Abstract
The aim of this study was to evaluate the pharmacokinetics of paclitaxel-loaded nanosponges (PLN) in rats. The study also evaluates the intrinsic effect of the dosage form on the improvement of paclitaxel oral bioavailability. Paclitaxel-loaded nanosponges were prepared and characterized in terms of size distribution, drug solubilization, and the kinetics of paclitaxel sedimentation. Taxol® and paclitaxel-loaded nanosponges were administered orally to rats. The plasma concentration of paclitaxel was determined using liquid chromatography. The average size of PLN was 350 ± 25 nm. The drug payload of paclitaxel was 500 ± 0.27 mg/g of lyophilized powder. The encapsulation efficiency was 99.1 ± 1.0%, and 1.7 ± 0.2% of paclitaxel was crystallized after 48 h. The relative oral bioavailability of paclitaxel-loaded nanosponges was 256. After oral administration of paclitaxel-loaded PLN, the area under the plasma concentration time curve was significantly increased (∼ 3-fold) in comparison to the control group (p < 0.05). The results indicated that PLN provided a promising new formulation to enhance the oral bioavailability of paclitaxel while avoiding the use of cremophore El: Ethanol in Taxol®.
Introduction
Paclitaxel (Taxol®) is an anti-neoplastic agent derived from the bark of the Pacific yew tree (Taxus brevifolia). Paclitaxel is primarily metabolized in the liver and undergoes biliary excretion (CitationHarris et al., 1994; CitationRahman et al., 1994). The fecal excretion level in humans approximates 70% of the administered paclitaxel dose, with 6-hydroxypaclitaxel being the major metabolite (CitationWalle et al., 1995). Orally administered paclitaxel presents a major therapeutic problem because of low bioavailability due to poor solubility and the first-pass metabolism that occurs in the liver and intestine. Therefore, paclitaxel is currently dissolved in a mixture of olyoxyethyleneglycerol triricinoleate 35 (Cremophor EL) and dehydrated ethanol (1:1 v/v) for the intravenous (I.V.) dosage form. Cremophor EL, however, is itself toxic and causes vasodilatation, labored breathing, lethargy, and hypotension (CitationRowinsky, 1993). When taken orally, paclitaxel is a substrate for P-glycoprotein (P-gp) efflux in the intestine. P-gp is a member of the ATP-binding cassette (ABC) super family (CitationSparreboom et al., 1997). Several studies have reported that the low bioavailability of paclitaxel following oral administration would result from metabolism by the enzymes or counter-transport processes due to P-gp in the intestinal wall. It was reported that when concomitantly administered with cyclosporine A (a P-gp and CYP 3A inhibitor) the oral bioavailability of paclitaxel was markedly higher than paclitaxel alone in humans (CitationMeerum et al., 1999) and mice (Citationvan Asperen et al., 1998). Paclitaxel is also known to be a substrate for the efflux of other ABC super-family members, such as BCRP (CitationDoyle & Ross, 2003) and MRP2 (CitationHuisman et al., 2005). The ABC family of transport proteins plays a central role in the defense of organism against toxic compounds and a diverse number of anti-cancer agents (CitationHiggins, 1992). Moreover, the MRPs and Pgp are co-localized with CYP3A4, glutathione-S-transferases, and UDP-glucuronosyltransferases (CitationSutherland et al., 1993), which are thought to act synergistically in regulating the bioavailability of many orally ingested compounds.
Oral administration of paclitaxel is thus very attractive because it improves a patient’s quality-of-life. Indeed, oral drug treatment is practical and convenient to patients, and it facilitates the development of chronic treatment schedules. Finally, with the aim of decreasing costs of anti-cancer therapy, oral treatment of cytotoxic agents is interesting because oral administration eliminates the need for hospitalization, medical and nursing assistance, and infusion equipment. Unfortunately, paclitaxel has a very low level of oral bioavailability, at less than 10% (Citationvan Asperen et al., 1998; CitationBardelmeijer et al., 2000; CitationMalingre et al., 2001; CitationGao et al., 2003). This poor bioavailability level results from limited aqueous solubility and dissolution, affinity for the intestinal and liver cytochrome P450 (like CYP3A4) metabolic enzymes, and the multidrug efflux pump P-glycoprotein (P-gp), which is present abundantly in the gastrointestinal tract (CitationRowinsky et al. 1993; CitationMalingre et al., 2001; CitationZuylen et al., 2001; CitationKruijtzer et al., 2002). In a proof-of concept study, the oral bioavailability of paclitaxel increased from only 11% in wild-type mice to 35% in mice that were homozygous for a disruption of the mdr1a gene, demonstrating that P-gp plays a major role in reducing the bioavailability of this drug (CitationSparreboom et al., 1997).
‘Nanosponge’, a novel derivative of cyclodextrins for nanoparticulate drug delivery, was developed in our laboratory (CitationTrotta & Tumiatti, 2003; CitationCavalli et al., 2006; CitationVavia et al., 2007; CitationCavalli & Trotta 2009; CitationSwaminathan et al., 2009). The polymeric derivative of beta cyclodextrin more commonly called ‘Nanosponge’ takes its name by its property of forming a sponge-like structure when lyophilized and high capacity to entrap small molecules in its matrix. The nanosponge is highly capable of rapidly forming suspension when dispersed in water, thereby forming a matrix-like structure in aqueous media and allowing a free transfer of entrapped drug molecules (CitationVavia et al., 2007; CitationSwaminathan et al., 2009). The objective of the current study was to evaluate in rats the pharmacokinetics of paclitaxel-loaded nanosponges to assess the intrinsic effect of the dosage form on the improvement of paclitaxel oral exposure.
Materials and methods
Materials
Nanosponges were synthesized from β-cyclodextrin in our lab (CitationTrotta & Tumiatti, 2003). Paclitaxel used for PLN formulation was from Indena (Milan, Italy). An injectable solution of paclitaxel at 6 mg/mL (Taxol®) was obtained from Bristol-Myers Squibb (Rome, Italy). Water was obtained from a Milli RO System (Millipore, Milan, Italy). Methanol and tert-butylmethylether were of high-performance liquid chromatography (HPLC) grade and were from Sigma-Aldrich (Milan, Italy).
Preparation of paclitaxel-loaded nanosponges
The preparation of paclitaxel-loaded nanosponges was done by freeze-drying where the nanosponges and paclitaxel powders were mixed geometrically in a ratio (1:1 w/w) and the resultant powder (1 g) is suspended in 200 ml distilled water under constant stirring. The suspension was kept stirring for 1 h and then filtered to remove any undissolved particles. The filtrate was then lyophilized to get paclitaxel-loaded nanosponges which were assayed for paclitaxel content.
Size distribution
For determination of particle size a 90 plus particle sizer instrument (Brookhaven Instruments Corporation), with MAS OPTION particle sizing software, was used at the fixed angel of 90° and at a temperature of 25°C. The samples were diluted in physiological phosphate buffer (pH = 7.4) prior to measurements. Each sample was analyzed in triplicate. The mean hydrodynamic diameter (Dh) and polydispersity index, PI, of the particles were calculated in intensity using the cumulant analysis after averaging the three measurements. Dh was derived from the measured translational diffusion coefficient of particles moving under Brownian motion. The MSD software used to generate graphs of size distribution uses Non Negatively Constrained Least Squares (NNLS) algorithm for fitting the data. The light source was a SP 162 argon laser, working at 100 mW.
Drug payload and entrapment efficiency
Reversed-phase HPLC methods were used for the analysis of paclitaxel payload in PLN. Drug content analysis was performed using a C18 column (Lichrosorb 18, 5 μ, spherical, 4.6 mm × 250 mm, Merck KGaA, Germany) equipped with a C18 guard column at 25°C with a mobile phase containing a mixture of methanol, acetonitrile, and acetate buffer pH 5 (10/40/50, v/v/v) at a flow rate of 1 ml/min. Sample injection volumes were 20 µl and paclitaxel detection was performed using a Waters 2487 dual wavelength UV detector at a wavelength of 227 nm. Peak area was recorded and processed on a Waters Breeze software version 3.30 spa. 2002. The HPLC method was validated for parameters linearity, limit of detection, and quantitation. The method was found to be linear in the range of 1–20 µg of paclitaxel with a regression coefficient of 0.9737. Limit of detection and quantification were found out to be 0.07 µg and 0.5 µg, respectively.
For encapsulation efficiency, paclitaxel-loaded nanosponges were usually separated from free paclitaxel by following the procedure below; in brief, the PLN was diluted with water, centrifuged for 15 min at 10,000 rpm, and the precipitation was taken out, dried at vacuum, added with methanol, and the free paclitaxel existing in the precipitate was measured using the analysis method described above for drug payload. The drug encapsulation efficiency was calculated by the following formula:
Paclitaxel precipitation
Possible drug precipitation 24 h after preparation (storage at 7 ± 2°C) was followed up using the same analytical conditions as for drug payload determination. A sample of paclitaxel-loaded nanosponges dispersion was filtered with a nylon Microcon YM 30 membrane filter (Millipore). Paclitaxel remaining on the filter was dissolved in 5 mL of a 95:5 (v/v) methanol/tetrahydrofurane solution. Filtration was performed using a Microcon YM 30 0.45-µm filter (Millipore). A 20-µL aliquot of filtrate was injected into the HPLC column.
Animal study
Animals in this study were handled in accordance with the provisions of the Principles of Laboratory Animal Care (NIH publication #85-23, revised 1985). This experiment was also carried out in accordance with the Good practice guide to the administration of substances and removal of blood, including routes and volumes, adopted by the European Federation of Pharmaceutical Industries Associations (EFPIA) and the European Centre for the Validation of Alternative Methods (ECVAM) in 2001 (CitationDiehl et al., 2001). Experiments were performed on group of eight male Sprague Dawley (SD) rats weighing 200 ± 20 g (mean ± standard deviation) that were given no food overnight for 12–14 h, but had access to water.
Paclitaxel-loaded nanosponges (paclitaxel:nanosponges in ratio 1:1) were suspended in a PBS to achieve a concentration of 2 mg/ml. Taxol® was diluted with PBS to achieve the concentration 2 mg/ml.
Administration in rats
For oral administration (Taxol® and PLN), preparations were administered by gastric intubation after weighing in a syringe to achieve a target dose of paclitaxel of 10 mg/kg, i.e. a dosing volume of 1–1.5 mL. For intravenous bolus experiment, Taxol® and PLN were administered into the tail vein with an approximate volume of 1 ml of preparation to achieve a target dose of paclitaxel of 10 mg/kg.
Blood (0.5 ml) was collected via orbital sinus venipuncture method at 0, 1, 2, 4, 5, and 6 h after injection. Blood samples were placed into heparinized tubes. After centrifugation for 15 min at 10,000 rpm, the obtained plasma was stored at /4–8°C until determination.
Determination of paclitaxel in plasma
Serum samples were harvested by centrifugation by placing an aliquot of 0.5 ml blood sample into a centrifuge tube and supernatant was extracted with 2 ml tert-butyl methyl ether on a vortex mixer for 2 min, centrifugation, the organic layer was transferred to another clean test tube and evaporated under gentle nitrogen gas. The residue was reconstituted by 250 µl mobile phase, and 100 µl was injected into the HPLC system. The peak area of paclitaxel was recorded and the content of paclitaxel was determined from standard curve.
The HPLC system was Jasco Intelligent HPLC system consisting of Jasco PU 2010 Intelligent HPLC pump, Jasco 2015 UV-VIS multiwavelength Detector, and processing was done by Chrompass chromatography software.
The HPLC condition is listed below: Licrosphere C18 column (250 mm × 4.6 mm, 5 µm), the mobile phase was composed of methanol:acetonitrile:acetate buffer pH 5.0 (10:40:50 v/v), flow rate of 1.0 ml/min, detecting wavelength of 227 nm, and sensitivity of 0.02 AUFS.
Pharmacokinetic data analysis
The concentration-time data were analyzed by Kinetica (v4.2; Innaphase, Philadelphia, PA) using Wagner-Nelson analysis to obtain the pharmacokinetic parameters of paclitaxel. The conditions for use were one compartment, with an intravenous reference with first order elimination. The area under the plasma paclitaxel concentration-time curve (AUC) and the area under the first moment curve (AUMC) were calculated using the linear trapezoidal method. Then the AUC was calculated by dividing the concentration of last point (C300) by the elimination rate constant (λz) as follows:
The percentage of extrapolated AUC was below 30%. The AUMC was calculated as follows:
The mean residence time (MRT), the plasmatic clearance (Cl), and the apparent volume of distribution at steady state (Vss) were calculated using the IV data set.
The terminal elimination half-life (t1/2) observed for paclitaxel following IV administration was calculated using the slope from linear regression analysis of log-transformed concentration values from the last four time data points.
Results and discussion
Characterization of paclitaxel-loaded nanosponges
Nanosponges loaded with paclitaxel had a monomodal particle size distribution of ∼ 350 ± 25 nm, with a narrow distribution (polydispersity index, p < 0.2). The mean drug payload of all batches was 500 ± 0.27 mg of paclitaxel/g of PLN lyophilized powder. The incorporation of paclitaxel was very efficient, as demonstrated by the high encapsulation efficiency (99.1 ± 1.0%). After storage for 24 h at 7 ± 2°C, 1.7 ± 0.2% of paclitaxel was crystallized in the aqueous phase.
Pharmacokinetic analysis
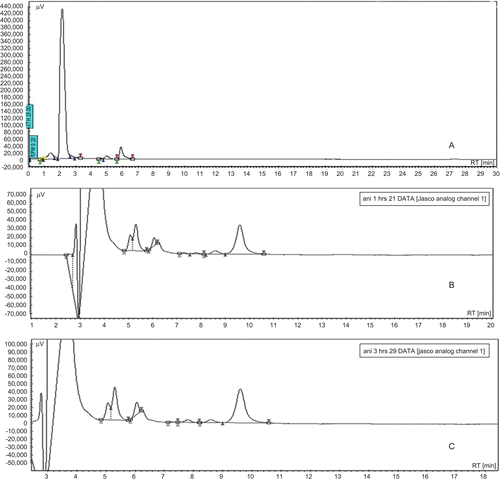
The plasma concentrations of paclitaxel after the oral and I.V. administration of paclitaxel (10 mg/kg) and paclitaxel-loaded nanosponges (10 mg/kg) are shown in and , respectively. shows chromatograms of plasma blank (a), paclitaxel in plasma by oral PLN administration (b), and IV Taxol® administration (c).
Figure 1. Paclitaxel: plasma concentration vs time.
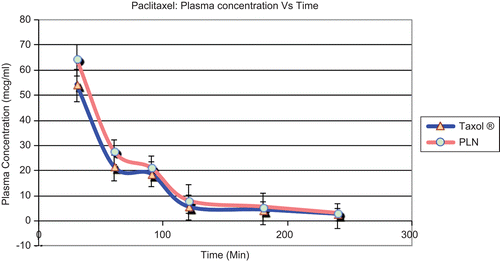
Figure 2. Absorption kinetics of paclitaxel.
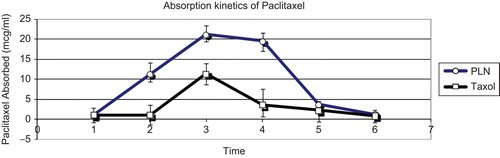
Figure 3. Chromatograms of plasma blank (A), paclitaxel in plasma by oral PLN administration (B), and IV Taxol® administration (C).
The plasma concentrations of paclitaxel by the oral nanosponge formulation were significantly (p < 0.01) higher than the Taxol® at the same dose of paclitaxel. The bioavailability and pharmacokinetic parameters of paclitaxel after the oral administration of Taxol® (10 mg/ kg) and the PLN (10 mg/kg) are shown in .
Table 1. Pharmacokinetic parameters of PLN and Taxol® (10 mg/kg).
The area under the plasma concentration–time curve (AUC) of paclitaxel by the oral Taxol® (10 mg/kg) was 5975.25 ± 485 μg/ml h. The AUC of paclitaxel by the oral PLN (10 mg/kg) was 6520.31 ± 752 μg/ml h, RB% of paclitaxel from the PLN was 256.
The AUC of Taxol® after the intravenous administration (10 mg/kg) was 6477.88 ± 382 μg/ml h and elimination rate constant was 0.017, on the other hand, AUC of PLN (10 mg/kg) was 6516.46 ± 412 μg/ml and elimination rate constant was 0.023. The mean absolute bioavailability (AB) of paclitaxel by Taxol® group was 0.203. AB of oral PLN was 0.521, which increased 2.5-fold compared to paclitaxel parent drug in rats. The peak plasma concentration of paclitaxel (Cmax) by the oral Taxol® (10 mg/kg) was 11.41 ± 2.5 μg/ml. The peak plasma concentration of paclitaxel (Cmax) by the PLN (10 mg/kg) was 21.2396 ± 3.5 μg/ml. The time to reach the maximum plasma concentration (Tmax), mean residence time, and the terminal half-life (t1/2) of paclitaxel in the Taxol® (10 mg/kg) showed no significant changes compared to those of the PLN.
This study introduced a β-cyclodextrin derivative commonly called a nanosponge which is capable of forming a matrix-like structure when suspended in water, thereby solubilizing the water-insoluble drugs like paclitaxel by forming a nanosuspension (CitationCavalli et al., 2006; CitationVavia et al., 2007; CitationCavalli & Trotta, 2009). At the same dose of paclitaxel, the AUC of paclitaxel in the 20 mg of PLN (equivalent to 10 mg paclitaxel) resulted in a remarkable increase compared to 10 mg of Taxol®. Therefore, the mean absolute bioavailability of PLN was 2.5-fold higher than paclitaxel. It was reported that paclitaxel is metabolized by cytochrome p-450 (CYP3A) both in the liver and in the epithelial cells of the small intestine, and, in addition, the absorption of paclitaxel was inhibited by the P-gp efflux pump in the intestinal mucosa (CitationRahman et al., 1994). The increased AB% of paclitaxel by nanosponges might have resulted from the physicochemical properties of the nanosponge, which is a polymeric derivative of beta cyclodextrin.
Many excipients often used in pharmaceutical formulations have been reported to have inhibitory effects on P-glycoprotein, an important membrane-associated transport protein (CitationWang et al., 2004). Because inhibition of efflux transporters can have an effect on drug bioavailability, identification of these excipients and their extent of inhibition are therefore important for pharmaceutical development. The enhanced bioavailability of paclitaxel can be related to cyclodextrin, a building block of nanosponges, possessing a high-solubilizing capacity and exhibiting modulating impact on membrane integrity and absorption systems such as passive diffusion and carrier-mediated permeation. Although there are no reports of nanosponges having an inhibitory effect on P-glycoprotein, the absence of literature is merely due to the fact that the present work is just one of the preliminary efforts of investigation on nanosponges for their possible use in drug delivery. However, more specific work for investigating P-gp and its possible inhibition by nanoaponges is under consideration.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Bardelmeijer, H.A., Beijnen, J.H., Brouwer, K.R., Rosing, H., Nooijen, W.J., Schellens, J.H., van Tellingen, O. (2000). Increased oral bioavailability of paclitaxel by GF120918 in mice through selective modulation of P-glycoprotein. Clin Cancer Res. 6:4416–21.
- Cavalli, R., Trotta, F. (2009). Characterization and applications of new hyper-cross-linked cyclodextrins. Compos Interface. 16:39–48.
- Cavalli, R., Trotta, F., Tumiatti, W. (2006). Cyclodextrin-based nanosponges for drug delivery. Incl Phenom Macrocycl Chem. 56:209–13.
- Diehl, K.H., Hull, R., Morton, D., Pfister, R., Rabemampianina, Y., Smith, D., Vidal, J.M., Vorstenbosch, C. (2001). A good practice guide to the administration of substances and removal of blood, including routes and volumes. J Appl Toxicol. 21:15–23.
- Doyle, L.A., Ross, V. (2003). Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene. 22:7340–58.
- Gao, P., Rush, B.D., Pfund, W.P., Huang, T., Bauer, J.M., Morozowich, W., Kuo, M.S., Hageman, M.J. (2003). Development of a supersaturable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailability. J Pharm Sci. 92:2386–98.
- Harris, J.W., Rahman, A., Kim, B.R., Guengerich, F.P., Collins, J.M. (1994). Metabolism of taxol by human hepatic microsomes and liver slices: participation of cytochrome P450 3A4 and an unknown P450 enzyme. Cancer Res. 54:4026–35.
- Higgins, C.F. (1992). ABC transporters: from microorganisms to man. Annu Rev Cell Biol. 8:67–113.
- Huisman, M.T., Chhatta, A.A., Tellingen, O., Beijnen, J.H., Schinkel, A.H. (2005). MRP2 (ABCC2) transports taxanes and confers paclitaxel resistance and both processes are stimulated by probenecid. Int J Cancer. 116:824–9.
- Kruijtzer, C.M., Beijnen, J.H., Schellens, J.H. (2002). Improvement of oral drug treatment by temporary inhibition of drug transporters and/or cytochrome P450 in the gastrointestinal tract and liver: an overview. Oncologist. 7:516–30.
- Malingre, M.M., Beijnen, J.H., Schellens, J.H. (2001). Oral delivery of taxanes. Invest New Drugs. 19:155–62.
- Meerum, J.M., Terwogt, M.M., Malingre, J.H., Beijnen, W.W., Huinink, H., Rosing, F.J., Koopman van, Tellingen, Swart, O.M., Schellens, J.H. (1999). Coadministration of oral cyclosporin A enables oral therapy with paclitaxel. Clin Cancer Res. 5:3379–84.
- Rahman, A., Korzekwa, K.R., Grogan, J., Gonzalez, F.J., Harris, J.W. (1994). Selective biotransformation of taxol to 6 alpha-hydroxytaxol by human cytochrome P4502C8. Cancer Res. 54:5543–6.
- Rowinsky, E.K. (1993). Clinical pharmacology of Taxol. J Natl Cancer Inst Monogr. 15:25–37.
- Rowinsky, E.K., Eisenhauer, E.A., Chaudhry, V., Arbuck, S.G., Donehower, R.C. (1993). Clinical toxicities encountered with paclitaxel (Taxol). Semin Oncol. 20:1–15.
- Sparreboom, A., van Asperen, J., Mayer, U., Schinkel, A.H., Smit, J.W., Meijer, D.K., Borst, P., Nooijen, W.J., Beijnen, J.H., van Tellingen, O. (1997). Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc Natl Acad Sci USA. 94:2031–5.
- Sutherland, L., Ebner, T., Burchell, B. (1993). The expression of UDP glucuronosyltransferases of the UGT1 family in human liver and kidney and in response to drugs. Biochem Pharmacol. 45:295–301.
- Swaminathan, S., Pastero, L., Serpe, L., Troota, F., Vavia, P., Aquilano, D., Trotta, M., Zara, G., Cavalli, R. (2009). Cyclodextrin-based nanosponges encapsulating camptothecin: physicochemical characterization, stability and cytotoxicity. Eur J Pharm Biopharm. 74:193–201.
- Trotta, F., Tumiatti, W. (2003). Cross-linked polymers based on cyclodextrin for removing polluting agents. World patent: WO 03/085002.
- van Asperen, J., Tellingen, O., van der Valk, M.A., Rozenhart, M., Beijnen, J.H. (1998). Enhanced oral absorption and decreased elimination of paclitaxel in mice cotreated with cyclosporin A. Clin Cancer Res. 4:2293–7.
- Vavia, P.R., Swaminathan, S., Trotta, F., Torne, S.J. (2007). Formulation of beta cyclodextrin based nanosponges of itraconazole. Incl Phenom Macrocycl Chem. 57:89–94.
- Walle, T., Walle, U.K., Kuma, G.N., Bhalla, K.N. (1995). Taxol metabolism and disposition in cancer patients. Drug Metab Disp. 23:506–12.
- Wang, S., Monagle, J., Mcnulty, C., Putnam, D., Chen, H. (2004). Determination of P-glycoprotein inhibition by excipients and their combinations using an integrated high-throughput process. J Pharm Sci. 93:2755–67.
- Zuylen, L., Verweij, J., Sparreboom, A. (2001). Role of formulation vehicles in taxane pharmacology. Invest New Drugs. 19:125–41.