Abstract
The stability and activity of the HIV cell-penetrating TAT peptide (TATp) on the surface of TATp-modified micelles and liposomes in relation to its proteolytic cleavage was investigated. TATp moieties were attached to the surface of these nanocarriers using TATp modified with a conjugate of phosphatidyl ethanolamine with a ‘short’ PEG (PEG-PE). Following pre-incubation with trypsin, elastase, or collagenase, the proteolytic stability of TATp on the surface of these modified carriers was studied by HPLC with fluorescence detection using fluorenylmethyl chloroformate (FMOC) labeling. All tested enzymes produced a dose-dependent cleavage of TATp as shown by the presence of TATp Arg-Arg fragments. Inhibition of TATp cleavage occurred when these TATp-micelles were modified by the addition of longer PEG-PE blocks, indicating an effective shielding of TATp from proteolysis by these blocks. TATp-modified carriers were also tested for their ability to accumulate in EL-4, HeLa, and B16-F10 cells. Trypsin treatment of TATp-modified liposomes and micelles resulted in decreased uptake and cell interaction, as measured by fluorescence microscopy and fluorescence-activated cell sorting techniques. Furthermore, a decrease in the cytotoxicity of TATp-modified liposomes loaded with doxorubicin (Doxil) was observed following trypsin treatment. In conclusion, steric shielding of TATp is essential to ensure its in vivo therapeutic function.
Introduction
Cell penetrating peptides (CPPs) have been successfully used to deliver a large variety of cargoes, from small particles to proteins, peptides, nucleic acids, and even pharmaceutical nanocarriers to the cell interior both in vitro and in vivo (CitationAbes et al., 2007; CitationJarvert et al., 2007; CitationFoged & Nielsen, 2008; CitationKersemans et al., 2008; CitationTorchilin, 2008b; CitationSawant & Torchilin, 2009). In terms of their composition, all CPPs can be divided into two classes: the first consists of amphipathic helical peptides (e.g. transportan and model amphipathic peptide) with lysine as the main contributor to their positive charge, while the second includes arginine-rich peptides, (e.g. TAT peptide (TATp), Antp or penetratin) (CitationHallbrink et al., 2001).
A representative CPP is TATp47–57, which is derived from the transactivator of transcription (TAT) of human immunodeficiency virus (CitationGreen & Loewenstein, 1988; CitationChauhan et al., 2007). It contains the two positively-charged amino acids, arginine and lysine (YGRKKRRQRRR), which makes it highly sensitive to cleavage by proteolytic enzymes. Since cell penetration of CPPs relies on their basic amino acid sequence, intracellular delivery of therapeutic molecules with TATp-modified carriers may be limited by cleavage with proteolytic enzymes present in the circulation or in the cell cytoplasm. Surprisingly, only a few papers deal with the enzymatic stability of CPPs. A few reports suggested that CPP-mediated drug delivery may be hindered by the rapid metabolic degradation of the peptide by cells or body fluids (CitationElmquist & Langel, 2003; CitationTrehin et al., 2004; CitationFoerg et al., 2008). Using radioactive TATp, CitationFrankel and Pabo (1988) demonstrated that, although TATp becomes localized in the nucleus after cellular uptake, it appears to go through the proteolytic degradation process. The researchers used chloroquine as a protective and stabilizing agent. Furthermore, as shown by CitationElmquist and Langel (2003), CPPs stability could be enhanced with protection of the C-terminus of peptides by amidation. Synthesized D-CPPs were resistant against degradation and retained their translocation potential.
Thus, when used in a free form, chemically coupled or physically conjugated with a variety of active molecules or drug carriers, TATp and other CPPs should be considered potential substrates for various proteolytic enzymes. To date, the role of these peptide–enzyme interactions in the context of using CPPs as surface modifiers of sterically shielded (PEGylated, long-circulating) drug delivery systems has been little explored. Recently, we investigated the kinetics of the proteolytic cleavage of free TATp, a TATp-PEG-PE conjugate and TATp-modified PEG-PE micelles (CitationGrunwald et al., 2009). It was clearly demonstrated that the TATp moiety becomes completely protected against trypsinolysis when TAT-PEG-PE conjugate is incorporated into self-assembling PEG-PE micelles if the PEG block in PEG-PE is longer than the PEG block in the TATp-PEG-PE conjugate.
In the current study, we have investigated the extent to which the proteolytic cleavage of TATp influences the in vitro cellular internalization of TATp-modified pharmaceutical nanocarriers (liposomes and polymeric PEG-PE micelles), when the TATp moiety is openly exposed on their surface or when partially shielded by long PEG-PE blocks. By using flow cytometry and fluorescence microscopy techniques, we were able to demonstrate that TATp cleavage diminished particle incorporation by cells. When using TATp-modified carriers, temporary peptide steric protection is required to achieve an efficient carrier-to-cell interaction. To gain insight into the specifics of the TATp cleavage following nanoparticle modification with long PEG-PE blocks, we also manipulated the nanoparticle structures by varying the levels of PEGylation. We have shown that when designing multifunctional drug delivery systems with a cell-penetrating function (e.g. with TATp), steric protection of CPP moieties against proteolytic degradation is essential.
Materials and methods
Materials
TAT-cysteine peptide (TATp-Cys 12-mer: CysTyrGlyArgLysLysArgArgGlnArgArgArg; molecular mass 1663 Da with one reactive thiol group) was synthesized by the Tufts University Core Facility (Boston, MA). 1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), NHS-PEG1000-maleimide, diacyllipid polyethylene glycols (PEG750-PE, PEG1000-PE, PEG2000-PE, and PEG5000-PE), rhodamine-phosphatidylethanolamine (Rh-PE), and phosphatidylthioethanol (DPPE-SH) were purchased from Avanti Polar Lipids (Alabaster, AL). Triethylamine (TEA) and fluorenylmethyloxycarbonyl chloride (FMOC-Cl) were purchased from Fluka (AG, Switzerland). 4-Acetyl phenyl maleimide (APmal) was from Acros Organics (Fairlawn, NJ), mPEG-SH- from Laysan Bio, Inc. (Huntsville, AL), and 4-(4-N-maleimidophenyl) butyric acid hydrazide hydrochloride (MPBH) from Pierce Biotechnology, Inc. (Rockford, IL). Doxil was from Ben Venue Laboratories Inc. (Bedford, OH), Fluoromount-G was from Southern Biotechnology Associates Inc. (Birmingham, AL), and CellTiter Blue cell viability assay was from Promega (Madison, WI). TATp-PEG1000-PE (TATp-conjugate) was synthesized in-house (see below). Trypsin from porcine pancreas (type IX-S), chymotrypsin-trypsin inhibitor, heparinase-I from Flavobacterium heparinum, and lyophilized human plasma (reconstituted with deionized water) were purchased from Sigma-Aldrich (St. Louis, MO). Collagenase (type II) was obtained from Worthington Biochemical Corporation (Lakewood, NJ). Elastase (Grade II) from pig pancreas was obtained from Roche Applied Science (Indianapolis, IN). Cell lines (B16-F10, EL-4, and HeLa) were purchased from the American Type Culture Collection (Manassas, VA). All cell culture media, DMEM, heat-inactivated fetal bovine serum (FBS), and concentrated solutions of penicillin/streptomycin stock solutions were from Cellgro® (Herndon, VA). All other chemicals and solvents were of analytical grade, purchased from Fisher Scientific, and used without further purification.
Methods
Synthesis of TATp-PEG1000-PE (TATp conjugate)
The TATp-PEG1000-PE conjugate was synthesized as described previously (CitationTorchilin et al., 2001) with some modifications. Briefly, an ~ 1.5-fold molar excess of NHS-PEG1000-maleimide was reacted with DOPE by stirring for 2 h in chloroform at room temperature with a 3-fold molar excess of triethylamine. A 2-fold molar excess of TATp-Cys was then added, and the reaction was continued with stirring overnight. The solvent was evaporated, and the product was freeze-dried overnight. The excess of TATp-Cys was separated from the product by gel filtration chromatography. The solvent was evaporated, product was freeze-dried overnight, and the dried product was dissolved in 0.5 mL of water and loaded onto a Sephadex G25 column (length, 10–15 cm; diameter, 0.5–1 cm). Fractions were collected and monitored by TLC using silica plates (mobile phase of chloroform/methanol 80:20% v/v), and TATp-PEG-PE was visualized with phosphomolybdic acid and Dragendorff spray reagents.
Preparation of TATp-micelles
For all micelle preparations, a lipid film was first obtained from a mixture of diacyllipid-PEGs (PEG750-PE, PEG1000-PE, PEG2000-PE, and PEG5000-PE) in different ratios and 5 mol% of a TATp-PEG1000-PE conjugate in chloroform. One mol% of rhodamine-PE (λex568/λem590) was added to fluorescently labeled micelles. Chloroform was evaporated with N2 gas followed by freeze-drying on a Freeze Dry System Freezone 4.5 (Labconco, Kansas City, MO). The film was hydrated with PBS pH 7.4 and vortexed at room temperature for 5 min. Preparations were pre-exposed to the various proteolytic enzymes or inhibitors at 37°C for 1 h before further analysis or before adding them to cell preparations.
Preparation of TATp-liposomes
For all liposome preparations, a lipid film was first obtained from a mixture of egg-phosphatidylcholine, cholesterol, and rhodamine-PE (7:3:0.1 molar ratio) in chloroform. For TATp-modified liposomes, 5 mol% of TATp-PEG1000-PE was added. Chloroform was removed using N2 gas followed by freeze-drying on a Freeze Dry System Freezone 4.5 (Labconco, Kansas City, MO). The film was hydrated with an appropriate buffer and vortexed at room temperature for 5 min. The lipid dispersion was extruded 21 times through polycarbonate filters (pore size 200 nm) with a hand-held extruder (Avanti, Alabaster, AL). Preparations were pre-exposed to the various proteolytic enzymes or trypsin inhibitor at 37°C for 1 h before further analysis or before adding them to cell preparations.
Preparation of TATp-modified Doxil
The technique for anchoring a peptide (CitationWang et al., 2010) and the monoclonal antibody, 2C5 (CitationGupta & Torchilin, 2007), into liposomal bilayers of doxorubicin-loaded PEGylated liposomes (Doxil) was previously described. TATp conjugate (5 mol%) was incubated with the Doxil liposomal preparation overnight to form a TATp-Doxil® complex. Doxil and TATp-Doxil liposome size determined by dynamic light scattering (see below) showed size for both preparations of 54–90 nm.
Preparation of PEGylation shielded micelles
We have designed a series of nanoparticles, containing TATp moieties shielded with PEG2000-PE or PEG5000-PE blocks in different concentrations. We analyzed the cleavage of TATp Arg-Arg groups by fluorenylmethyl chloroformate (FMOC) labeling using HPLC with fluorescence detection. Micelles were prepared using PEG750-PE as the main micelle-forming component with the addition of 5 mol% TATp-PEG1000-PE. This formulation was considered ‘non-shielded’. Additional micellar formulations were prepared using longer PEG-PE blocks (PEG2000-PE and PEG5000-PE). Mol% of the longer PEG-PE blocks was increased from zero (‘non-shielded’) to 95mol% (‘fully shielded’). We analyzed the percentage of TATp Arg-Arg group cleavage by fluorenylmethyl chloroformate (FMOC) labeling using HPLC with fluorescence detection.
Size and zeta-potential measurements
Micelle/liposome size measurements and size distribution analysis were performed by dynamic light scattering (DLS) using a Coulter® N4-Plus Submicron Particle Sizer (Coulter Corporation, Miami, FL). Particles dispersions were diluted with de-ionized distilled water until a concentration providing a light scattering intensity of 5 × 104–1 × 106 counts/s was achieved. Micelle/liposome surface charge analysis was performed using a Zeta Phase Analysis Light Scattering (PALS) UltraSensitive Zeta Potential Analyzer instrument (Brookhaven Instruments, Holtsville, NY). Each sample was diluted with de-ionized distilled water to adjust the signal intensity within the limits required by the instrument. The zeta-potential of each sample was determined from five independent measurements.
HPLC analysis
The Hitachi Elite Lachrom system (Tokyo, Japan) composed of a fluorescence detector and EZChrom Elite (Agilent, Pleasanton, CA) data software was used for the determination of TATp cleavage on the surface of TATp-PEG1000-PE-containing micelles and liposomes. The chromatographic separation was performed on a reversed phase symmetric C-18 column (pore size, 100 Å; particle size, 5 μm; dimension, 4.6 × 150 mm, Waters, Milford, MA) with a binary linear gradient elution program. The binary gradient consisted of eluent A (0.1% formic acid in acetonitrile) and eluent B (0.1% formic acid in deionized water). The column was equilibrated with 30% eluent A, and the separation was performed with the [eluent A]:[eluent B] gradient containing the break points at 0 min (30:70, v/v), 10 min (90:10), 15 min (90:10), and 20 min (30:70). TATp, attached to carrier surfaces, was labeled with fluorenylmethyl chloroformate (FMOC) after incubation with enzymes. As previously shown by MALDI-TOF MS (CitationGrunwald et al., 2009), the sequence of TATp can be cleaved at six sites by trypsin. Using HPLC fluorescence detection, we followed the TATp Arg-Arg (RR) pair proteolysis fragments formed as a result of its endocleavage. The degree of this pair’s cleavage was correlated with TATp cleavage and was measured following the exposure of the TATp-containing liposomes and micelles to the various proteolytic enzymes: trypsin, elastase, and collagenase. Nanoparticles were incubated with the various enzymes for 10 min in PBS, pH 7.4, at 37°C before HPLC analysis. For samples treated with trypsin, an excess of trypsin inhibitor (1 mg/ml) was used simultaneously to hinder trypsin proteolytic properties and prevent peptide trypsinolysis.
Cell cultures
The cell lines mouse melanoma (B16-F10), human adenocarcinoma (HeLa), and mouse lymphoma (EL-4) were grown in DMEM with 2 mM L-glutamine supplemented with 10% (v/v) heat-inactivated fetal bovine serum, 100 units/ml penicillin G, and 100 μg/ml streptomycin. Cultures were maintained at 37°C in a humidified 5% CO2 incubator. In all studies, the cells were sub-cultured every 2–3 days and were used for experiments at passages 5–20.
Interaction of micelles/liposomes with cells
The cellular uptake of the various carriers was studied visually using trypsin-treated micelles and liposomes labeled with 1 mol% rhodamine-PE. Adherent B16-F10 and HeLa cells were grown on glass cover slips placed in six-well tissue culture plates. When the cells reached a confluency of 60–70%, they were washed twice with PBS, medium was replaced, and a further incubation for 1 h at 37°C, 5% CO2 with pre-treated samples of micelles and liposomes was conducted. The micelle and liposome samples were pre-exposed to trypsin (0.1 mg/ml) or trypsin and trypsin inhibitor (1 mg/ml) prior to their addition to the cells. Hoechst nuclear stain (2.5 μg/ml) was added for the last 30 min of incubation. Cells were washed three times with PBS and mounted individually cell-side down on glass slides using fluorescence-free glycerol-based mounting medium (Fluoromount-G). Since EL-4 cells were grown in suspension, the exposure procedure with the various formulations was slightly different. Briefly, EL-4 cells were placed onto plastic six-well culture plates at 5 × 105 cells/ml. Following the incubation with trypsin-treated rhodamine-labeled micelles or liposomes and with Hoechst nuclear stain (see above), cells were washed with PBS three times and then mounted individually on glass slides using the mounting medium. All mounted slides were studied with a Nikon Eclipse E400 microscope under fluorescence using a Rh/TRITC filter for Rh-labeled micelles/liposomes (red) and a UV-2B filter for Hoechst labeled nuclei (blue).
FACS analysis
To assess the cell binding of TATp-modified liposomes and micelles compared to trypsin-treated carriers, flow cytometry (FACS) analysis of the different cell types incubated with different formulations was performed. HeLa and B16-F10 cells (5 × 104 cells/ml) were plated in a six-well plate (35 mm diameter) in 1.5 ml medium and incubated for 24 h before further experiments. Equal numbers of the suspension-grown EL-4 cells were used for FACS experiments. After the replacement of the medium with fresh serum-free medium, the pre-treated TATp-modified rhodamine-labeled micelles/liposomes were added to the cells. Rhodamine-labeled formulations were pre-exposed to trypsin (0.1 mg/ml) and trypsin inhibitor (1 mg/ml) for 1 h. The final lipid concentration was 0.3 mg/ml. After 1 h of incubation, the medium was discarded and the cells were washed three times with cold PBS. B16-F10 and HeLa cells were detached following 5 min of incubation with trypsin solution (0.25%) at 37°C and then washed with cold PBS, centrifuged at 1000 rpm, and re-suspended three times. The cellular uptake/attachment efficiency was determined by FACScan™ (Beckton Dickinson biosciences, San Jose, CA), acquiring 10,000 events per histogram.
Cytotoxicity
The cytotoxicity of TATp-modified liposomes, loaded with doxorubicin (TATp-Doxil), against B16-F10 and HeLa cells was studied. Liposomal formulations were pre-treated with trypsin (0.1 mg/ml) or trypsin (0.1 mg/ml) with trypsin inhibitor (1 mg/ml) for 1 h before addition to cells. Cells were seeded into 96-well microplates at a density of 5 × 103 cells/well. Following 24 h incubation and growth to ~ 50% confluency, trypsin-treated formulations containing doxorubicin were added to cells in triplicate to give the final concentration of 1 μg/ml. After 24 h incubation at 37°C, 5% CO2, plates were washed three times with PBS, followed by the addition of 20 μl/well of CellTiter Blue solution to assess viability. After 1 h incubation at 37°C, 5% CO2, the cell survival was estimated by measuring the fluorescence intensity using a microplate reader (Synergy HT multimode microplate reader, BioTek Instrument, Winooski, VT) with 525/590 nm excitation/emission wavelengths.
Statistical analysis
Statistical analysis was performed using GraphPad Prism (GraphPad Software, San Diego, CA). Unless mentioned, the results are presented as the mean ± SD. Unpaired Student’s t-tests were performed as specified in figure legends (differences were considered significant when p ≤ 0.05).
Results and discussion
Particles size and zeta potential
The results of the zeta potential measurements are presented in , which shows the effect of trypsin on TATp-modified liposomes/micelles zeta potential values. Our results suggest that the positive charge of the TATp-modified particles decreases following the incubation with 0.1 mg/ml of trypsin for 10 min at 37°C. Since TATp is rich in arginine, which contributes to its cationic properties, it is also the main contributor to the total positive charge of both micelles and liposomes. The decrease in zeta potential values from −6.7 ± 3.0 mV to −15.7 ± 1.4 mV for TATp-modified micelles and from 4.3 ± 2.7 mV to −15.9 ± 2.0 mV for TATp-modified liposomes supports our conclusion that TATp was cleaved by trypsin. When particles were incubated simultaneously with trypsin and trypsin inhibitor the positive charge of TATp-modified particles was retained, confirming that trypsinolysis resulted in TATp cleavage and charge decrease. Particle size analysis of both micelles and liposomes showed no significant changes following incubation with trypsin, retaining a magnitude of 20 nm and 150 nm for micelles and liposomes, respectively. Thus, TATp degradation does not affect the size characteristics of the peptide-modified particles.
Table 1. Zeta potential of TATp-modified liposomes and micelles. Nanocarriers Incubated with trypsin (0.1mg/ml) for 10 min in PBS, pH 7.4, at 370C. Trypsin inhibitor, 1mg/ml. (*p≤0.05 vs. plain nanocarriers, n=5).
Enzymatic cleavage of TATp-modified micelles and liposomes
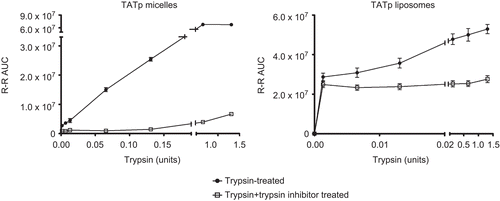
The fragments of the surface-attached TATp formed as the result of its proteolysis by various enzymes were followed by HPLC analysis, as previously described (CitationGrunwald et al., 2009). The TATp cleavage was monitored by tracking the amount of the cleaved Arg-Arg (RR) groups using FMOC fluorescence labeling. demonstrates that trypsin cleaved TATp on the surface of TATp-modified micelles and liposomes in a dose-dependent manner. While for liposomes the concentration needed to reach 50% peptide cleavage was 0.001 units (0.1 μg/ml), TATp-modified micelles required higher trypsin amounts in order to achieve the same cleavage percentage (0.1 units, equivalent to 10 μg/ml). These results show higher shielding for TATp on PEG-PE micelles vs Chol-PC liposomes, probably due to the membrane structure of PEGylated micelles vs liposomes. Furthermore, when particles were pre-incubated with chymotrypsin-trypsin inhibitor TATp cleavage levels were reduced by ~ 50% for TATp-modified liposomes and ~ 85% for TATp-modified micelles (see ). In order to explore whether the TATp cleavage by trypsin is exclusive for trypsin or can occur with other proteolytic enzymes, we used elastase and heparinase treatments. It was previously shown by mass spectroscopy techniques (CitationGrunwald et al., 2009) that TATp is susceptible for cleavage in plasma, suggesting that other proteolytic enzymes degrade the peptide and influence its properties. Additional enzymes tested heparinase and elastase (not shown), cleaved the TATp in a dose-dependent manner. This endocleavage occurred in both TATp-modified liposomes and micelles.
Figure 1. HPLC TATp RR fragment analysis of enzymatic cleavage following incubation of TATp-modified micelles/liposomes with trypsin/trypsin inhibitor (0.1mg/ml and 1mg/ml respectively). Particles were incubated with enzymes for 10 min in PBS, pH 7.4, at 370C before HPLC analysis. (n=3)
Steric shielding of TATp-modified particles
When designing a ‘smart’ multifunctional drug delivery platform that responds to specific characteristics of a targeted pathological area such as increased temperature or lowered pH, which are typical of inflamed, ischemic, and neoplastic tissues, TATp-containing drug carriers should be designed in such a way that, during the first phase of delivery, the non-specific cell-penetrating function (CPP moiety) is sterically shielded by a polymer or targeting antibody. This shielding is of great importance in view of the presence of various proteolytic enzymes in the circulation and the possible cleavage of TATp by their action. Upon accumulation in the target area, this stimulus-sensitive steric shielding ideally becomes detached under the local cellular-pathological conditions to expose the TATp and allow delivery of the carrier and its cargo within cells. TATp-modified stimuli-sensitive polymeric micelles and liposomes with an enhanced ability to interact with cells at low pH have been previously described (CitationTorchilin, 2008a; CitationBae & Kataoka, 2009; CitationKale & Torchilin, 2010).
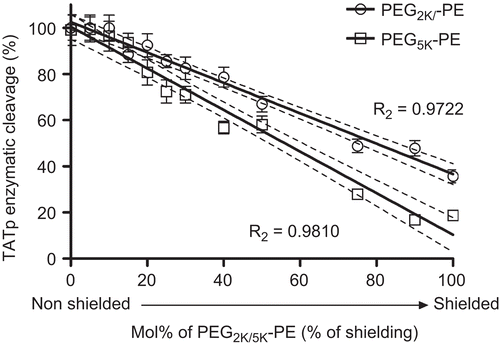
We have designed a series of TATp-modified PEGylated micelles (see Materials and methods). As shown in , higher PEG shielding resulted in lower TATp enzymatic cleavage. Furthermore, the longer the PEG chain used for shielding (PEG2k-PE vs PEG5k-PE) the less the enzymatic cleavage. Our results suggest that stimuli-sensitive architecture of intelligent drug delivery platforms should also take into consideration the need for the shielding of TATp from enzymatic cleavage.
Figure 2. PEG shielding of TATp-micelles. HPLC TATp RR fragments analysis of TATp-modified micelles, shielded with increasing Mol% of long PEG2k-PE or PEG5k-PE blocks. TATp enzymatic cleavage was measured following incubation of TATp-modified micelles with trypsin (0.1mg/ml) and presented as percent of cleavage. Particles were incubated with enzyme for 10 min in PBS, pH 7.4, at 370C. (n=3)
In vitro studies
Trypsin was used to confirm that when TATp is attached to the surface of micelles or liposomes, TATp-to-cell interaction is influenced by TATp exposure to proteolytic enzymes. TATp-modified micelles and liposomes, treated with trypsin for 1 h, were analyzed for their cell-interaction capabilities by fluorescence microscopy and FACS techniques. In addition, TATp-modified liposomes, loaded with doxorubicin (TATp-Doxil) and pre-incubated with trypsin, were monitored for their cytotoxicity properties.
Fluorescence microscopy
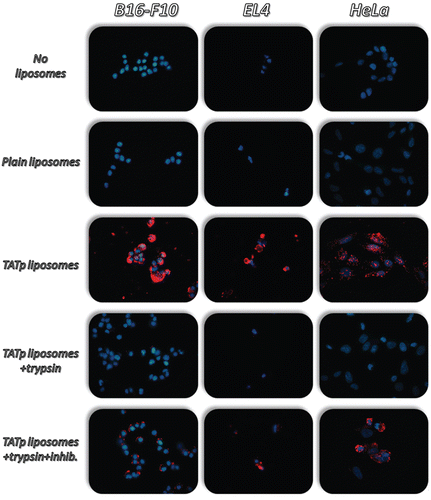
TATp-modified liposomes were tested for their interaction with cells and for the effect of a proteolytic agent on this interaction. Fluorescence microscopy showed that non-rhodamine-labeled TATp-modified liposomes were significantly internalized by HeLa, B16-F10, and EL-4 cells (). Diminished cell internalization was observed following the pre-incubation of formulations with trypsin. This decrease was prevented by pre-incubation of trypsin inhibitor with the formulation solutions. Similar results were obtained using rhodamine-labeled TATp-modified micelles (data not shown). These results suggest that proteolytic enzymes, like trypsin, will prevent or reduce the interaction of TATp-modified liposomes and micelles with cells by degrading TATp moieties on their surface and that those carriers should be shielded in order to maintain their function.
Figure 3. Fluorescence microscopy showing the internalization of rhodamine-PE-labeled TATp-containing liposomes (Red-rhodamine; Blue-Hoechst nuclei staining) by B16-F10, EL4 and HeLa cells following incubation with trypsin (0.1mg/ml) for 1hr at 370C. Both TATp-modified liposomes and TATp-modified liposomes pre-treated with trypsin and with trypsin inhibitor (1mg/ml) enhanced internalization by cells.
Fluorescence activated cell sorting (FACS) analysis
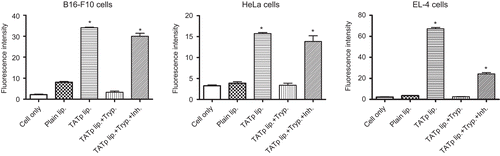
The in vitro cell interaction of the TATp-modified particles was confirmed using FACS (). As expected, the analysis of the geometric mean fluorescence revealed a 5–30-fold stronger binding of TATp-modified liposomes to cells compared with plain liposomes. As before, binding experiments with liposomes pre-treated with trypsin (0.1 mg/ml for 1 h) showed an absence of cell binding. Again, when trypsin inhibitor (1 mg/ml) was added during the pre-incubation time, a significant signal recovery occurred, indicating prevention of TATp cleavage. Similar results were obtained with TATp-modified micelles (data not shown). Furthermore, when TATp-modified liposomes were pre-incubated with human plasma for 30 min and then added to cells, a 40% decrease in fluorescence signal was observed, indicating less interaction of liposomes with cells (data not shown). This suggests that TATp was cleaved by plasma’s proteolytic enzymes, even after a short incubation and prevented liposome binding to cells.
Figure 4. Flow cytometry. Average fluorescence intensity of B16-F10, EL4 and HeLa cells incubated with rhodamine-labeled TATp liposomes following exposure to trypsin/trypsin inhibitor (0.1mg/ml and 1mg/ml respectively) for 1hr. (*p≤0.05 vs. plain liposomes, n=5)
Cytotoxicity of doxorubicin-loaded TATp liposomes
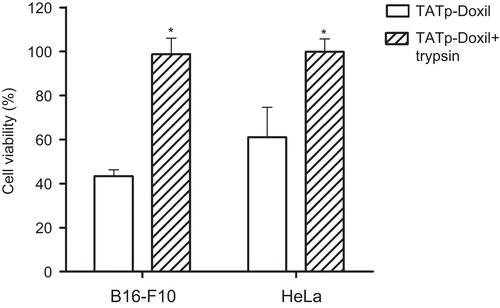
The cytotoxicity of TATp-modified liposomal preparations loaded with doxorubicin (TATp-Doxil) was investigated using B16-F10 and HeLa cells. Formulations were pre-treated with trypsin to explore its effect on the TATp moieties’ ability to enhance cellular cytotoxicity. presents the data obtained at 1 μg/ml final doxorubicin concentrations following 24 h incubation of the various formulations with cells. While TATp-modified liposomes formulation demonstrated a significant high toxicity against B16-F10 and HeLa cancer cell lines (43% and 61% viability, respectively, p ≤ 0.05), significantly less cytotoxicity was shown if the formulation had been exposed to trypsin for 1 h before addition to the cells.
Figure 5. In vitro cytotoxicity of a TATp-Doxil formulation towards B16-F10 and HeLa cells. The formulation was pre-treated with 0.1mg/ml trypsin for 1 hr. followed by 24 hr. incubation with cells at 370C before the viability assay. (*p≤0.05 vs. TATp-Doxil, n=5)
Conclusion
The proteolysis of TATp on the surface of TATp-modified liposomes and PEG-PE micelles was investigated. Proteolytic enzymes degrade TATp even when TATp is attached to the surface of liposomes or micelles. EL4, B16-F10, and HeLa cells had little internalization of trypsin-treated TATp-modified nanocarriers compared to non-treated nanocarriers. As expected, trypsin inhibition decreased TATp degradation and left TATp in its active form. Degradation of CPPs before reaching their target, whether in the cytoplasm, nucleus, or extracellular fluids, must be avoided for CPPs to serve as an efficient moiety that promotes intracellular delivery of a carrier or cargo. Steric shielding of CPPs is essential to ensure its effective in vivo function.
Declaration of Interest
This work was supported by the NIH grants RO1 CA121838 and RO1 CA128486 to Vladimir P. Torchilin.
References
- Abes, R., Arzumanov, A.A., Moulton, H.M., Abes, S., Lvanciva, G.D., Lversen, P.L., Gait, M.J., Lebleu, B. (2007). Cell-penetrating-peptide-based delivery of oligonucleotides: an overview. Biochem Soc Trans. 35:775–9.
- Bae, Y., Kataoka, K. (2009). Intelligent polymeric micelles from functional poly (ethylene glycol)-poly (amino acid) block copolymers. Adv Drug Deliv Rev. 61:768–84.
- Chauhan, A., Tikoo, A., Kapur, A.K., Singh, M. (2007). The taming of the cell penetrating domain of the HIV Tat: myths and realities. J Contr Rel. 117:148–62.
- Elmquist, A., Langel, U. (2003). In vitro uptake and stability study of pVEC and its all-D analog. Biol Chem. 384:387–93.
- Foerg, C., Weller, K.M., Rechsteiner, H., Nielsen, H.M., Fernandez-Carneado, J., Brunisholz, R., Giralt, E., Merkle, H.P. (2008). Metabolic cleavage and translocation efficiency of selected cell penetrating peptides: a comparative study with epithelial cell cultures. AAPS J. 10:349–59.
- Foged, C., Nielsen, H.M. (2008). Cell-penetrating peptides for drug delivery across membrane barriers. Exp Opin Drug Deliv. 5:105–17.
- Frankel, A.D., Pabo, C.O. (1988). Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 55:1189–93.
- Green, M., Loewenstein, P.M. (1988). Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell. 55:1179–88.
- Grunwald, J., Rejtar, T., Sawant, R., Wang, Z., Torchilin, V.P. (2009). TAT peptide and its conjugates: proteolytic stability. Bioconjug Chem. 20:1531–7.
- Gupta, B., Torchilin, V.P. (2007). Monoclonal antibody 2C5-modified doxorubicin-loaded liposomes with significantly enhanced therapeutic activity against intracranial human brain U-87 MG tumor xenografts in nude mice. Cancer Immunol Immunother. 56:1215–23.
- Hallbrink, M., Floren, A., Elmquist, A., Pooga, M., Bartfai, T., Langel, U. (2001). Cargo delivery kinetics of cell-penetrating peptides. Biochim Biophys Acta. 1515:101–9.
- Jarvert, P., Langel, K., El-Andaloussi, S., Langel, U. (2007). Applications of cell-penetrating peptides in regulation of gene expression. Biochem Soc Trans. 35:770–4.
- Kale, A., Torchilin, V. (2010). Environment-responsive multifunctional liposomes. Meth Mol Biol. 605:213.
- Kersemans, V., Kersemans, K., Cornelissen, B. (2008). Cell penetrating peptides for in vivo molecular imaging applications. Curr Pharm Des. 14:2415–27.
- Sawant, R.R., Torchilin, V.P. (2009). Enhanced cytotoxicity of TATp-bearing paclitaxel-loaded micelles in vitro and in vivo. Int J Pharm. 374:114–8.
- Torchilin, V. (2008a). Multifunctional pharmaceutical nanocarriers: development of the concept. Multifunctional Pharm Nanocarriers. Vol. 4: 1–32.
- Torchilin, V.P. (2008b). Cell penetrating peptide-modified pharmaceutical nanocarriers for intracellular drug and gene delivery. Biopolymers. 90:604–10.
- Torchilin, V.P., Rammohan, R., Weissig, V., Levchenko, T.S. (2001). TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc Natl Acad Sci USA. 98:8786–91.
- Trehin, R., Nielsen, H.M., Jahnke, H.G., Krauss, U., Beck-Sickinger, AG., Merkle, H.P. (2004). Metabolic cleavage of cell-penetrating peptides in contact with epithelial models: human calcitonin (hCT)-derived peptides, Tat(47–57) and penetratin(43–58). Biochem J. 382:945–56.
- Wang, T., D’souza, G.G., Bedi, D., Fagbohun, O.A., Potturi, L.P., Papahadjopoulos-Sternberg, B., Petrenko, V.A., Torchilin, V.P. (2010). Enhanced binding and killing of target tumor cells by drug-loaded liposomes modified with tumor-specific phage fusion coat protein. Nanomedicine (Lond). 5:563–74.