a). Environmental fate and transport modeling for perfluorooctanoic acid emitted from the Washington Works Facility in West Virginia. Environ Sci Technol, 45, 1435–42.
- Shin HM, Vieira VM, Ryan PB, Steenland K, Bartell SM. (2011b). Retrospective exposure estimation and predicted versus observed serum perfluorooctanoic acid concentrations for participants in the C8 Health Project. Environ Health Perspect, 119, 1760–5.
- Sibinski LJ. (1987). Two year oral (diet) toxicity/carcinogenicity study of fluorochemical FC-143 in rats. Experiment No. 0281CR0012. Report prepared for 3M, St. Paul, Minnesota by Riker Laboratories, Inc. U.S. EPA Public Docket AR-226–0437, Washington, DC.
- Sigurdson AJ, Doody MM, Rao RS, Freedman DM, Alexander BH, Hauptmann M, et al. (2003). Cancer incidence in the US radiologic technologists health study, 1983–1998. Cancer, 97, 3080–9.
- Steenland K, Woskie S. (2012). Cohort mortality study of workers exposed to perfluorooctanoic acid. Am J Epidemiol, 176, 909–17.
- Sykes G. (1987). Two-year toxicology/carcinogenicity study of fluorochemical FC-143 in rats. Memo. From G Sykes to C Reinhardt, Haskell Lab. Toxicol Ind Med. Dated October 29, 1987. As cited in: Draft Risk Assessment of the Potential Human Health Effects Associated with Exposure to Perfluorooctanoic Acid and its Salts. U.S. EPA Office of Pollution Prevention and Toxics Risk Assessment Division, Washington, DC, January 4, 2005.
- Tan YM, Clewell HJ III, Andersen ME. (2008). Time dependencies in perfluorooctylacids disposition in rat and monkeys: a kinetic analysis. Toxicol Lett, 177, 38–47.
- U.S. EPA. (2006). 2010/2015 PFOA Stewardship Program. Available at: http://www.epa.gov/opptintr/pfoa/pubs/stewardship/. Last updated: January 16, 2013.
- Ubel FA, Sorenson SD, Roach DE. (1980). Health status of plant workers exposed to fluorochemicals–a preliminary report. Am Ind Hyg Assoc J, 41, 584–9.
- Vassiliadou I, Costopoulou D, Ferderigou A, Leondiadis L. (2010). Levels of perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA) in blood samples from different groups of adults living in Greece. Chemosphere, 80, 1199–206.
- Vieira VM, Hoffman K, Shin HM, Weinberg JM, Webster TF, Fletcher T. (2013). Perfluorooctanoic acid exposure and cancer outcomes in a contaminated community: a geographic analysis. Environ Health Perspect, 121, 318–23.
- Woskie SR, Gore R, Steenland K. (2012). Retrospective exposure assessment of perfluorooctanoic acid serum concentrations at a fluoropolymer manufacturing plant. Ann Occup Hyg, 56, 1025–37.
- Yeung LW, Guruge KS, Taniyasu S, Yamashita N, Angus PW, Herath CB. (2013). Profiles of perfluoroalkyl substances in the liver and serum of patients with liver cancer and cirrhosis in Australia. Ecotoxicol Environ Saf, 96, 139–46.