
Abstract
Famotidine HCl has low bioavailability (40–45%) due to its narrow absorption window and low solubility in intestinal pH. Lipids were utilized in the formulation of novel gastroretentive dosage forms to increase the availability of famotidine HCl at its absorption site. Novel non-swellable gastroretentive lipid disks (D) and swellable compression coated tablets with a lipid core (T) were prepared. Formulae were characterized by friability testing, in-vitro buoyancy, in-vitro drug release and scanning electron microscopy (SEM). Factorial designs of 22 × 31 and 32 were planned for the optimization of disks and tablets, respectively, using Design-Expert® software. X-ray imaging was used for the in-vivo visualization of the selected formula in human gastrointestinal tract (GIT). Moreover, a bioavailability study was performed in healthy human volunteers using the optimized disk formula (D10). Results showed that formulae D10 (containing stearyl alcohol and polyethylene glycol in a ratio of 9:1 w/w) and T7 (containing polyethylene oxide only) had highest desirability values (0.684 and 0.842, respectively). Lipids achieved instantaneous floating and sustained the release of famotidine HCl over a prolonged period of time with significant bioavailability enhancement.
Introduction
Oral administration is a preferred route for drug delivery due to its higher patient compliance. However, the bioavailability of orally administered drugs is always restricted by their low solubility in the physiological conditions of the gastrointestinal tract (GIT), low permeability through the intestinal wall, and in some cases, extensive pre-systemic metabolism in the gut and liver (Han & Wang, Citation2005; Sarparanta et al., Citation2012).
During the past few decades, numerous oral controlled drug delivery systems have been investigated and developed to act as drug reservoirs for release of drugs over a defined period of time at a controlled rate. However, it is a challenge to develop an oral controlled-release drug delivery system with a sustained drug release as well as prolonged presence of the dosage form within the GIT until the drug is completely released within the desired period of time (Prajapati et al., Citation2008; Hooda et al., Citation2012). Anyhow, typical controlled release formulations are limited by inadequate retention in the stomach (Darandale & Vavia Citation2012). To extend the residence time of dosage forms in the stomach, a number of strategies have been developed, including floating systems (Singh & Kim Citation2000), high density systems (Hwang et al., Citation1998), mucoadhesive systems (Pund et al., Citation2011) and expandable systems (Deshpande et al., Citation1997; Klausner et al., Citation2003). Each of these approaches has its advantages and disadvantages (Darandale & Vavia, Citation2012).
Floating gastroretentive drug delivery systems are of particular interest for drugs that are locally acting or primarily absorbed in the stomach, exhibiting poor solubility at an alkaline pH or having a narrow absorption window in the upper part of GIT (Deshpande et al., Citation1997; Singh & Kim, Citation2000; Tadros, Citation2010; Zhang et al., Citation2012). The floating systems can prolong the retention time of a dosage form in the stomach, and hence improve the oral bioavailability of the drug (Rao et al., Citation2013).
Famotidine HCl is a histamine H2-receptor antagonist (Gray, Citation2011). It is used in gastric ulcers, duodenal ulcers, Zollinger-Ellison syndrome and gastro-esophageal reflux disease. Famotidine HCl is classified as BCS class III (high solubility and low permeability) (Dahan & Amidon, Citation2009). The incomplete absorption of famotidine HCl from the GIT, low bioavailability (40–45%) and short biological half-life (2.5–3.5 h) make it a good candidate for its formulation as a floating system (Jaimini et al., Citation2007).
The floating of famotidine HCl using different techniques has been extensively studied. For example, the common reported techniques are microballoons (Gupta & Pathak, Citation2008; Ramachandran et al., Citation2010), oil entrapped calcium alginate beads (Satishbabu et al., Citation2010) and effervescent tablets (Jaimini et al., Citation2007). Effervescent tablets suffer from floating lag time and limited floating duration (Jaimini et al., Citation2007). On the other hand, beads and microballoons have the drawbacks of incomplete floating and a difficulty in scaling up (Murata et al., Citation2003; Mishra & Pathak, Citation2008). Moreover, some of these techniques suffer from poor drug encapsulation due to curing in acidic gelation medium and rapid drug release in acidic environment of the stomach (Ostberg et al., Citation1994; Aslani & Kennedy, Citation1996; Kikuchi et al., Citation1997).
In this manuscript, novel non swellable lipid disks and swellable compression coated tablets were formulated for the gastric retention of famotidine HCl. We hypothesize that lipids can be utilized for promoting floating of both tablets and disks with no lag time. In addition, although non swellable, lipids can achieve sustained drug release from the disks over a prolonged period of time. On the other hand, the sustained drug release from tablets will be achieved using different swellable polymers.
To verify this hypothesis, the novel lipid disks and compression coated tablets were formulated and statistically analyzed by response surface model using Design-Expert® software. Furthermore, the in-vivo performance of the optimized formula was assessed by visualization using X-ray imaging as well as bioavailability determination in healthy human volunteers.
Experimental
Materials
Famotidine HCl was kindly supplied by EPCI pharmaceutical company (Cairo, Egypt). Cetyl alcohol (CA), stearyl alcohol (SA), polyethylene oxide 5 M (PEO), polyethylene glycol 6000 (PEG) and liquid paraffin were purchased from Sigma-Aldrich Co. (St. Louis, CA). Hydroxypropyl methyl cellulose (HPMC) K100 and K4M were purchased from Alfa Aesar (Ward Hill, MA). Pluronic F127 was purchased from BASF (Florham Park, NJ). Avicel PH 102 (microcrystalline cellulose) was purchased from FMC Corporation (Philadelphia, PA).
Preparation of the lipid disks
Famotidine HCl lipid disks were prepared by melt-molding method (Ndindayino et al., Citation2002). The lipid (CA or SA), either alone or mixed with the release modifier (PEG or Pluronic), was melted on a thermostatic hotplate stirrer (Daihan scientific Co., Korea) at 90 °C. Famotidine HCl (10% w/w of the lipid or the lipid – release modifier mixture) was dispersed in the melt with continuous stirring. The heater was turned off and the whole melt was poured, just before congealing, into a handmade metal mould composed of a 10 mm die and a movable lower punch. Disks weight was adjusted to be 200 mg. The discs were allowed to congeal and then ejected from the mould. The different ratios of the lipid to the release modifier are shown in .
Table 1. Composition of the lipid disks formulae.
Preparation of the compression coated tablets
Famotidine HCl compression coated tablets were prepared in two steps. Firstly, the tablet core composed of a lipid disk was prepared by melting CA either alone or with liquid paraffin in a ratio of 1:0.1 or 1:0.25 w/w, respectively. The prepared melt was poured into a handmade metal mould composed of a 7 mm die and a movable lower punch. The melt was allowed to congeal and then the core discs were ejected.
Secondly, the coat mixtures were prepared using a pestle and a mortar by homogenously mixing the drug and the swellable polymer (HPMC K100, HPMC K4M or PEO) in a ratio of 1:9 w/w, respectively. The polymer was used either alone or in combination with Avicel (20% or 40% w/w of the total coat weight). Fifty percent of the coat weight (100 mg) was placed in the die (diameter: 10 mm) and the prepared core was centered. Finally, the rest of the coat weight (100 mg) was placed and compressed (Yehia et al., Citation2009). The composition of the tablets is shown in .
Table 2. Composition of compression coated tablets of the different formulae.
Physical characterization of the prepared formulae
Friability and weight variation of disks and tablets formulae were determined according to the British pharmacopoeia (Commission, Citation2008). Tablets formulae were also tested for their hardness using tablet hardness tester (Copley Scientific, Nottingham, UK).
In-vitro buoyancy study
The in-vitro buoyancy was evaluated by measuring floating lag times as well as duration of buoyancy according to the method described earlier (Jiménez-Castellanos et al., Citation1994). The formulae were placed in a beaker containing 100 ml of 0.1 N HCl. The time taken by the unit to rise to the surface and float was recorded as the floating lag time. The time for which discs or tablets kept floating was termed as buoyancy time.
In-vitro drug release
The release study was performed using a USP II dissolution apparatus in 900 ml of 0.1 N HCl as dissolution medium at 37 °C and stirring rate of 50 rpm. The drug release from the prepared formulae was compared to that from the market product (Pepcid®). At predetermined time intervals (0.5, 1, 1.5, 2, 3, 4, 5, 6, 8 and 12 h), aliquots of 5 ml were withdrawn, filtered and replaced by fresh medium. The samples were analyzed spectrophotometrically at λmax of 264 nm. The experiments were run in triplicate and the mean was calculated.
Kinetic study of the release data
The data obtained from the dissolution studies were kinetically analyzed using Microsoft Excel 2007 to determine the order and mechanism of drug release from the different formulae. Generally, zero-order kinetics, first-order kinetics and Higuchi diffusion were used for the analysis of the dissolution mechanism (Higuchi, Citation1963; Peppas, Citation1985). In addition, Korsmeyer–Peppas model was utilized for the elaboration of the drug release from the swellable coat of the tablets formulae (Ritger & Peppas, Citation1987):
where
represents the drug dissolved fraction at time t, K is a kinetic constant and n is the diffusional exponent which depends on the release mechanism and the shape of the drug delivery device (Sasa et al., Citation2006). For the case of cylindrical disks and tablets, in particular, n ≤ 0.45 corresponds to a Fickian diffusion release (case I diffusional), 0.45 < n ≤ 0.89 to an anomalous (non-Fickian) transport, n = 0.89 to a relaxation (case II) release kinetics and n > 0.89 to a super case II transport (Lotfipour et al., Citation2004; Vueba et al., Citation2004).
The approximate contribution of the diffusional and relaxational mechanisms to the anomalous release process is calculated by fitting the release data from tablets formulae to the model proposed by Peppas & Sahlin (Citation1989):
where m is the diffusional exponent, the first term of the right-hand side (k1) represents the Fickian contribution and the second term (k2) is the relaxational contribution (Karatas & Baykara, Citation2001). When k1 > k2, the release is mainly controlled by diffusion and when k2 > k1, the release is predominantly controlled by matrix swelling/erosion (Kim & Fassihi, Citation1997).
Statistical analysis
A factorial design of 22 × 31 was used in case of the lipid discs to study the effect of type of lipid (A), type and concentration of release modifier (B and C, respectively) on friability, burst release and t50%. On the other hand, a factorial design of 32 was used in case of the compression coated tablets to study the effect of type of polymer (A) and concentration of Avicel (B) on friability, hardness, floating lag time, burst release and t50%. The designs were analyzed using Design-Expert® v8 software (Stat-Ease, Burlington, MA).
Scanning electron microscopy
The surface characteristics of optimized disks and tablets formulae were observed using a SEM (JXA-840; JEOL, Tokyo, Japan). The samples were gold-coated under vacuum and then examined. The morphology of the formulae was studied before and after 12 h of the in-vitro release study (Chia et al., Citation2001).
X-ray imaging in human GIT
X-ray imaging technique was used for monitoring the selected formula in the human GIT. Four healthy male volunteers, with a mean age of 29 years (range 25–35 years) and a mean body weight of 78 kg (range 70–90 kg), have participated in the study. The subjects were presented with full details of the investigation and their written consent had been taken. The protocol of the study was reviewed and approved by the Ethical Committee of the Faculty of Pharmacy, Cairo University, Cairo, Egypt. The selected formula was loaded with barium sulfate (20% w/w) to make it visible in the GIT by the X-ray (Gande & Rao, Citation2011). After an overnight fast period, subjects were fed with low calories breakfast. Then, three subjects ingested barium sulfate containing formula (selected formula) orally with 200 ml water. The fourth subject ingested barium sulfate tablet E-Z-Disk® (200 mg) available in the market as a control. The formula was visualized using plain X-ray. Abdominal radiographs were taken after 1, 4, 8 and 12 h. The volunteers were served with food, before dosage form administration (breakfast) and 4 h (lunch) after the administration (Yehia et al., Citation2011).
Bioavailability and pharmacokinetic study in healthy human volunteers
Study design and subjects selection
The study was conducted according to a randomized, two period crossover design under fed condition. Six healthy adult male volunteers between 22 and 29 years old (mean 25.98 years, SD 2.69 years) and weighing from 75 to 92 kg (mean 83.33 kg, SD 6.47 kg) participated in the study. They were judged to be healthy and were not receiving any other medication during the study period. The volunteers were given information on the drug as well as nature of the study in advance of the trial and they had given their written consent. The study was done in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) and the GCP/GLP standards. Before study, the protocol was approved by the institutional review board of Genuine Research Center, Cairo, Egypt.
Drug administration and sample collection
Six volunteers were randomly selected to receive either the optimized famotidine HCl floating dosage form (2 units each containing 20 mg famotidine HCl) or one conventional famotidine HCl tablet (Pepcid®, 40 mg) with 250 ml of water after a low fat meal. Blood samples (4 ml) were collected into tubes (containing sodium heparin as an anticoagulant) at proper time after dosing. Blood collection time points for the two formulae were 0, 0.33, 0.66, 1.00, 1.33, 1.66, 2.00, 2.50, 3.00, 3.50, 4.00, 6.00, 8.00, 12.00 and 24.00 h. Blood samples were centrifuged and the plasma was then transferred to polypropylene tubes, frozen immediately and stored at −20 °C until analysis. The study was repeated after a washing out period of 7 d to complete the crossover study design.
Chromatographic conditions
A sensitive, selective and accurate LC-MS/MS method was developed and validated before the study for determination of famotidine HCl in plasma. The method was validated following the international guidelines (Shah et al., Citation1991). All chemicals and reagents were of analytical grade, solvents used were of HPLC grade. Loratadine internal standard (IS) stock solution was prepared by dissolving 10 mg in methanol and serially diluted with mobile phase to give a final working concentration of 50 ng/ml. A Shimadzu Prominence (Shimadzu, Japan) series LC system equipped with degasser (DGU-20A3), solvent delivery unit (LC-20AB) along with auto-sampler (SIL-20 AC) was used to inject 10 µl aliquots of the processed samples on a Luna C18 (Phenomenex, Torrance, CA) (50 × 4.6) mm, 5 μm particle size. All analyses were carried out at room temperature. The isocratic mobile phase (pH 4.5) consisted of acetonitrile and (0.02 M) ammonium acetate buffer (70%: 30%, v/v, respectively) and 0.1% formic acid which was delivered at a flow rate of 0.50 ml/min into the mass spectrometer’s electrospray ionization chamber. Quantitation was achieved by MS/MS detection in positive ion mode for both famotidine HCl and IS, using an API-3200 mass spectrometer (MDS Sciex, Foster City, CA) equipped with a Turbo ionsprayTM interface at 300 °C. The ion spray voltage was set at 5500 V. The common parameters: nebulizer gas, curtain gas, auxillary gas and collision gas were set at 15, 27, 32 and 13 psi, respectively. The compound parameters: declustering potential (DP), collision energy (CE), entrance potential (EP) and collision exit potential (CXP) were 48, 35, 5 and 4 V for famotidine HCl and 60, 50, 10 and 4 V for loratadine (IS), respectively. Detection of the ions was performed in the multiple reaction monitoring (MRM) mode, monitoring the transition of the m/z 337.91 precursor ion to the m/z 189.10 for famotidine HCl and m/z 383.04 precursor ion to the m/z 337.10 for IS. Quadrupoles Q1 and Q3 were set on unit resolution. The analytical data were processed by Analyst software version 1.5 (Applied Biosystems Inc., Foster City, CA).
Sample preparation
All frozen human plasma samples were thawed at ambient temperature. Human plasma samples (0.5 ml) were placed in 7 ml glass tubes, and 100 µl of IS solution was added to each and vortexed for 1 min. Four milliliters of methyl-t-butyl-ether (MTBE) were then added and samples were then vortexed for 2 min. The tubes were then centrifuged for 10 min at 4000 rpm (1790 g). Then, the upper organic phases were transferred to glass tubes and evaporated to dryness using centrifugal vacuum concentrator Vacufuge® 5301 (Eppendorf, Hamburg, Germany) at 40 °C. Dry residues were then dissolved in 200 µl of mobile phase and vortexed for 1 min to reconstitute residues, and 10 µl was injected using the autosampler.
Pharmacokinetic and statistical analysis
Plasma concentration–time data of famotidine HCl were analyzed for each subject by non-compartmental pharmacokinetic models using Kinetica® software version 4.4.1 (Thermo Fisher Scientific Inc., Waltham, MA). The peak plasma concentrations (Cmax) and the time of their occurrence (Tmax) were directly obtained from the concentration–time data. The area under the plasma concentration–time curve (AUC) from time zero to last measured concentration (AUC0–t) was calculated according to the linear trapezoidal rule. Linear regression of the terminal portion of the ln (concentration)–time curve was used to estimate the terminal elimination rate constant (λz), and the elimination half-life was calculated. AUC0–∞ was the corresponding area extrapolated to infinity by AUC + Ct/λz, where Ct was the last measurable drug concentration. The sustained release characteristics of the tested formulation (HVDt50%Cmax) were evaluated by the time span during which the plasma concentrations were at least 50% of the Cmax value (Meier, Nuesch et al., Citation1974; Steinijans, Citation1990). The HVD values were determined from the individual plasma concentration–time profiles. The ratio between the HVDt50%Cmax values of a test formulation and the immediate release reference formulation (Pepcid®) expressed as RDt50%Cmax indicates sustained release effect: a ratio of 1.5, 2 and >3 indicates, low, intermediate and strong sustained release effect, respectively (Meier et al., Citation1974). One-way analysis of variance was used to assess the effect of formulation on the pharmacokinetic parameters. Differences between two related parameters were considered statistically significant for p value equal to or less than 0.05.
Results and discussion
Physical characterization of the prepared formulae
The weight variation testing showed that all formulae were within the pharmacopeial limits. The friability of the lipid disks was in the range of 0.74–4.14% (). No disks formulae were excluded based on friability since it is not a pharmacopeial parameter for the evaluation of disks. However, the lowest friability value was taken as a selection criterion for the optimized formula. It was observed that formulae containing SA and PEG (D9–D11) had the lowest friability testing values (0.74–0.93%).
Table 3. Responses data used in the analysis of the factorial design of the disks formulae.
In case of tablets, friability was in the range of 0.03–0.98% (). All formulae were below the pharmacopeial limit (Commission, Citation2008). The lowest friability values (0.02–0.03%) were observed in case of formulae containing PEO in the coat (T7–T9).
Table 4. Responses data used in the analysis of the factorial design of the tablets formulae.
The hardness of the compression coated tablets ranged from 0.73 to 3.65 kg (). Avicel was added to increase the hardness values of the prepared tablets formulae (Kuentz & Leuenberger, Citation2000; Jamzad & Fassihi, Citation2006). The highest hardness value was observed when PEO was combined with 40% Avicel (T9). This might be due to the presence of the lipid core in the prepared compression coated tablets which decreased the hardness of the formulae.
In-vitro buoyancy study
The buoyancy time of all formulae was more than 12 h. In addition, lipid disks formulae floated immediately without any lag time. This might be due to the low density of CA and SA compared to the dissolution medium (0.908 and 0.906 g/cm3, respectively).
On the other hand, the tablets formulae (T1, T4 and T7) were optimized to achieve immediate floating. This was done by adding liquid paraffin (density = 0.83–0.89 g/cm3) to the core in different ratios (1:0.1 and 1:0.25 w/w, CA to liquid paraffin). The ratio of 1:0.25 w/w resulted in a zero lag time in case of HPMC K100 (T1) and HPMC K4M (T4) while the immediate floating was achieved with the ratio of 1:0.1 w/w in case of PEO (T7). This might be due to the slightly higher density of HPMC polymers (1.326 g/cm3) compared to PEO (1.3 g/cm3) (Rowe et al., Citation2009). Furthermore, the increase in lag time by increasing the Avicel percentages () could be explained by the high density of Avicel (1.42–1.46 g/cm3) (Rowe et al., Citation2009).
In-vitro drug release
and show the drug release from the lipid disks, compared to the market product (Pepcid®). Drug release from the market product was rapid and finished within one hour. On the other hand, drug was slowly and incompletely released from formulae D1 and D8 (containing CA and SA, respectively). This could be attributed to the high hydrophobicity of the used fatty alcohols which hindered water diffusion (Li et al., Citation2006). In addition, lower rate and extent of drug release was observed in D8 compared to D1. This could be explained by the longer hydrocarbon chain and consequently the higher hydrophobicity of SA compared to CA (O’Keefe, Citation2008).
Figure 1. Release profiles of famotidine HCl from disks formulae containing cetyl alcohol in different ratios with PEG (a) or Pluronic (b), compared to the market product (Pepcid®), in 0.1 N HCl at 37 °C.
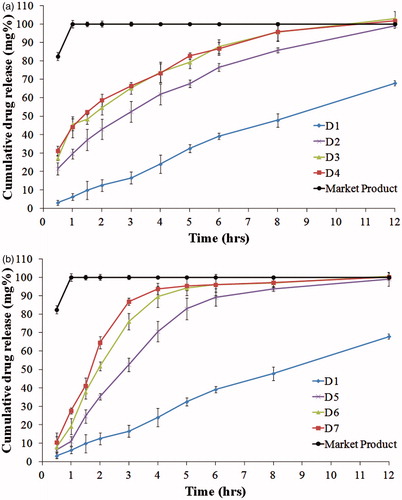
Figure 2. Release profiles of famotidine HCl from disks formulae containing stearyl alcohol in different ratios with PEG (a) or Pluronic (b), compared to the market product (Pepcid®), in 0.1 N HCl at 37 °C.
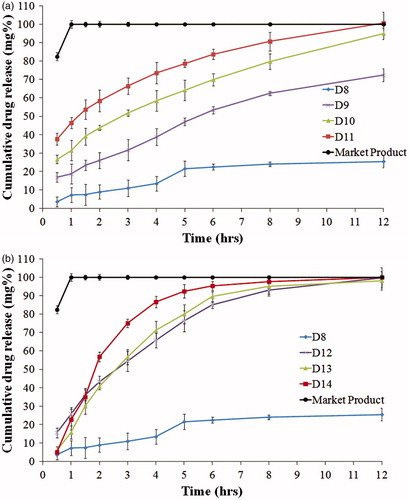
Upon addition of PEG or pluronic (release modifiers) to the lipid disks, complete drug release was observed from all formulae except D9 (SA and PEG, in a ratio of 9.5:0.5 w/w, respectively). The rate of the drug release increased when the ratio of the release modifier increased. This could be explained by the high water solubility of PEG and pluronic which formed pores in the lipid matrix upon dissolving (Mishra & Mishra, Citation2010; Kaul et al., Citation2011). These pores facilitated water access to the entrapped drug. The release half-life values (t50%) in the presence of the release modifier ranged from 0.47 to 5.15 h. It was observed that formulae containing SA and PEG (D9–D11) showed the highest t50% values (). This could also be explained by the higher hydrophobicity of SA compared to CA as mentioned earlier (Rowe et al., Citation2009). In addition, pluronic, being a non ionic surfactant with hydrophilic–lipophilic balance (HLB) greater than 20, might solubilize the fatty alcohols leading to higher drug release rate compared to PEG (Grube & Langguth, Citation2008).
show the drug release from the compression coated tablets. When comparing formulae T1, T4 and T7 (containing HPMC K100, HPMC K4M and PEO, respectively), T7 showed the slowest release rate and the highest t50% (2.59 h), as shown in . On the other hand, T1 showed the fastest release rate. This might be due to the higher nominal viscosity of PEO gel formed during dissolution (5500–7000 cps) compared to HPMC K4M (4000 cps) and HPMC K100 (100 cps) (Rowe et al., Citation2009). Similar results were reported by Gao et al. when studying the effect of HPMC viscosity on the release of adinazolam mesylate from matrix tablets (Gao et al., Citation1996).
Figure 3. Release profiles of famotidine HCl from tablets formulae containing HPMC K100, compared to the market product (Pepcid®), in 0.1 N HCl at 37 °C.
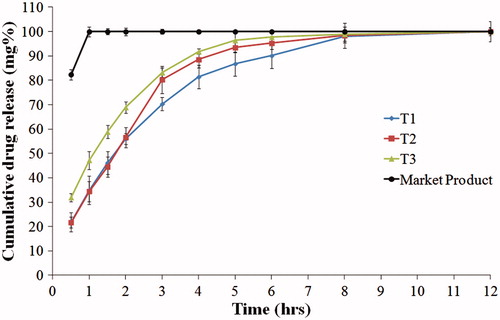
Figure 4. Release profiles of famotidine HCl from tablets formulae containing HPMC K4M, compared to the market product (Pepcid®), in 0.1 N HCl at 37 °C.
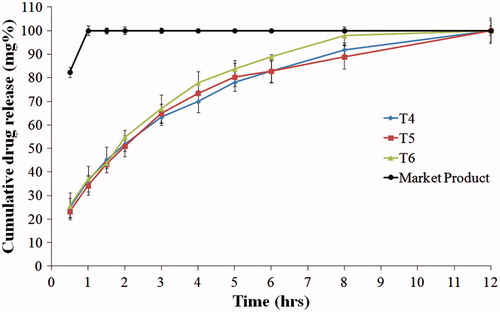
Figure 5. Release profiles of famotidine HCl from tablets formulae containing PEO, compared to the market product (Pepcid®), in 0.1 N HCl at 37 °C.
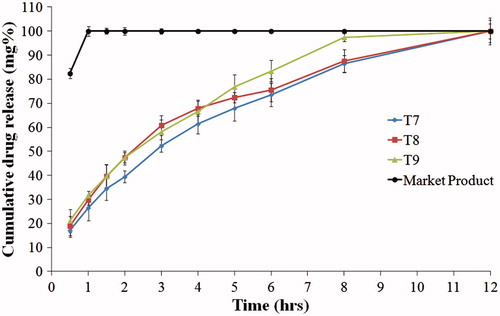
Incorporation of 20% Avicel, in formulae T2, T5 and T8, increased the release rate of the drug. Further increase in the drug release rate was observed with increasing Avicel concentration to 40% in case of formulae T3, T6 and T9. This might be due to the presence of lower percentage of the swellable polymer which formed a weaker gel with lower ability for sustaining the drug release.
Kinetics analysis of the release data
In order to determine the release model, the in-vitro release data were analyzed according to the equations of zero order, first order and Higuchi diffusion (Higuchi, Citation1963). The drug release of all formulae followed the diffusion mechanism except D1 and D5 which showed zero-order release ().
Table 5. Kinetic analysis of drug release from lipid disks formulae.
According to Korsmeyer–Peppas and Peppas and Sahlin models, drug release from all tablets formulae was anomalous with greater diffusional contribution, as shown in . Anomalous release was previously reported by Tajiri et al. and Tritt-Goc et al. from tablets containing high percentages of PEO and HPMC, respectively (Tritt-Goc & Kowalczuk, Citation2005; Tritt-Goc et al., Citation2005; Tajiri et al., Citation2010).
Table 6. Kinetic analysis of drug release from compression coated tablets formulae.
Statistical analysis
D-optimal response surface model was utilized to analyze the factorial design data. The predicted R2 values were in a reasonable agreement with the adjusted R2 in all responses except the tablets burst release (). The latter had a negative predicted R2 value implying that the overall mean was a better predictor of the response (Quinten et al., Citation2009; Basalious et al., Citation2011). Adequate precision measured the signal to noise ratio to ensure that the model can be used to navigate the design space (Lima et al., Citation2011). A ratio greater than 4 (the desirable value) was observed in all responses, except tablets burst release.
Table 7. Output data of the factorial analysis.
The selection criteria of the optimized disks formulae were the lowest friability, the lowest burst release and the highest release t50%. The combination of SA and PEG with a ratio of 9.5:0.5 w/w (D9) had the highest desirability value (0.841) as shown in (a). However, this formula was excluded as it released only 72% of the drug after 12 h. Hence, D10 (containing SA and PEG in a ratio of 9:1 w/w) with a desirability value of 0.684 was selected as the optimum formula.
Figure 6. The desirability of the prepared disks (a) and tablets (b) formulae.
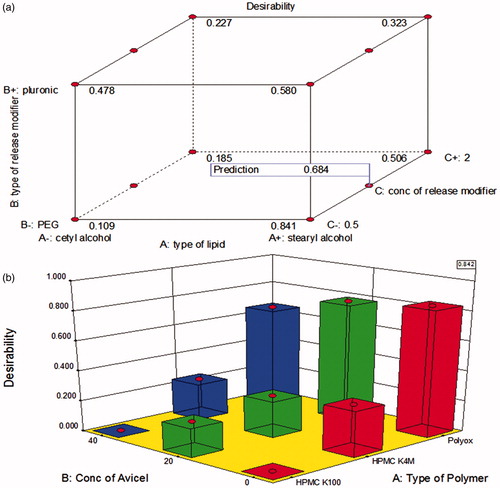
The optimized tablet formula was selected according to the same criteria as lipid disks in addition to the highest hardness and the lowest floating lag time. Formula T7 (containing PEO only) showed the highest desirability value (0.842) as shown in (b).
Scanning electron microscopy
It was observed from (a) and (c) that the optimized formulae (D10 and T7) had uniform non porous surfaces. On the other hand, porous surfaces were observed after 12 h of release (b and d).
Figure 7. Scanning electron micrograph of D10 (a and b) and T7 (c and d) before (a and c) and after (b and d) drug release.

X-ray imaging in human GIT
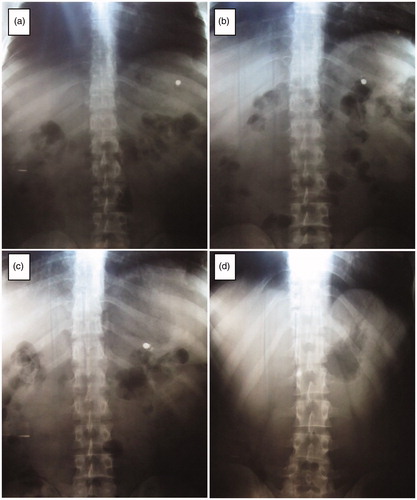
Disk formula (D10) was selected for the in-vivo imaging by X-ray due to its instantaneous floating and higher release half-life (3.92 ± 0.08 h), when compared to the tablet formula (T7). Abdominal radiographs of D10 showed that the disk formula remained intact in the stomach in all the subjects for eight hours as shown in . On the other hand, the control formula occurred in the antrum part of the stomach after one hour. Moreover, it failed to remain in the stomach and was visualized in the intestine after four hours. This ensured the applicability of the lipid disks in vivo considering the gastric retention and floating duration.
Figure 8. Localization of disk formula (D10) in the human GIT after 1 (a), 4 (b), 8 (c) and 12 (d) h.
Bioavailability and pharmacokinetic study in healthy human volunteers
The LC-MS/MS assay has been validated and has a good linearity from 0.5 to 500 ng/ml with acceptable within- and between-day reproducibility. The lower limit of famotidine HCl quantification in plasma was 0.5 ng/ml.
shows famotidine HCl mean (n = 6) plasma time concentrations after administration of disk formula (D10) and Pepcid® tablets to the healthy human volunteers. The pharmacokinetic parameters of the test and reference formulations are summarized in .
Figure 9. Mean plasma concentrations of famotidine HCl after oral administration of disc formula (D10) (the test product) and Pepcid® tablets (the reference product) to healthy human volunteers.
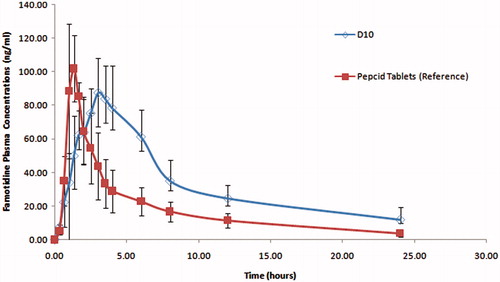
Table 8. Pharmacokinetic parameters of famotidine after oral administration of disc formula (D10), the test product and Pepcid tablets, the reference product to healthy volunteers.
The plasma concentration–time profiles showed that the disk formula (D10) sustained the oral absorption of famotidine HCl, expressed by the non significant lower Cmax of 96.80 ng/ml compared to 114.83 ng/ml for the reference product, significant delayed Tmax of 3.33 h in contrast to 1.28 h for the reference product and the significant higher HVDt50%Cmax of 5.12 h compared to 1.62 h for the reference product. The prepared disc (D10) showed a higher bioavailability expressed by the higher AUC0–t which is found to be 800.13 (ng.h/ml) compared to 443.60 (ng.h/ml) for the reference tablets which in turns resulted in 180.37% relative bioavailability. D10 formula proved to be superior in sustaining the oral absorption of famotidine HCl expressed in its higher RD (3.15).
The higher drug bioavailability might be due to the presence of lipids which promoted floating of the discs with no lag time and hence prolonged the residence time in the major absorption site. It is worthy to note that the study was done in fed condition to enhance the dosage form floating and hence improve drug clinical effectiveness. Moreover, low fat breakfast was employed to prevent the meal effect on the gastric emptying rate (Yin et al., Citation2013).
Conclusion
Techniques used in this study were characterized by simplicity and ease of scaling up. The results showed that lipids achieved immediate floating with zero lag time in all disks formulae and some compression coated tablets formulae. In addition, sustained release of famotidine HCl over a prolonged period of time was achieved with lipids and their release modifier in case of disks or with the swellable polymers in case of tablets. Response surface analysis was utilized for the selection of the best formulae. X-ray imaging investigated the gastric retention of the selected disk formula over eight hours. The gastroretentive system improved the drug bioavailability since it prolonged the residence time in the major absorption site. Finally, lipid disks were found to be superior over swellable compression coated tablets regarding absence of floating lag time, in all formulae, and ease of drug release modulation with avoidance of the previously mentioned disadvantages of the common reported gastroretentive techniques.
Declaration of interest
The authors report no declaration of interest.
References
- Aslani P, Kennedy RA. (1996). Effect of gelation conditions and dissolution media on the release of paracetamol from alginate gel beads. J Microencapsul 13:601-14
- Basalious EB, El-Sebaie W, El-Gazayerly O. (2011). Application of pharmaceutical QbD for enhancement of the solubility and dissolution of a class II BCS drug using polymeric surfactants and crystallization inhibitors: development of controlled-release tablets. AAPS PharmSciTech 12:799–810
- Chia HH, Yang YY, Chung TS, et al. (2001). Auto-catalyzed poly(ortho ester) microspheres: a study of their erosion and drug release mechanism. J Control Release 75:11–25
- Commission BP. (2008). The British Pharmacopoeia. London: The Stationary Office
- Dahan A, Amidon GL. (2009). Segmental dependent transport of low permeability compounds along the small intestine due to P-glycoprotein: the role of efflux transport in the oral absorption of BCS class III drugs. Mol Pharm 6:19–28
- Darandale S, Vavia P. (2012). Design of a gastroretentive mucoadhesive dosage form of furosemide for controlled release. Acta Pharm Sin B 2:509–17
- Deshpande AA, Shah NH, Rhodes CT, Malick W. (1997). Development of a novel controlled-release system for gastric retention. Pharm Res 14:815–19
- Gande S, Rao Y. (2011). Sustained-release effervescent floating matrix tablets of baclofen: development, optimization and in vitro-in vivo evaluation in healthy human volunteers. Daru 19:202–9
- Gao P, Skoug JW, Nixon PR, et al. (1996). Swelling of hydroxypropyl methylcellulose matrix tablets. 2. Mechanistic study of the influence of formulation variables on matrix performance and drug release. J Pharm Sci 85:732–40
- Gray J. (2011). Therapeutic choices. Ottwa, Ontario, Canada: Canadian Pharmacist Association
- Grube S, Langguth P. (2008). Excipients as modulator of drug-carrier mediated absorption in intestine. In: Mashkevich BO, ed. Drug delivery research advances. NY: Nova Science Pub Inc, 77–116
- Gupta R, Pathak K. (2008). Optimization studies on floating multiparticulate gastroretentive drug delivery system of famotidine. Drug Dev Ind Pharm 34:1201–8
- Han C, Wang B. (2005). Drug delivery – principles and applications. Factors that impact the developability of drug candidates. In: Wang B, Siahaan TJ, Soltero R, eds. NJ: Wiley Interscience, 1–14
- Higuchi T. (1963). Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci 52:1145–9
- Hooda A, Nanda A, Jain M, et al. (2012). Optimization and evaluation of gastroretentive ranitidine HCl microspheres by using design expert software. Int J Biol Macromol 51:691–700
- Hwang SJ, Park H, Park K. (1998). Gastric retentive drug-delivery systems. Crit Rev Ther Drug Carrier Syst 15:243–84
- Jaimini M, Rana AC, Tanwar YS. (2007). Formulation and evaluation of famotidine floating tablets. Curr Drug Deliv 4:51–5
- Jamzad S, Fassihi R. (2006). Development of a controlled release low dose class II drug-Glipizide. Int J Pharm 312:24–32
- Jiménez-Castellanos MR, Zia H, Rhodes CT. (1994). Design and testing in vitro of a bioadhesive and floating drug delivery system for oral application. Int J Pharm 105:65–70
- Karatas A, Baykara T. (2001). Studies on indomethacin inserts prepared by water-soluble polymers. II. The relation between dissolution rate and swelling behaviour. Farmaco 56:197–202
- Kaul G, Huang J, Chatlapalli R, et al. (2011). Quality-by-design case study: investigation of the role of poloxamer in immediate-release tablets by experimental design and multivariate data analysis. AAPS PharmSciTech 12:1064–76
- Kikuchi A, Kawabuchi M, Sugihara M, et al. (1997). Pulsed dextran release from calcium–alginate gel beads. J Control Release 47:21–9
- Kim H, Fassihi R. (1997). Application of a binary polymer system in drug release rate modulation. 1. Characterization of release mechanism. J Pharm Sci 86:316–22
- Klausner EA, Lavy E, Friedman M, Hoffman A. (2003). Expandable gastroretentive dosage forms. J Control Release 90:143–62
- Kuentz M, Leuenberger H. (2000). A new theoretical approach to tablet strength of a binary mixture consisting of a well and a poorly compactable substance. Eur J Pharm Biopharm 49:151–9
- Li FQ, Hu JH, Deng JX, et al. (2006). In vitro controlled release of sodium ferulate from Compritol 888 ATO-based matrix tablets. Int J Pharm 324:152–7
- Lima LSd, Araujo MDM, Quináia SP, et al. (2011). Adsorption modeling of Cr, Cd and Cu on activated carbon of different origins by using fractional factorial design. Chem Eng J 166:881–9
- Lotfipour F, Nokhodchi A, Saeedi M, et al. (2004). The effect of hydrophilic and lipophilic polymers and fillers on the release rate of atenolol from HPMC matrices. Farmaco 59:819–25
- Meier J, Nuesch E, Schmidt R. (1974). Pharmacokinetic criteria for the evaluation of retard formulations. Eur J Clin Pharmacol 7:429–32
- Mishra M, Mishra B. (2010). Design and evaluation of microporous membrane coated matrix tablets for a highly water soluble drug. Chem Pharm Bull (Tokyo) 58:995–1000
- Mishra SK, Pathak K. (2008). Formulation and evaluation of oil entrapped gastroretentive floating gel beads of loratadine. Acta Pharm 58:187–97
- Murata Y, Kofuji K, Kawashima S. (2003). Preparation of floating alginate gel beads for drug delivery to the gastric mucosa. J Biomater Sci Polym Ed 14:581–8
- Ndindayino F, Vervaet C, Van den Mooter G, Remon JP. (2002). Direct compression and moulding properties of co-extruded isomalt/drug mixtures. Int J Pharm 235:159–68
- O’Keefe SF. (2008). Nomenclature and classification of lipids. In: Akoh CC, Min DB, eds. Food lipids: chemistry, nutrition, and biotechnology. FL: CRC Press, 3–37
- Ostberg T, Lund EM, Graffner C. (1994). Calcium alginate matrixes for oral multiple unit administration: IV. Release characteristics in different media. Int J Pharm 112:241–8
- Peppas NA. (1985). Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv 60:110–11
- Peppas NA, Sahlin JJ. (1989). A simple equation for the description of solute release. III. Coupling of diffusion and relaxation. Int J Pharm 57:169–72
- Prajapati ST, Patel LD, Patel DM. (2008). Gastric floating matrix tablets: design and optimization using combination of polymers. Acta Pharm 58:221–9
- Pund S, Joshi A, Vasu K, et al. (2011). Gastroretentive delivery of rifampicin: in vitro mucoadhesion and in vivo gamma scintigraphy. Int J Pharm 411:106–12
- Quinten T, Gonnissen Y, Adriaens E, et al. (2009). Development of injection moulded matrix tablets based on mixtures of ethylcellulose and low-substituted hydroxypropylcellulose. Eur J Pharm Sci 37:207–16
- Ramachandran S, Shaheedha SM, Thirumurugan G, Dhanaraju MD. (2010). Floating controlled drug delivery system of famotidine loaded hollow microspheres (microballoons) in the stomach. Curr Drug Deliv 7:93–7
- Rao GK, Mandapalli PK, Manthri R, Reddy VP. (2013). Development and in vivo evaluation of gastroretentive delivery systems for cefuroxime axetil. Saudi Pharm J 21:53–9
- Ritger PL, Peppas NA. (1987). A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J Control Release 5:37–42
- Rowe RC, Sheskey PJ, Quinn ME. (2009). Handbook of pharmaceutical excipients. NY: Pharmaceutical Press
- Sarparanta MP, Bimbo LM, Makila EM, et al. (2012). The mucoadhesive and gastroretentive properties of hydrophobin-coated porous silicon nanoparticle oral drug delivery systems. Biomaterials 33:3353–62
- Sasa B, Odon P, Stane S, Julijana K. (2006). Analysis of surface properties of cellulose ethers and drug release from their matrix tablets. Eur J Pharm Sci 27:375–83
- Satishbabu BK, Sandeep VR, Ravi RB, Shrutinag R. (2010). Formulation and evaluation of floating drug delivery system of famotidine. Indian J Pharm Sci 72:738–44
- Shah VP, Midha KK, Dighe S, et al. (1991). Analytical methods validation: bioavailability, bioequivalence and pharmacokinetic studies. Conference report. Eur J Drug Metab Pharmacokinet 16:249–55
- Singh BN, Kim KH. (2000). Floating drug delivery systems: an approach to oral controlled drug delivery via gastric retention. J Control Release 63:235–59
- Steinijans VW. (1990). Pharmacokinetic characterization of controlled-release formulations. Eur J Drug Metab Pharmacokinet 15:173–81
- Tadros MI. (2010). Controlled-release effervescent floating matrix tablets of ciprofloxacin hydrochloride: development, optimization and in vitro-in vivo evaluation in healthy human volunteers. Eur J Pharm Biopharm 74:332–9
- Tajiri T, Morita S, Sakamoto R, et al. (2010). Release mechanisms of acetaminophen from polyethylene oxide/polyethylene glycol matrix tablets utilizing magnetic resonance imaging. Int J Pharm 395:147–53
- Tritt-Goc J, Kowalczuk J. (2005). Spatially resolved solvent interaction with glassy HPMC polymers studied by magnetic resonance microscopy. Solid State Nucl Magn Reson 28:250–57
- Tritt-Goc J, Kowalczuk J, Pislewski N. (2005). MRI study of Fickian, case II and anomalous diffusion of solvents into hydroxypropylmethylcellulose. Appl Magn Reson 29:605–15
- Vueba ML, Batista de Carvalho LA, Veiga F, et al. (2004). Influence of cellulose ether polymers on ketoprofen release from hydrophilic matrix tablets. Eur J Pharm Biopharm 58:51–9
- Yehia SA, Elshafeey AH, Elsayed I. (2011). Pulsatile systems for colon targeting of budesonide: in vitro and in vivo evaluation. Drug Deliv 18:620–30
- Yehia SA, Elshafeey AH, Sayed I, Shehata AH. (2009). Optimization of budesonide compression-coated tablets for colonic delivery. AAPS PharmSciTech 10:147–57
- Yin L, Qin C, Chen K, et al. (2013). Gastro-floating tablets of cephalexin: preparation and in vitro/in vivo evaluation. Int J Pharm 452:241–8
- Zhang C, Xu M, Tao X, et al. (2012). A floating multiparticulate system for ofloxacin based on a multilayer structure: in vitro and in vivo evaluation. Int J Pharm 430:141–50