Abstract
The aim of the present study was to design a depot delivery system of ropivacaine hydrochloride using multivesicular liposomes (RP-MVLs) to overcome the limitations of conventional therapies and to investigate it’s in vivo effectiveness for sustained delivery. RP-MVLs were prepared by the multiple emulsion method. Appearance, particle size, encapsulation efficiency, zeta potential, and initial stability of RP-MVL were also studied. The in vitro release of RP-MVLs formulations was found to be in a sustained manner. Three batches of RP-MVLs were prepared and the release profile in vitro fitted to a first-order equation. RP-MVLs releasing mechanism was also studied and it was indicated that the drug released from MVLs by diffusion and erosion. Following subcutaneous administration to rats, the time to reach maximum (Tmax) of RP-MVLs formulations was significantly (p < 0.01) later than that of RH solution. The concentrations of RP-MVLs have steadily changed in the low level, which significantly (p < 0.01) lower than RH solution. T1/2 and MRT were significantly prolonged (p < 0.01). Besides, AUC was also increased significantly (p < 0.01). The increase in AUC and decrease in Cmax reflects that the MVL formulations could reduce the toxic complications and limitations of conventional injected formation therapies.
Introduction
Ropivacaine, a new long-acting aminoamide local anesthetic agent, is produced as a pure enantiomer. It is structurally related to bupivacaine with a duration of action almost as long as for bupivacaine. The sensory block provided by ropivacaine is similar to that produced by an equivalent dose of bupivacaine in extradural and peripheral nerve block. The motor block produced by ropivacaine is slower in onset, less intense and shorter in duration than that after an equivalent dose of bupivacaine. Motor block intensifies as the dose of ropivacaine is increased in extradural anaesthesia. This, together with its lower toxicity compared with bupivacaine, enables ropivacaine to be used for surgical anaesthesia in concentrations up to 1%. The profile of block with low doses of ropivacaine in terms of its greater sensory-motor separation and higher clearance than bupivacaine make it suitable for use (CitationMcClure, 1996). Preparations of ropivacaine are all injections (e.g., Naropin®). It is needed to give repeated multiple doses to maintain the efficacy, for the short action time after a single dose. Therefore, there may be clinical demand for a longer duration of surgical analgesia after the administration. The present study was about a sustainable release preparation of ropivacaine.
Multivesicular liposomes (MVL) were first prepared by CitationKim et al. (1983). They are a new kind of liposome prepared by Depofoam technology (DepoFoam™). MVL are distinct from unilamellar and multilamellar liposomes by the characteristic structure and composition of MVL. MVL are governed by general topological constraints in analogy to gas–liquid foams, simple liquids, and metallic glasses (CitationMantripragada, 2002). Also, neutral oil, as the essential component, became part of corners or edges where membranes meet each other, thus stabilizing membrane boundaries analogous to planar black lipid membranes that also require a neutral lipid. Only breaches in the outermost membranes of an MVL result in release of encapsulated drug to the external medium, and release of drug from the internal vesicles results in a redistribution of drug inside the particle instead of drug release from the particle (CitationKim et al., 1996; CitationSpector & Zasadzinski, 1996; CitationKatre et al., 1998; CitationLangston et al., 2003; CitationJain et al., 2005). The interconnected network of the multi-vesiculated structure also ensures that the vesicles can rearrange themselves internally without release of drug by internal fusion and division. Therefore, the non-concentric nature of the arrangement of lipid layers confers an increased level of stability and longer duration of drug release (CitationMantripragada, 2002; CitationDavidson et al., 2010; CitationHe et al., 2010; CitationZhao et al., 2010).
In this study, MVL formulations of ropivacaine hydrochloride (RP-MVL) for its sustained delivery were prepared. In vitro release behavior and in vivo pharmacokinetics characteristics of RP-MVL following subcutaneous injection to rats were investigated and compared with those of RP-solution.
Materials and methods
Materials and equipments
Ropivacaine Hydrochloride (Shandong Keyuan Pharmaceutical Company, Lot 0605004); Egg phospholipid (PC, Lipoid Company of Germany); Triolein (TO, Shanghai Chemicals); cholesterin (CH); L-lysine (Shanghai Huixing Biochemical Reagents); and Male Sprague–Dawley rats (190–210 g) were purchased from Qinglongshan Experimental Animal Center (Nanjing, China); FM200 high shear dispersing emulsifier (FLUKO, Shanghai, China); Mastersizer particle size analyzers (Malvern, 2000); Zetasizer 3000HS Laser Diffraction Particle Size Analyzer (Malvern); pH meter (Leici Instrument of Shanghai); and Olympus (BH-2) microscope (Olympus Optical Co., Tokyo, Japan).
Preparation of ropivacaine hydrochloride MVL (RH-MVL)
RH-MVL were prepared by the multiple emulsion method. The emulsions were prepared with a two-step-procedure. In the first step a W/O emulsion was formulated using 50% water phase containing ropivacaine hydrochloride and glucose, and 50% oil phase (CHCl3:EtOH = 1:1) containing PC,CH, and TO. Both phases were mixed together and then emulsified using an FM200 high shear dispersing emulsifier for 8 min (10,000 r/min). In the second step, 4% glucose solution containing 40 mmol/L lysine (the second water phase) was added into the primary W/O emulsion. The W/O/W emulsion was carried out with high shear dispersing emulsification (4000 r/min). In order to get suspension of RH-MVL, the second water phase was added into the emulsion, and then the organic solvent removed with nitrogen. The resulting MVLs were washed with PBS (phosphate-buffered saline) to remove any unencapsulated RH, and harvested by centrifugation at 600 × g. After washing, the RH-MVL were resuspended in PBS. Blank DepoFoam, containing no RH in the first aqueous solution, was also prepared using the same process.
Characterization of RH-MVL
The morphology of RH-MVL suspension was estimated by an optical microscope (×500). Particle size was measured by a Mastersizer particle size analyzer, zeta potential was determined by a Zetasizer 3000HS Laser Diffraction Particle Size Analyzer, and pH was determined by a pH meter.
Determination of encapsulation efficiency
Exactly 0.1 mL of the mixture containing 1 mL RH-MVL suspension and 1 mL of normal saline was added isopropanol 2 mL and diluted to 10 mL. The total content of drug (Dtot) in the preparations was determined by HPLC. The free drug (Dfree) was separated by centrifugation (2000 rpm, 5 min) and quantified with HPLC. Encapsulation efficiency (En%) of RH-MVL was estimated by the following equation:
In vitro release and releasing mechanism of RH-MVL
In vitro release
RH-MVL suspensions were diluted with saline solution and centrifugated for 5 min (2000 rpm). The precipitation was well-dispersed in PBS. Aliquots of the suspension (1 mL) were pipetted into the bag filters (5.0 cm × 2.5 cm, Mw = 14,000), which was in a flask containing 30 ml PBS (0.01 M). The flasks were incubated at 37°C under constant rotation at 100 rpm. Samples were collected at each time point (0.5, 1, 2, 4, 6, 12, 24, 36, 48, 60, and 72 h). Releases of RH solution and the mixture of RH solution and blank MLV suspension were studied the same way. Besides, common liposome suspensions were prepared using a film dispersion method with the same ratio of drug and lipoids, and their release behavior in vitro was also evaluated. The RH concentrations were determined using HPLC assay.
Releasing mechanism of RH-MVL
Aliquots of the suspension (1 mL) were pipetted into the bag filters (5.0 cm × 2.5 cm, Mw = 14,000), which was in a flask containing 30 ml PBS (0.01 M). The flasks were incubated at 37°C under constant rotation at 100 rpm. Samples were collected at each time point (0.5, 1, 2, 4, 7, 10, 24, 36, and 48 h), and observed using an optical microscope (×500).
In vivo pharmacokinetic studies
Rats were randomized into groups of two and six in each. The animals were fasted for 12 h with free access to water and treated subcutaneously with RH injection (group A) or RH-MVL suspension (group B). Both doses were 10 mg. Samples were from group A at each time point (5 min, 15 min, 30 min, 1 h, 1.5 h, 2 h, 4 h, 6 h, 8 h, and 10 h), while from group B at 0.5, 1, 2, 4, 6, 12, 24, 36, 48, 60, and 72 h, from orbital veins (0.3 mL). The heparinized blood was immediately centrifuged at 10,000 rpm for 10 min. Rat plasma was obtained and stored at −20°C until analysis. A 0.1 mL sample of the plasma was dissolved in 0.1 mL mobile phase. The solution was separated by vortex for 1 min and centrifugation at 10,000 rpm for 10 min. A 20 μl aliquot of the solution was injected into the HPLC system for analysis. Plasma concentrations vs time data were analyzed by a compartmental and a non-compartmental model using the 3P87 computer program. The maximum plasma concentration (Cmax) and the time to reach this maximum (Tmax) were obtained directly from the concentration-time profile. Statistical analysis was performed using Student’s t-test with SPSS 11.5.
Results
Characterization of RH-MVL
In the previous study, the formulation of RH-MVL was optimized by Box-Behnken design. shows the morphology of RP-MVLs with an optical microscope (×500), from which we can find that the MVLs were spherical with multiple non-concentric lipid vesicles inside. The opaque parts were lipid membrane, while the transparent parts were aqueous rooms.
Figure 1. Light micrograph of RP-MVL suspension at 500× magnification.
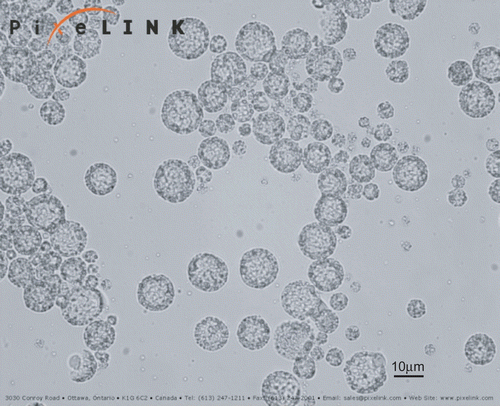
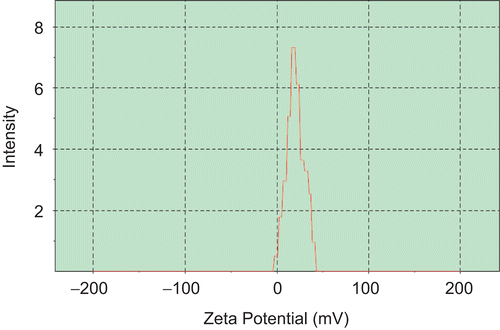
The mean particle size of RP-MVLs was 15.360 μm, and more than 85% of the particles were in the range of 7~30 μm (). The zeta potential of RH-MVL suspension was 20.7 mV (), and the pH was 7.43.
Figure 2. Particle size distribution of RP-MVL suspension.
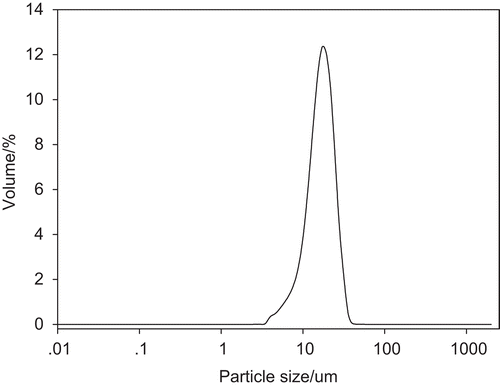
Figure 3. Zeta potential of RP-MVL suspension.
Encapsulation efficiency
shows the encapsulation efficiencies determined from three batches prepared with the optimal formulation. The encapsulation efficiencies of RP-MVLs were 90.20 ± 0.77%.
Table 1. Entrapment efficiency of RP-MVL.
In vitro drug release and approach of RH-MVL releasing mechanism
In vitro drug release
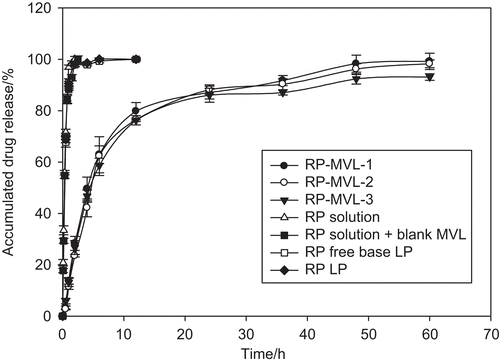
The release profile of ropivacaine from the multivesicular liposomes, ropivacaine solution, mixture of ropivacaine solution and blank multivesicular liposomes, ropivacaine free base liposomes, and ropivacaine hydrochloride liposomes in vitro are shown in . The drug release rate from RP-MVLs was much slower than that from ropivacaine free base liposomes, for the former released 90% within 48 h, while the latter released more than 90% within 6 h. Another two solutions released quickly, finishing in 2 h. The profiles of three batches of RH-MVL suspensions were well coincided with each other, which was due to reproducibility of the formulation and the technology. Fitting the cumulative release percentage (Q) with first order equation: Ln(1 − Q) = −kt; Higuchi equation: Q = kt1/2; Weibull equation: LnLn[1/(1 − Q)]= kLn(t − τ)-Lnt0, and Hixcon-Crowell equation: (1 − Q)1/3 = −kt. They fitted well with the first order equation ().
Table 2. Fitting equations and correlation coefficients.
Figure 4. The release profile of ropivacaine from the multivesicular liposomes, ropivacaine solution, mixture of ropivacaine solution and blank multivesicular liposomes, ropivacaine free base liposomes, ropivacaine hydrochloride liposomes in vitro (mean ± SD, n = 3).
Study of RH-MVL releasing mechanism
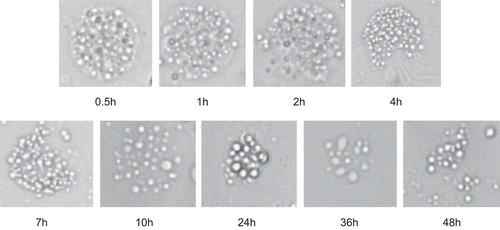
shows morphologic change of RP-MVL during the releasing period. With time passed, the integrated sphericity of RH-MVL appeared to be corrosion, and the particle size reduced for vacuoles broke up. This was probably because of rearrangement of lipid membranes. Vesicles inside may aggregate with each other for the number of compartments decreased and the compartments became smaller. Besides, some particles with irregular modality appeared and so did free single vesicles at the end of the releasing process. This indicated that the drug release from MVLs was by the effort of diffusion and erosion.
Figure 5. Morphologic change of RP-MVL during the releasing period (×500).
In vivo pharmacokinetic studies
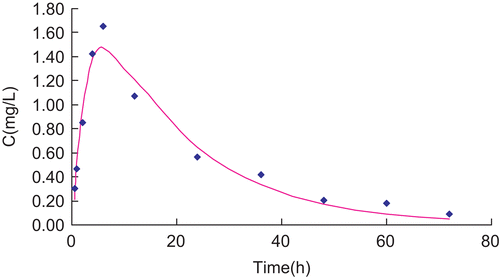
Plasma concentrations vs time data of RH solution and RH-MVL suspension were analyzed by a compartmental using the 3P87 computer program (, and ). Following the Akaike’s information criterion (AIC), both profiles well fitted the signal compartment model of first order absorption. R is 0.9819 and 0.9580, respectively. and showed the fitting curve of concentration-time profiles of RP in plasma after subcutaneous injection of RP solution and MVL in rats by 3P87. Statistical moment parameters, including AUC, AUMC, S2, MRT VRT, and K were calculated using the mean values of determined. It can be found that there were differences between each other ().
Table 3. Mean plasma concentration of RP after subcutaneous injection of RP solution and RP-MVL in rats (![]() ± SD, n = 6).
± SD, n = 6).
Table 4. Pharmacokinetics parameters of RP in plasma after subcutaneous administration in rats (![]() ± SD, n = 6).
± SD, n = 6).
Table 5. Parameters based on statistical moment theory for RP in plasma after subcutaneous administration in rats (![]() ± SD, n = 6).
± SD, n = 6).
Figure 6. Concentration-time profiles of RP in plasma after subcutaneous injection of RP solution and RP-MVL in rats (![]()
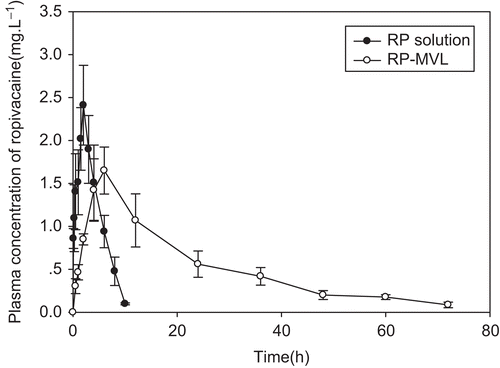
Figure 7. Fitting curve of concentration-time profiles of RP in plasma after subcutaneous injection of RP solution in rats.
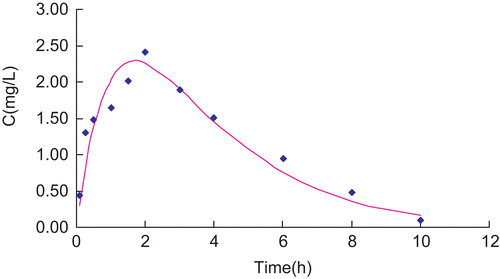
Figure 8. Fitting curve of concentration-time profiles of RP in plasma after subcutaneous injection of RP-MVL in rats.
shows concentration vs time profiles of RP in plasma after subcutaneous injection of RP solution and RP-MVL suspension in rats. For RH-MVL suspension, the time to reach maximum (Tmax) was 6.063 ± 0.830 h, which was significantly (p < 0.01) later than RH solution (Tmax = 1.696 ± 0.221 h). The concentrations of RH-MVL have steadily changed in the low level, and Cmax was 1.472 ± 0.268 mg/L, significantly (p < 0.01) lower than RH solution (Cmax = 2.289 ± 0.379 mg/L). T1/2 (12.746 ± 1.835 h vs 1.816 ± 0.266 h) and MRT (24.783 ± 4.151 h vs 3.749 ± 0.224 h) were significantly prolonged (p < 0.01). Besides, AUC was also increased significantly (p < 0.01).
The significant prolonged Tmax indicated that drug releasing from RH-MVL was delayed, while from solution was quick, and the drug were also eliminated swiftly. MVLs’ big size helped them escape from the absorption of the lymph or capillary network. In addition, MVLs had numerous aqueous chambers with drugs inside. Therefore, MVLs containing drugs could stay at the administration site as a drug-depot. The plasma concentration can be keep at a relative high level within a long time, for the drug diffused into systemic circulation slowly (CitationJain et al., 2005). Compared with common injections, the profile of MVL has lower concentrations, smaller changes, and later Tmax. The sustained-release of RH from MVLs could contribute to avoiding toxic reactions for exorbitant concentrations in a short time.
Conclusions
RP-MVLs were prepared in this study. Mean particle size of RP-MVLs was 15.360 μm, zeta potential was 20.7 mV, and the pH was 7.43, which made them have biocompatibility. In addition, MVLs, as a drug carrier, can form a drug store at the administration site. In vitro release profile of RP-MVLs fitted the first order equation. The morphologic changes of RP-MVL during the releasing period indicated that the drug release mechanism from MVLs contributed to the effort of diffusion and erosion. In vivo pharmacokinetic studies demonstrated that drug released sustainedly from MVLs, and the differences between parameters were significant (p < 0.01). Clearly, the prepared RP-MVLs are able to overcome the shortcoming of common injections of RH, such as toxic actions, short half-life, and low bioavailability. Taken together, the sustained-delivery durations of RH-MVL both in vitro and in vivo prolonged significantly compared with those of RH-LP and RH solution, which offers a safe and convenient sustained-release platform to deliver systems.
Declaration of Interest
The authors thank the partial financial support from Ministry of Science and Technology of the People’s Republic of China under project 2009ZX09310-004.
References
- Davidson, E.M., Barenholz, Y., Cohen, R., Haroutiunian, S., Kagan, L., Ginosar, Y. (2010). High-dose bupivacaine remotely loaded into multivesicular liposomes demonstrates slow drug release without systemic toxic plasma concentrations after subcutaneous administration in humans. Anesth Analg. 110:1018–23.
- He, S., Zhu, J., Xie, F. (2010). Preparation and characterization of tramadol PEG-coated multivesicular liposomes for sustained release. Pharmazie. 65:467–70.
- Jain, S.K., Jain, R.K., Chourasia, M.K., Jain, A.K., Chalasani, K.B., Soni, V., Jain, A. (2005). Design and development of multivesicular liposomal depot delivery system for controlled systemic delivery of acyclovir sodium. AAPS PharmSciTech. 6:35–41.
- Katre, N.V., Asherman, J., Schaefer, H., Hora, M. (1998). Multivesicular liposome (DepoFoam) technology for the sustained delivery of insulin-like growth factor-I (IGF-I). J Pharm Sci. 87:1341–6.
- Kim, S., Turker, M.S., Chi, E.Y., Sela, S., Martin, G.M. (1983). Preparation of multivesicular liposomes. Biochim Biophys Acta. 728:339–48.
- Kim, T., Murdande, S., Gruber, A., Kim, S. (1996). Sustained-release morphine for epidural analgesia in rats. Anesthesiology. 85:331–8.
- Langston, M.V., Ramprasad, M.P., Kararli, T.T., Galluppi, G.R., Katre, N.V. (2003). Modulation of the sustained delivery of myelopoietin (Leridistim) encapsulated in multivesicular liposomes (DepoFoam). J Contr Rel. 89:87–99.
- Mantripragada, S. (2002). A lipid based depot (DepoFoam technology) for sustained release drug delivery. Prog Lipid Res. 41:392–406.
- McClure, J.H. (1996). Ropivacaine. Br J Anaesth. 76:300–307.
- Spector, M.S., Zasadzinski, J.A. (1996). Topology of multivesicular liposomes, a model biliquid foam. Langmuir. 12:4704–8.
- Zhao, Y., Liu, J., Sun, X., Zhang, Z.R., Gong, T. (2010). Sustained release of hydroxycamptothecin after subcutaneous administration using a novel phospholipid complex-DepoFoam technology. Drug Dev Ind Pharm. 36:823–31.