
Abstract
To develop a novel PEGylated ibuprofen tablet formulations and evaluate its anti-inflammatory activity and pharmacokinetics profile in an animal model. Six batches of PEGylated ibuprofen tablets were prepared by direct compression using Avicel® and lactose as the binder diluents. In vivo anti-inflammatory activity of the tablets was carried out as well as the pharmacokinetics profiles. The PEGylated ibuprofen tablet reduced carrageenan-induced inflammation in experimental animals and sustained its anti-inflammatory action for over 10 h. The pharmacokinetics profile of the optimized formulations were greater than that of the marketed sample and the pure drug sample. In conclusion, PEGylation of ibuprofen conferred a high level of anti-inflammatory activity and slowed plasma clearance level, indicating sustained release. Thus, further exploration of this novel formulation to be used as an alternative carrier for this drug is required.
Introduction
PEGylation has been widely and successfully applied to improve the solubility, dissolution rates and consequently improve the bioavailability of poorly water soluble drugs and peptides. Several studies have described a number of techniques of PEGylation of drug molecules including molecular interaction, functional group complexation and a combination of the two techniques. These techniques have been shown to be effective in PEGylation of both water soluble and water insoluble molecules. In our previous study, PEGylated metformin hydrochloride was evaluated in vivo in diabetic rat models, and the blood glucose lowering effect was found to be higher than that of conventional tablet formulations of metformin.
Ibuprofen is a widely used non-steroidal anti-inflammatory drug used in the treatment of rheumatoid arthritis, ankylosing spondylitis and osteoarthritis (Lamprecht et al., Citation2004), and it is also effective in the relief of pain. It is used at the daily dosage of 600 mg to 1.2 g given in two to three divided administrations. In order to minimize the incidence of gastric mucosal damage resulting from the administration of ibuprofen and provide an effective blood level for a reasonably long period, it has been formulated as sustained release tablets (Pang et al., 2012). The sustained released formulations still have some major defects such as poor solubility, which ultimately affect its bioavailability (Pang et al., 2012; Newa et al., Citation2007). All these lead to poor disease management and subsequently cause therapeutic failures. In addition, because of it poor bioavailability and short half-life, multiple doses are needed to maintain minimum effective plasma concentration; this also leads to patient non-compliance. To overcome these obstacles, a drug solubilization enhancer is very important in formulation or carrier for ibuprofen delivery system. Several polymers have been evaluated for this purpose, and the responses were very impressive, especially in the area of oral delivery formulation of poorly water soluble materials (Rupp et al., Citation2010).
Among the carriers used in the formation of PEGylation, polyethylene glycol (PEG) is the most commonly used in the field of drug design and delivery. PEG is a hydrophilic water soluble polymer that has gained wider applications in the field of drug delivery, especially for drugs with poor solubility problems (Attama et al., Citation2009). It is a used in drug solubilizer and is often used as vehicle because of its low toxicity and high aqueous solubility (Attama et al., Citation2009), availability in various molecular weights, economic cost and physiological tolerance (Amit & Samarpreet, Citation2011). More so, PEG is one of the acceptable materials in the United States for the preparation of oral and parenteral formulation for human consumption. Additional attractive features of PEGs include their ability to solubilize some compounds and improve compound wettability. These and other properties make PEG a suitable vehicle in the formulation of dosage forms (Franco et al., Citation2001; Teresa et al., Citation2002; Moreshwar et al., Citation2009).
Thus, the aim of this investigation was to develop a novel PEGylated-ibuprofen using solvent interaction method (Manimaran et al., Citation2010), which is a molecular PEGylation as no covalent link was formed as found in particle PEGylation and evaluate its anti-inflammatory activity in an animal model. The pharmacokinetic properties of the prepared drug were also evaluated after oral administration.
Materials and methods
Materials
Ibuprofen (Spectrum, Houston, TX), PEG (PEG-8000) (Carl Roth, Karlsruhe, Germany), Avicel® powder PH101 (Brussels, Belgium), polyvinyl alcohol Mw 2200 (Sigma-Adrilch, St. Louis, MO), lactose (Merck, Darmstadt, Germany), acetone, ethanol, (BDH, Poole, England) and magnesium stearate (May & Baker, England). Distilled water was collected from an all glass still. All other reagents were of analytical grade and used as such without further purification.
Methods
Preparation of PEGylated ibuprofen
The ratios of ibuprofen to PEG used in preparing the PEGylated-matrices were (1:0, 0:1, 1:1, 1:2 and 2:1). Briefly, 50 mg quantities of ibuprofen and PEG-8000 were dispersed in distilled water in separate 100 ml beakers and stirred with a glass rod for 5 min and then allowed to stand for 12 h to produce homogeneous dispersions of both ibuprofen and PEG-8000. Thereafter, the contents of both beakers were mixed according to the ratio, ibuprofen: PEG (1:0, 1:1, 1:2 and 2:1), respectively, and homogenized using a Kenwood mixer (Kenwood Ltd., Upper Saddle River, NJ) set at a stirring speed of 200 rpm. The PEGylated-ibuprofen matrices were then precipitated by slow addition of diethyl ether and the PEGylate was collected, dried, pulverized and kept in an air-tight container until use. The ibuprofen-PEGylated-matrices were mixed with other excipients needed for tablets compression in a tubular mixer (Erweka, Dreieich, Germany) for 25 min, in their respective quantities as shown in to obtain the final powder mixtures and compressed at 46–48 kgf using a 9.0 mm punch and die set fitted into an automated F3 Manesty Single Punch tabletting machine.
Table 1. Composition of the formulated batches of ibuprofen tablets (mg).
Swelling studies
The swelling properties of the tablet formulations were determined by water sorption method (Sinye et al., Citation2013). The water-uptake study of the tablets was carried out using a modified dissolution apparatus. In all cases, each tablet was weighed and then placed in 10 ml of distilled water in a glass vial at room temperature throughout the study. After 1, 2, 4, 6 and 8 h, the tablet was withdrawn, blot dried and weighed. The percentage increase in weight at each time interval due to absorbed liquid or water uptake was calculated using Equation (1):
In vivo evaluation of tablets in an animal model
Pharmacodynamic study: Animals Healthy Albino male rats weighing between 210 and 230 g were obtained from the Department of Pharmacology laboratory of our institution. The animals were kept in metallic cages and housed in an animal care facility, which was maintained at 28 ± 5 °C on a 12-h light/12-h dark cycle and fed with standard laboratory diet, while water was provided ad libitum. All the animal experiments in this work were approved by the Faculty Animal Ethical Committee and were in accordance with the Federation of European Laboratory Animal Science Association and the European Community Council Directive of 24 November 1986 (86/609/EEC).
Anti-inflammatory activity of the prepared tablets
Thirty male Albino rats were randomly divided into five groups of five animals per group. Group 1 (control) received normal saline, groups 2–5 received batches A–D, respectively; while groups 6 received market sample (MKT). All tested samples and controls were given orally, 1 h before subcutaneous injection of γ-carrageenan into the right hind paws according to an earlier study (Anosike et al., Citation2009). The paw volume was measured before drug administration and then at 1, 2, 3, 4, 5 and 6 h after γ-carrageenan injection, using a digital plethysmometer (Basile, Comerio, Italy). The amount of paw swelling was determined from time to time and expressed as follows:
Pharmacokinetics studies: Animals. Fifteen mice weighing between 21.0 and 25.0 g were obtained from the Department of Pharmacology Laboratory, University of Nigeria, Nsukka. Animals were housed under unidirectional airflow rooms with controlled temperature (22 ± 2 °C) with 12-h light/dark cycles. Food and water were applied as discussed earlier. The animals were divided into three groups of five rats per group. Animals in group A received the optimized formulation of ibuprofen tablet (batch C), group B received MKT as the reference drug, while the last group received the pure sample (PS) as the control. All administrations were done using oral route.
Analysis of pharmacokinetic parameters
Pharmacokinetic analysis was performed using a bioavailability Calc 2002 pharmacokinetic analysis computer program (Korea Food & Drug Administration, Korea). Area under the curve (AUC) was calculated using the linear trapezoidal rule. Maximum plasma concentration (Cmax) and the time needed to reach the maximum plasma concentration (Tmax) were determined directly from the concentration-time data. The AUC was used as an index to measure the bioavailability of prepared ibuprofen tablets and the reference drug as discussed earlier (Yang et al., Citation1999). All experimental data points were expressed as the mean (± SD). The corresponding mean pharmacokinetic parameters (± SD) were calculated from individual estimates in the animal group. Significant differences between the experiments were calculated using a Student’s t test (unpaired) (Sigma plot®, RockWare Inc., Golden, CO), using a two-tailed distribution and a two-sample unequal variance (heteroscedastic) method. A value of p < 0.05 was adopted to indicate statistical significance.
Statistical analysis
All experiments were conducted in replicates, and data generated were expressed as mean ± SEM, and differences between means were determined by one-way analysis of variance followed by Tukey–Kramer multiple comparison test; and p values < 0.05 were considered significant.
Results
Anti-inflammatory properties
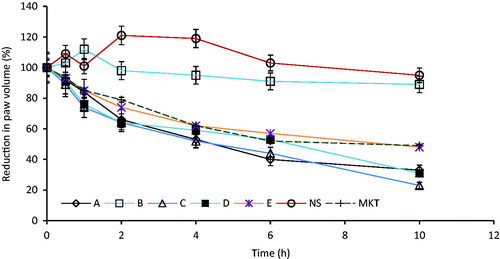
The anti-inflammatory results, presented in , showed that PEGylated ibuprofen tablet exhibited good anti-inflammatory activity that was significantly higher than that of the reference drug (MKT) and the pure drug sample (p < 0.05). The maximum edema inhibition were 67.2 ± 0.1, 77.4 ± 1.0, 69.1 ± 0.3 and 52.3 ± 0.3 % for A, C, D and E, respectively, for the prepared PEGylated ibuprofen tablets, while that of reference drug gave a maximum inhibition of 51.5 ± 0.2%.
Figure 1. Anti-inflammatory properties of prepared-ibuprofen tablet after oral administration: batches (A–E), normal saline (NS) and the commercial tablet (MKT), (n = 3).
Pharmacokinetics analysis
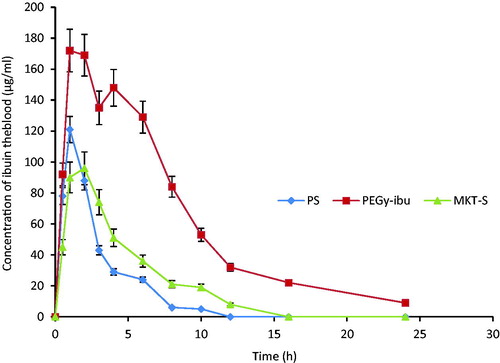
The plasma concentration–time profiles of the drug are shown in . The mean plasma concentrations after oral administration of PEGylated ibuprofen tablets increased at broader peaks than the plasma concentrations of the reference tablets. The mean Cmax value for the PEGylated ibuprofen tablets was 169 µg/ml compared with 89 µg/ml for the reference drugs. The mean Tmax for PEGylated ibuprofen tablets and reference drug were 2.00 and 3.00 h, respectively; however, the Cmax and Tmax values were not significantly different (p > 0.05). The T1/2 for the PEGylated ibuprofen tablets was 6 h, whereas the T1/2 value for the reference drug was 2 h. The mean AUC value for the prepared PEGylated ibuprofen tablet was significantly greater than those for the reference drugs (p < 0.005).
Figure 2. Plasma concentration-time profiles of ibuprofen after oral administration of the formulated PEGylated ibuprofen tablet (PEGy-ibu), pure drug sample (PS) and the commercial tablet (MKT-S) to rat (n = 3).
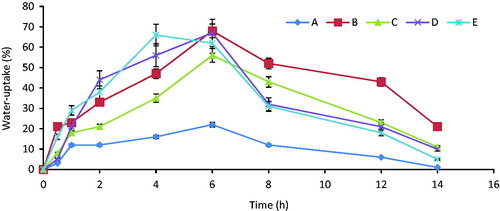
The result of swelling studies () revealed that the capability of the various tablet formulations to absorb water depend largely on the concentration of the PEG in the formulation and to a less extent on the drug content. The results of the swelling studies () showed that water-uptake (%) increased with increase in the concentration of PEG-8000 used in the PEGylating; maximum water-uptake (%) of 22.4, 68.2, 56.4, 67.2 and 66.7% were obtained for A, B, C, D and E, respectively. Thereafter, the swelling index decreased after attaining the saturated point. So, the PEGylated tablets formulated with 50 mg of PEG resulted in higher water uptake (%), while those formulated with no PEG (batch A) gave the least. However, all the batches formulated with PEGylated material had good water-uptake in the range of 22.4–66.7%. The concentration of active pharmaceutical ingredient had a direct effect on the water uptake; the batches that contained higher quantities of ibuprofen were greatly retarded in their water-uptake compared with batches that contain no drug or less quantity of the drug. Batches A, C and D that each contained 100 mg of ibuprofen were retarded greatly after 6 h of water uptake (reaching the maximum equilibrium) compared to batches B and E whose water uptake decreased gradually at a slight rate to the lowest valued on reaching maximum water uptake at equilibrium.
Figure 3. Effects of the PEG and drug concentration on water-uptake of PEGylated ibuprofen tablets. Bars represent mean ± S.D (n = 3).
Discussion
The swelling study is a function of the amount of PEG-8000 used in the PEGylated of ibuprofen as evaluated, based on the amount of water-uptake. The result of the swelling study indicated that the PEGylated tablets were capable of absorbing water and subsequently gradually released the incorporated drug. High values of the percentage of water uptake were observed in all the formulation with the exception of batch A that contained no PEG-8000, this is a strong indication that the formulation technique adopted will be able to undergo disintegration over a longer period of time thereby releasing its content for possible absorption in the biological system for the therapeutic activity. The percentage water-uptake across the batches increased with increase in the PEG and slightly retarded with increase in drug content. It was observed that the percentage of water-uptake of all the formulations gradually increased until reaching equilibrium and then gradually decreased. The maximum percentage water-uptake of formulations A–E were 22, 68.2, 56.4, 67.2 and 66.7%, respectively. The results were in conformation with the in vitro release studied carried out on such formulations where it was observed that the increase in the release of the formulations depended on the concentration of the PEG. Our result in this study is in agreement with the earlier research on the use of PEG as solubilizing agent according to Sinha and Rohera (Citation2002).
The anti-inflammatory properties of PEGylated ibuprofen tablets was evaluated and were compared among the various batches and also compared with MKT in rat model using carrageenan as the physiologic agent. The inhibition of inflammation in the PEGylated was observed to be higher compared to the control groups (). The prepared PEGylated ibuprofen tablets showed promising anti-inflammatory activities in all cases, the effect were comparably higher and more sustained than the reference tablets. The percentage changes in the paw volume produced by batch A and commercial tablets (MKT) were almost the same and showed a similar pattern over a period of time. However, maximum decrease of 77% was recorded for batch C, while batch B had 11% edema inhibition at 10 h. Other formulated batches (A, D and E, respectively) showed 67, 69 and 52% edema inhibition at 10 h, while that of negative and positive controls showed 5 and 51% inhibition, respectively. These results indicated that batch A and the commercial sample showed similar activities in exhibiting normal release pattern, while batches C and D exhibited slow and prolonged release characteristics. This release pattern, which is attributable to the pharmacodynamic parameter as observed from the in vivo study, might strongly correlate with the in vitro dissolution study.
Pharmacokinetic parameters for the prepared tablets, such as the maximum blood concentration, peak time and the area under the concentration-time curve, are presented in . Pharmacokinetic results shown in demonstrated that the concentration of ibuprofen in the blood increased rapidly and reached peak values in rats dosed with either PEGylated tablet or marketed sample. Reference sample gave a maximum at 4.0 h, which was less than the formulated tablets. The AUC of the PEGylated ibuprofen tablet was significantly (p < 0.05) higher than those of the references tablets and the PS. The Cmax was higher in the formulated tablets than the reference drug and the PS, an indication that the PEGylated system could deliver the drug orally. The peak plasma concentration was reached 3 h after oral administration of the PEGylated ibuprofen tablets and sustained its effect for additional 2 h, before it declined at 24 h. The importance of this evaluation was to enable the pharmaceutical industries to determine the exert concentration of the drug in the system over a period of time and also to predict the frequency of dosage administration as evaluated by Engers et al. (Citation2010).
The swelling indices were high in batches B, C and E. This was probably because weight gained by tablet was increased proportionally with rate of hydration up to certain limit. Later on, it decreases gradually due to dissolution of outermost gelled layer of tablet into dissolution medium. The direct relationship was observed between swelling index and polymer concentration, and as polymer concentration increases, swelling index increased (Sujja-areevath et al., Citation1998). There was no significant difference in batches B, C and E. However, batch D showed a slight significant p < 0.005 from batch A that contained no PEG. The results in this batch A confirmed that PEG-8000 played an important role in the swelling as well as in dissolution of the tablet.
Conclusion
In designing a drug delivery device, such as those based on PEGylated materials, the choice as well as the combination ratio of PEG and other excipients should be considered based on the desired release pattern, route of administration, stability and other physicochemical considerations. In this research work, the effect of PEG-8000 concentration on the anti-inflammatory and pharmacokinetics characteristics was directly related. It was observed that the most concentrated formulation had the highest anti-inflammatory activities. One can suggest that the optimized formulation (batch C) containing 50 mg of PEG-8000 in the PEGylated tablets offer the most promising combination as regards anti-inflammatory and prolong plasma concentration and could be further investigated for oral delivery of ibuprofen.
Declaration of Interest
The authors state no conflicts of interest and have received no funding for the research or in the preparation of this manuscript.
References
- Amit KA, Samarpreet S. (2011). Physicochemical characterization and dissolution study of solid dispersions of diacerein with polyethylene glycol 6000. Drug Dev Ind Pharm 37:1181–91
- Anosike AC, Onyechi O, Ezeanyika LU, Nwuba MM. (2009). Anti-inflammatory and anti-ulcerogenic activity of the ethanol extract of ginger (Zingiber officinale). Africa J Biochem Res 3:379–84
- Attama AA, Okafor CE, Builders PF, Okorie O. (2009). Formulation and in vitro evaluation of a PEGylated microscopic lipospheres delivery system for ceftriaxone sodium. Drug Deliv 16:448–57
- Engers D, Teng J, Jimenez-Novoa J, et al. (2010). A solid-state approach to enable early development compounds: selection and animal bioavailability studies of itraconazole amorphous solid dispersion. J Pharm Sci 99:3901–22
- Franco MT, Latrofa AT, Provenzano MR, Serra M. (2001). Dissolution properties and anticonvulsant activity of phenytoin-polyethylene glycol 6000 and polyvinylpyrrolidone K-30 solid dispersions. Int J Pharm 225:63–73
- Lamprecht A, Saumet JL, Roux J, Benoit JP. (2004). Lipid nanocarriers as drug delivery system for ibuprofen in pain treatment. Int J Pharm 278:407–14
- Manimaran V, Damodharan N, Mothilal M, et al. (2010). Enhancement of dissolution rate of glibenclamide by solid dispersion technology. Int J Curr Pharm Res 2:14–17
- Moreshwar P, Patil MP, Gaikwad NJ. (2009). Preparation and characterization of gliclazide-polyethylene glycol 4000 solid dispersions. Acta Pharm 59:57–65
- Newa M, Bhandari KH, Li DX. (2007). Preparation, characterization and in vivo evaluation of ibuprofen binary solid dispersions with poloxamer 188. Int J Pharm 343:228–37
- Pang J, Luan Y, Li F. (2010). Ionic liquid-assisted synthesis of silica particles and their application in drug release. Mater Lett 64:2509–12
- Rupp C, Steckel H, Muller BW. (2010). Solubilization of poorly water-soluble drugs by mixed micelles based on hydrogenated phosphatidylcholine. Int J Pharm 395:272–80
- Sinha DR, Rohera BD. (2002). Comparative evaluation of rate of hydration and matrix erosion of HEC and HPC and study of drug release from their matrices. Eur J Pharm Sci 16:193–9
- Sinye AB, Salome AC, Anthony AA, et al. (2013). In vitro and In vivo characterisation of piroxicam-loaded dika wax lipospheres. Trop J Pharm Res 12:33–8
- Sujja-Areevath J, Munday DL, Cox PJ, Khan KA. (1998). Relationship between swelling, erosion and drug release in hydrophilic natural gum minimatrix formulations. Eur J Pharm Sci 1998;6:207–17
- Teresa MM, Victoria MM, Gloria ES. (2002). Characterization and solubility study of solid dispersions of flunarizine and polyvinylpyrrolidone. Farmaco 57:723–7
- Yang SC, Lu LF, Cai Y, et al. (1999). Body distribution in mice of intravenously injected camptothecin solid lipid nanoparticles and targeting effect on brain. J Control Release 59:299–307