
Abstract
Context: Terbinafine hydrochloride is an antifungal drug for onychomycosis. Poor permeability of its external preparation leads to poor curative effect. Transfersomes, also known as flexible liposome, could improve transmission of drug for local external use. Terbinafine hydrochloride-loaded liposome is expected to become a breakthrough on the treatment of onychomycosis.
Objective: This study is aimed to prepare high skin penetration terbinafine hydrochloride transfersomes with high encapsulation efficiency, appropriate drug loading and good stability.
Materials and methods: Taking entrapment efficiency as the main indicator, the formulations and the processes of preparation were investigated. Transfersomes with different surfactants were prepared in the optimization processes, and the formulations were optimized through the transdermal test in vitro. As a result, a gel contained transfersomes was obtained with a brief evaluation. Its pharmacokinetic properties of going through the skin were studied by using the micro dialysis technology and liquid chromatography-mass spectrometry to assay the penetration behavior of terbinafine.
Results: Mean particle size of the terbinafine hydrochloride transfersomes was 69.6 ± 1.23 nm, and the entrapment efficiency was 95.4% ± 0.51. The content of the gel was 4.45 ± 0.15 mg/g. The accumulated permeation of the transfersomes gel in 12 h was 88.52 ± 4.06 µg cm−2 and the intracutaneous drug detention was 94.38 ± 5.26 µg cm–2. The results of pharmacokinetic studies showed the Cmax and area under the curve (AUC) were apparently higher than the commercial cream.
Discussion and conclusion: The terbinafine hydrochloride transfersomes was highly absorbed by the skin. The absorption rate was significantly higher than that of the commercial cream either in the transdermal test in vitro or in the pharmacokinetic studies in vivo.
Introduction
Onychomycosis (Odom et al., Citation2000) caused by fungal infection is the most common but intractable disease. It has a high incidence rate, which increases with age (Yu, Citation2004). Because of the low drug penetration of topical administration, oral administration of antifungal drug as an effective therapeutic way is preferential to date. However, it is worth noting that the adverse effects resulting from oral administration are also the problems needed to be solved.
Currently, the oral medications used to treat onychomycosis mainly include itraconazole, ketoconazole, terbinafine hydrochloride, etc., which need to be orally administrated constantly for more than three months to obtain ideal curative effect. Consequently, alimentary canal toxicity, hepatotoxicity, neurovirulence and immune system toxicity could occur due to long-time administration, which makes it difficult for patients to persist with the medication treatment, thus resulting in treatment failure finally. So, external preparations of these drugs are taken into account. External medications with acceptable effect are amorolfine and ciclopirox clinically. In addition, amorolfine and ciclopirox are only suitable for the plain type onychomycosis and mild distal and lateral subungual onychomycosis with a small and superficial infection or uninfected nail matrix, while for proximal subungual onychomycosis and all damaged armor type onychomycosis, treatment with single external medicine shows no obvious effect. Since these external preparations fail to permeate through skin and nail to reach the nail bed, therapeutic effect is poor. Thus, it will bring significant changes for treatment of onychomycosis if we can develop a novel external drug delivery system that could obviously improve the transmittance of the drug and get the drug absorbed by skin around the nail and distributed to nail bed.
Transfersomes (Qi, Citation2002; Ceve et al., CitationUS7820720), which is proposed by professor Ceve for the first time (Ceve et al., Citation1996), is obtained by the modification of conventional liposomes. Its main compositions are phospholipids (membrane) and surfactant (membrane softening agent). Because of the addition of surfactant, it has a high ability of deformation and is highly hydrophilic. It can penetrate the small skin pore of cutin layer depending on the skin hydration pressure. As a result, as a carrier for drug delivery, it can obviously improve the transmittance of the drug when using externally. Simultaneously, its mimic biological membrane structure makes it available to form drug storage within the epidermis and dermis, leading to a continuous therapy as the drug releases slowly (Ceve et al., Citation1998). Transfersomes have shown its unique advantages as transdermal carriers of drug delivery. Ceve et al. reported that transfersomes hold promise for effective topical delivery of insulin with skin transmittance above 80% and effective duration of 150 ∼ 180 min on human study (Ceve et al., Citation1998). Research on it has become one of the most popular fields in pharmacy.
As a new broad-spectrum antifungal drug, terbinafine is one of the most widely used antifungal drugs clinically (Wei, Citation2008). It has obvious inhibitory and killing effect for onychomycosis caused by dermatophytes after orally administrated more than three months. Terbinafine is lipophilic and could appropriately permeate through corneous layer into nail plate. Taking advantage of transfersomes as carrier for terbinafine could significantly improve drug transmittance through skin. In this case, external preparation for the treatment of severe onychomycosis will become a reality. Simultaneously, it will provide evident basis for transfersomes as transdermal drug carrier.
In this study, we adopt transfersomes gel to overcome transfersomes’ shortcomings of low viscosity and temporary remain in administration site. First, the terbinafine transfersomes formulation and preparation procedure was optimized with high drug loading and high entrapment efficiency obtained. Second, the particle size, concentration of gel and dosage and types of surfactants were investigated by in vitro skin model, which made a contribution to deformability and penetrability of transfersomes. Finally, the in vivo transdermal test of terbinafine transfersomes gel was studied by means of liquid chromatography–tandem mass spectrometry (LC–MS) combined with micro dialysis technique. With this technique, we can accurately determine the real-time concentration of drug through the skin quickly.
Materials and methods
Materials
Terbinafine hydrochloride was purchased from Zhuhai City Pharmaceutical Co., Ltd. (Zhuhai, China). Phospholipid was purchased from German Lipoid Co. (Ludwigshafen, Germany). Tween-80 (Tianjin Chemical Reagent Factory, Tianjin, China). Polyoxyethylene 20 stearyl ether (Brij S20, Sigma Chemical Co., St. Louis, MO). Polyoxyethylene fatty alcohol (9) ether (AEO-9), polyoxyethylene castor oil (EL-35) and polyoxyethylene hydrogenated castor oil (RH-40) were supplied from BASF (Ludwigshafen, Germany). Sodium caprate (SC), deoxysodium cholate (NaDC) and sodium dodecyl sulfate (SDS) were supplied from Aladdin Reagent Database Inc. (Shanghai, China). Hydroxy propyl methyl cellulose (HPMC) 100 000 cp was supplied from Shin-Etsu Chemical Industry Co., Ltd. (Tokyo, Japan). All the other chemicals, reagents and solvents used were of analytical grade. Lamisil cream was purchased from the pharmacy.
Animals
Male Sprague-Dawley rats (200–250 g) were supplied by the Animal Experimental Center of Guangzhou University of Chinese Medicine. The rats were kept in the animal facility at 22–25 °C, 50–60% relative humidity under a 12/12 h light/dark cycle. Rats were allowed free access to food and water, fasted 2 d prior to experiment.
Preparation of terbinafine hydrochloride transfersomes and gel
The organic phase was produced by dissolving the drug, phospholipids, sodium cholesteryl sulfate and liposoluble surfactant into ethanol, to form an optically isotropic phase that appears as a clear solution. Furthermore, the aqueous phase was produced by dissolving some hydrosoluble surfactants into the phosphate buffer (PBS). After heating both of them up to 45 °C, the organic phase was slowly injected into the aqueous phase under stirring at a constant temperature of 45 °C. After injection, the stirring process continued for 20 min. Then, the transfersomes was formed by removing the ethanol with vacuum evaporation, and then dispersed by ultrasonic probe (UH-500 A, OTT thain instrument co., Ltd., Tianjin, China) for four times with 20 s each to reduce the particle size.
Several transfersomes samples containing different kinds of surfactants (NaDC, SDS, SC, Tween-80, BrijS20, AEO-9, RH-40 and EL-35) were prepared. Anionic surfactants like NaDC, SDS, SC were added to the water phase during preparation. Nonionic surfactants such as Tween-80, BrijS20, AEO-9, RH-40 and EL-35 were added to the organic phase during preparation.
The HPMC (100 000 cp) was weighed into a beaker, then, an appropriate volume of water was added to make it fully swelled into a clear blank gel with a content of 3% w/v. The medicated gel was formed by mixing the terbinafine transfersomes with the blank gel at the volume ratio of 1:1 after stirring.
Content determination
The terbinafine hydrochloride transfersomes gel was weighed into a suitable container, some methanol added to dissolve it. Then the upper clear liquid was taken as the sample after centrifuging. The concentration of terbinafine hydrochloride was determined by high performance liquid chromatography (HPLC) (LC-10 A, Shimadzu Ltd., Tokyo, Japan) with an Agilent Tc-C18 (Agilent Technologies Ltd., Santa Clara, CA) chromatographic column. The mobile phase was methanol–ammonium acetate buffer (dissolve 1.32 g ammonium acetate into 100 mL water) solution (95:5), the flow rate was 1 mL/min and the UV absorption wavelength was 283 nm. The injection volume is 20 µL.
Entrapment efficiency
The transfersomes suspension was dropped into the ultrafiltration membrane (EMD Millipore Co., Billerica, MA), then centrifuged at a speed of 3000 rpm for 3 min. The unentrapped terbinafine hydrochloride dissolved in transforsomes suspension was separated from transfersomes-entrapped terbinafine hydrochloride by centrifugal ultrafiltration. The filtrate obtained was determined by HPLC as the unentrapped terbinafine hydrochloride concentration. The total drug concentration was determined by HPLC.
The entrapment efficiency percent (%EE) can be calculated by using the following equation:
(1)
where %EE is the percentage of entrapment efficiency, C0 is the concentration of the total terbinafine hydrochloride and Cf is concentration of free terbinafine hydrochloride.
Particle size distribution
Terbinafine hydrochloride transfersomes suspension was diluted to a suitable concentration with water, and then it was subjected to the laser particle size analyzer (ZATA SIZER1000H, Malvern Instr., Worcestershire, UK) for measurement of the particle size.
In vitro transdermal test of transfersomes
An accurately measured quantity of terbinafine hydrochloride transfersomes suspension, equivalent to 10 mg of terbinafine hydrochloride, was suspended in the Franz diffusion pool (Deng et al., Citation2011) (Shanghai Kaikai Technology Trade Co., Ltd, Shanghai, China), having the diameter of 2 cm and diffusion area 3.14 cm2. The isolated skin of the rat with thickness of 0.5 mm was fitted between the supply pool and the reception pool and was placed in the transdermal diffusion apparatus (TK-20 A, Shanghai Kaikai Technology Trade Co., Ltd, China). The reception pool was fulfilled with 8 mL physiological saline–ethanol (70:30) with constant stirring at a speed (200 rpm) at 32 ± 1 °C.
At predetermined time intervals (1, 2, 4, 6, 8 and 12 h), 1 mL of the reception liquid was withdrawn for analysis and replaced with equal volume of fresh reception solution at the same temperature to maintain a constant volume. After the last sample was collected, the skin was taken off and washed, and then the surface area of 3.14 cm2 was cut up and grinded into chyle. Methanol was added to extract terbinafine hydrochloride that remained in the skin with ultrasonic treatment for 30 min, followed by centrifugation at low temperature at a speed of 10 000 rpm for 10 min. The supernatant liquid samples were collected and filtrated with 0.22 µm filter membrane. The concentration of the samples was measured by HPLC.
The amount of drug permeated through and remained in the skin was obtained by the following equations:
(2)
(3)
(4)
where Q (µg) is the accumulative amount of drug permeated in 12 h, Cn (µg/mL) is the concentration of the sample corresponding to the time point, V0 (mL) is the sample volume, J (µg/cm2/h) is the amount of drug permeated in time t(h) per unit area A (cm2), Q0 (µg) is the amount of drug remained in the skin, C0 (µg/mL) is the concentration of the sample about skin remaining, the volume related is V′(mL).
In vivo studies of transfersomes
A LC–MS combined with micro dialysis technique was used for the in vivo studies of the terbinafine hydrochloride transfersomes gel (El-Nabarawi et al., Citation2013; Yua et al., Citation2013). The micro dialysis probe was implanted into the skin of rat to collect samples containing terbinafine hydrochloride permeated from the skin surface. LC–MS was chosen as the analytical equipment to detect the concentration of samples for such low drug concentration beyond the limit of HPLC-UV detection.
LC–MS analysis
Samples were analyzed on a LC–MS (TSQ Quantum Access Max HPLC-MS, Thermo Fisher Scientific Inc., Waltham, MA). The mass spectra were acquired on a “triple” quadruple instrument equipped with electro spray ionization source, which was operated in a positive mode. The column of the ultra performance liquid chromatography (UPLC) system was provided by Thermo (Particle size (µm): 1.9; pore size (A): 175; material: Hypersil GOLD; column dimensions: 100 × 2.1 nm). The mobile phase consisted of a mixture of methanol and 0.1% formic acid at a flow rate of 0.3 mL/min. The initial ratio (15:85, v/v) was kept for 1.5 min, and then changed to 98:2 (v/v) in 0.5 min; this ratio was kept for 3 min and then changed to the initial ratio in 0.5 min. The injection volume was 2 µL, and the analysis time was 2 min per sample.
Acquisition was performed in a selected reaction monitoring mode using m/z 292 → 140 (terbinafine hydrochloride), m/z 152 → 109 (acetaminophen). The sheath gas pressure was 40 Psi; the Aux gas pressure was 10 Psi. The spray voltage was 3.5 kV, the vaporizer temperature was 350 °C and the capillary temperature was 350 °C.
In vitro relative recovery of probe
With regard to dialysis (Sun, Citation2006), the linear probe (CMA30, membrane length 10 mm, molecular weight cut-off 6 kDa, Microbiotech Corporation, Stockholm, Sweden), which connected to a micro dialysis system (MD-1001, BASiCMA, Stockholm, Sweden), was immersed and fixed in the vessel that contained physiological saline solution in which the concentration of terbinafine hydrochloride was 10 µg/mL (C0), with constant stirring at 37 ± 1 °C. Blank physiological saline was used as perfusion liquid to perfuse the probe at three different speeds (1, 1.5 and 2 µL/min). The dialysis samples were collected at each 30 min for three times. The drug concentrations were analyzed by LC–MS. The in vitro recovery was calculated by the following equation:
(5)
where C0 and Cout are the concentrations of terbinafine hydrochloride outside the probe and the dialysate fluids, respectively.
Then, three different concentrations (1, 5 and 10 µg/mL) as C0 were choosen, the perfusion speed was 1.5 µL/min, and other conditions were the same as described above. The dialysis samples were collected at each 30 min for three times. The drug concentration in the dialysis samples (Cout) was determined. The in vitro recovery was calculated by Equation (5).
With regard to reverse dialysis (Wang, Citation2010), the linear probe was immersed and fixed in the vessel that contained blank physiological saline solution with constant stirring at 37 ± 1 °C. Physiological saline solution in which the concentration of terbinafine hydrochloride was 10 µg/mL (Cin) was used as perfusion liquid to perfuse the probe at three different speeds (1, 1.5 and 2 µL/min). The dialysis samples were collected at each 30 min for three times. The drug concentrations were analyzed by LC–MS. The in vitro recovery was then calculated by the following equation:
(6)
where Cin and Cout are the concentrations of terbinafine hydrochloride inside the probe and the dialysate fluids, respectively.
Then, three different concentrations (1, 5 and 10 µg/mL) as Cin were chosen, the perfusion speed was 1.5 µL/min, and other conditions were the same as described above. The dialysis samples were collected at each 30 min for three times. The drug concentration in the dialysis samples (Cout) was determined. The in vitro recovery was calculated by Equation (6).
In vivo relative recovery of probe
The rat’s abdomen with the hair removed was implanted with the linear micro dialysis probe. The probe was connected to the micro dialysis system. In vivo relative recoveries of terbinafine hydrochloride were estimated by determining the loss of the drug in vivo using reverse dialysis technique. The probes were inserted into the skin of the abdomen area about 15 mm long. After one hour of stabilization, a physiological saline solution containing terbinafine hydrochloride (10 µg/mL) was perfused through the probe at a rate of 1.5 µL/min. The dialysis samples were collected at each 30 min for 10 h. The dialysates entering (Cin) and leaving (Cout) the probe were analyzed by LC–MS. The in vivo recovery was then calculated by Equation (6).
Drug administration and sampling
Six rats were randomly divided into two groups (n = 3 per group). Anesthetized with urethane, the rat was implanted with the micro dialysis probe in the skin of the abdomen as mentioned previously. One group was smeared with the terbinafine hydrochloride transfersomes gel, while the other group was smeared with the Lamisil cream. The administration area was 3.14 cm2. The probe was perfused with blank physiological saline solution with a flow rate of 1.5 µL/min. The whole system was stabilized for 1 h before drug administration. The dialysis samples were collected at each 30 min for 10 h. After sampling, the drug was washed off, the system was stabilized for 2 h and then the perfusion liquid was changed to physiological saline solution containing a concentration of terbinafine hydrochloride 10 µg/mL. The dialysis samples were collected at each 30 min for four times after stabilization for 1 h. The dialysates samples were analyzed by LC–MS. The drug concentration was calculated by the following equation:
(7)
where Cs and Cd are the concentrations of terbinafine hydrochloride inside the skin and the sample, respectively. R is the in vitro recovery and calculated by Equation (6).
The drug concentration–time data were evaluated, and the following pharmacokinetic parameters were calculated: Cmax (ng/mL), Tmax (h), K (1/h), t1/2 (h), AUC0→t (ng h/mL) and AUC0→∞ (ng h/mL) using the DAS 3.0 software (Clinical drug research center of Shanghai university of traditional Chinese medicine, China).
Results and discussion
Preparation of terbinafine hydrochloride transfersomes
The ethanol injection method used for preparation had been compared with the film dispersion method, which has toxic solvent involved (Chen et al., Citation2009). They were both feasible for the preparation, but ethanol was a good and safer solution, so the ethanol injection method was chosen. The entrapment efficiency dropt down when preparing temperature above 45 °C as shown in . The suitable prepare temperature is 45 °C.
Table 1. The entrapment efficiency and particle size of transfersomes under different preparation degree.
During the preparation, the entrapment efficiency was taken as the indicator. As the amount of phospholipids increased, the entrapment efficiency improved. But the transfersomes suspension became so thick that affected the liquidity of it when the amount of phospholipids above 4.5% (w/v). The electronegative sodium cholesteryl sulfate could make a contribution to combine the electropositive drug with phospholipids membrane and significantly improve entrapment efficiency when the amount of it reached 1.0% (w/v). Particle precipitates when the amount of sodium cholesteryl sulfates above 1.0% (w/v).
The suitable amount of phospholipids (4.5%, w/v), surfactant (Tween-80, 1.5%, w/v), sodium cholesteryl sulfate (1.0%, w/v) and drug (1.0%, w/v) was selected.
Entrapment efficiency (%EE)
The results of entrapment efficiency of terbinafine hydrochloride transfersomes containing different kinds of surfactants but same amount of terbinafine hydrochloride are illustrated in ().
Table 2. The entrapment efficiency of transfersomes containing different kinds of surfactants.
Drug concentration of terbinafine hydrochloride transfersomes containing different kinds of surfactants is above 9 mg/mL, and its entrapment efficiency is above 90%, which means they both satisfy in vitro transdermal test.
In vitro transdermal test of transfersomes
The factors that would affect the transmittance were studied, such as the pH of the PBS buffer, dosage of administration, particle size, drug loading, types and dosage of the surfactants. Results show that the pH of the PBS buffer, drug loading and types of the surfactants significantly affect the transmittance; the effect of dosage of administration, particle size and dosage of the surfactants is not obvious.
The amount of permeation in low pH buffer was obviously higher than that in high pH as shown in . This could be the lower solubility of terbinafine hydrochloride in higher pH value of buffer. As our previous study showed, the solubility of terbinafine was 12.09 ± 0.56 mg/mL in pH 2.0,10.80 ± 0.49 mg/mL in pH 5.0, 7.04 ± 0.38 mg/mL in pH 7.0 and 5.41 ± 0.61 mg/mL in pH 9.0, respectively. It also may relate to the pH of the skin and the potential changes of the system. There would be skin irritation if the pH of the buffer is too low, so the pH of the PBS buffer was chosen as 5.00. Under the same condition, the higher the drug loading, the more the drug permeated in the same time as it shows in . As shown in , when the drug loading was higher than 10 mg/mL, the entrapment efficiency was low, so the drug loading was decided as 10 mg/mL. There are two kinds of surfactant (Tween-80 and AEO-9), which are relatively good enough for the transmission of the terbinafine hydrochloride in transfersomes as shown in . Finally, Tween-80 was chosen as the surfactant for the final formulation ().
Figure 1. (A) Accumulative amount of drug permeated in 12 h under different pH of the PBS buffer and (B) the amount of drug remained in the skin under different pH of the PBS buffer. Data represents mean with standard deviation at n = 3.
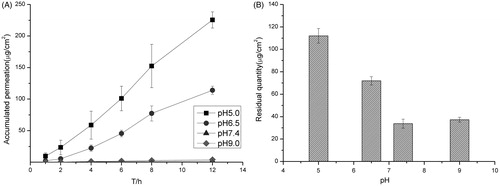
Figure 2. (A) Accumulative amount of drug permeated in 12 h under different drug loading and (B) the amount of drug remained in the skin under different drug loading. Data represents mean with standard deviation at n = 3.
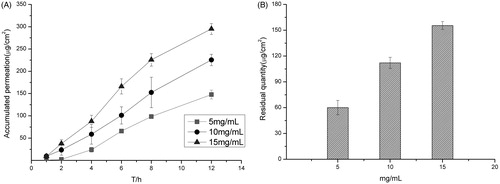
Figure 3. (A) Accumulative amount of drug permeated in 12 h under different surfactants and (B) the amount of drug remained in the skin under different surfactants. Data represents mean with standard deviation at n = 3.
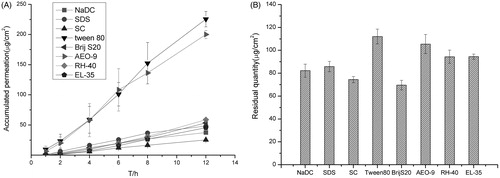
Table 3. The entrapment efficiency and particle size of transfersomes containing different amount of drug.
Table 4. The optimized formulation of terbinafine hydrochloride transforsomes.
Preparation and evaluation of transfersomes gel
The transfersomes finally selected containing Tween-80 as the surfactant and had a drug content of 9.81 mg/mL, entrapment efficiency of 94.8 ± 0.66% and with a pH 4.01 ± 0.01. The mean particle size of the system was 69.6 ± 1.23 nm.
Sodium carboxymethylcellulose (CMC-Na) and HPMC were compared as the gelling materials of the transfersomes, and the concentration of HPMC was studied, HPMC was a better material for the preparation of the gel because it had little prevention of the drug release. With the denseness of the substrate increased, the prevention of drug release was enhanced. However, the gel needs a suitable viscosity to be smeared, so the concentration of HPMC was chosen as 3%, and ratio of the transfersomes to gel substrate was volume ratio 1:1.
The transfersomes gel prepared had a drug content of 4.45 ± 0.16 mg/g and entrapment efficiency of 94.5 ± 0.45% and with a pH 4.38 ± 0.01.
The stability of the transfersomes gel was studied at 25 °C in the dark. The results in show that the transfersomes gel was stable with no obvious change in appearance, pH, content and entrapment efficiency. The transfersomes gel was compared with the Lamisil cream by transdermal test, and the results in show that the transmission of transfersomes gel was significantly higher than that of the Lamisil cream.
Figure 4. (A) Accumulative amount of drug permeated in 12 h and (B) the amount of drug remained in the skin of the transfersomes gel and the Lamisil cream. Data represents mean with standard deviation at n = 3.
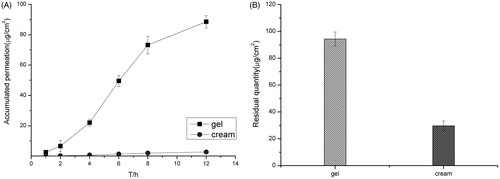
Table 5. The stability of the transfersomes gel in one month.
In vivo studies of transfersomes
The results of the in vitro relative recovery of dialysis and reverse dialysis are illustrated in . The effect of drug concentration of the perfusion liquid on the probe recovery was not obvious. The flow rate of the perfusion liquid had a certain influence to the probe recovery. The lower the flow rate, the higher the probe recovery is. When the flow rate of the perfusion liquid was 1.5 µL/min, the drug concentration of the perfusion liquid was 10 µg/mL, and the relative probe recovery obtained by dialysis and reverse dialysis were the closest. Therefore, in the condition of in vivo micro dialysis experiment, the recovery of the micro dialysis probe can be checked by the reverse dialysis method.
Table 6. The in vitro relative recovery of the probe.
The in vivo relative recovery of the probe was studied using the reverse dialysis method, and the results are shown in that the in vivo recovery of the probe was relatively stable in 10 h. The reverse dialysis method was used to check the recovery of the micro dialysis probe to calculate the concentration of the drug in the skin.
Figure 5. (A) In vivo recovery of the probe in reverse dialysis during 10 h and (B) the drug concentration-time curves of the transfersomes gel and the Lamisil cream. Data represents mean with standard deviation at n = 3.
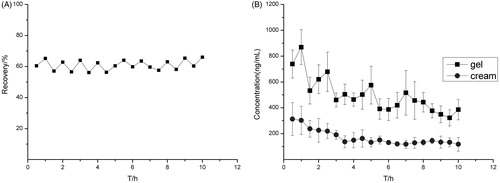
The in vivo transmission of the transfersomes gel and the Lamisil cream were studied, and the drug concentration–time curves are shown in . Furthermore, the pharmacokinetic parameters are shown in .
Table 7. The pharmacokinetic parameters.
The drug concentration–time curves fitted the one compartment model. The tmax and Cmax values were higher for the transfersomes gel than the Lamisil cream, and the difference indicates that the transmission of drug was faster in the transfersomes gel. The AUC0→t and AUC0→∞ values of the transfersomes gel were nearly three times higher than that of the Lamisil cream, which indicates that bioavailability of transfersomes gel is much better than that of Lamisil cream.
Conclusion and discussion
Terbinafine hydrochloride transfersomes () were successfully prepared with stable, high transmittance. The transmittance of the transfersomes gel was obviously higher than the Lamisil cream on sale both in the in vitro and in vivo studies.
Figure 6. Electron microscopy figure of terbinafine transfersomes (× 50 000).

Adoption of electronegative sodium cholesteryl sulfate significantly improved the drug loading and entrapment efficiency of transfersomes, which means that ionic surfactants has a certain significance in improving drug loading and entrapment efficiency of transfersomes. The transmission of terbinafine hydrochloride transfersomes was much higher than that of Lamisil cream, which illustrates the importance of transfersomes as drug carrier and partly verifies its ability to directly through skin. The results show that Tween-80 has evident effect on permeability of transfersomes. It gives us an inspiration that developing some novel surfactants will bring a positive effect on transmission of transfersomes through skin. The factors affecting transfersomes through skin and detailed mechanism deserve further validation to find out a better carrier for drugs.
Fluctuations of transdermal drug concentrations in vivo transdermal test may be due to the animals under anesthesia or also be affected by temperature changes. As for the transmission of transfersomes through nail, a further study is needed.
Furthermore, it is worthy to note that transfersomes is a good carrier for terbinafine hydrochloride transdermal delivery. The terbinafine hydrochloride transfersomes gel has a good application prospect for the treatment for onychomycosis.
Declaration of interest
No conflict of interest exits in the submission of this manuscript entitled “A novel drug delivery gel of terbinafine hydrochloride with high penetration for external use”, and the manuscript is approved by all authors for publication. The manuscript has not been previously published in any language anywhere and that it is not under simultaneous consideration by another journal.
References
- Ceve G, Gabiele B, Andress S, et al. (1996). The skinia pathway for systemic treatment with patches and lipid based agent carries. Adv Drug Deliv Rev 18:349–78
- Ceve G, Gebauer D, Stieber J, et al. (1998). Ultraflexible vesicles, transfersomes, have an extremely low pore penetration resistance and transport therapeutic amounts of insulin across the intact mammalian skin. Biochim Biophys Acta 1368:201–5
- Gregor C, Gauting DE, Ulrich V, et al. Topical terbinafine formulations and methods of administering same for the treatment of fungal infections. US7820720,-9-1
- Chen TK, Lin HQ, Li Y. (2009). The research progress of Flexible nanoliposomes as a new percutaneous drug delivery carrier. Prog Pharm Sci 12:732–4
- Deng H, Zhang S, Ling HQ, et al. (2011). Study of transdermal diffusion experiment of two kinds of terbinafine hydrochloride cream in vitro. China Pharm 22:2723–5
- El-Nabarawi MA, Bendas ER, El Rehem RT, Abary MY. (2013). Transdermal drug delivery of paroxetine through lipid-vesicular formulation to augment its bioavailability. Int J Pharm 443:307–17
- Odom RB, James WD, Berger TG. (2000). Andrews’ diseases of skin. 9th ed. : WB. Saunders Co., 376
- Qi HJ. (2002). The treatment of onychomycosis. China J Hosp Pharm 22:747–8
- Sun N. (2006). Cutaneous pharmacokinetics of azole antifungal drugs based on microdialysis and ultra-fast liquid chromatography. Shanghai: The Second Military Medical University
- Wang XP. (2010). The study on transdermal pharmacokinetics of podophyllotoxin-loaded solid lipid nanoparticles gel using microdialysis. : Southern Medical University
- Wei XQ. (2008). The synthesis process research of terbinafine hydrochloride. : Jilin University
- Yu RY. (2004). Onychomycosis therapy and thinking. J Dermatol Venereol 26:8–11
- Yua B, Ruan M, Cui XB, et al. (2013). Effects of borneol on the pharmacokinetics of geniposide in cortex, hippocampus, hypothalamus and striatum of conscious rat by simultaneous brain microdialysis coupled with UPLC–MS. J Pharm Biomed Anal 15:128–32