
Abstract
The use of pectin for colon-specific drug delivery has been extensively investigated; however, when used alone, pectin is often compromised due to its high solubility. This study explored the feasibility of using an in situ compression-coated crosslinking system, composed of pectin and calcium chloride, for colon-specific drug delivery. A pectin/calcium chloride (P/Ca) coating was compressed onto a core tablet. The colon specificity of the compression-coated tablet was verified by dissolution, pharmacokinetics and scintigraphy with 99mTc labeling. The in situ pectin and calcium chloride gel slowed the release of indomethacin. The lag time varied between 3 h and 7 h depending on the amount of calcium chloride and the coating weight. Pectinase triggered the release of indomethacin from the compression-coated tablet, which was then accelerated by the calcium chloride in the coating layer. The compression-coated tablet had a prolonged tmax and apparent t1/2, as well as a decreased Cmax and AUC0–t, compared with the core tablet counterpart. Evaluation with γ-scintigraphy verified colon-specific delivery of the compression-coated tablet. In conclusion, the P/Ca in situ crosslinking system worked well for colon-specific drug delivery.
Introduction
Colon-specific drug delivery systems (CSDDS) have been extensively investigated for use in the treatment of localized colon diseases. Among the different approaches that have been developed for the purpose of drug administration to the colon, polysaccharide-based CSDDS have demonstrated better colon-specificity when compared with either pH- or time-dependent delivery systems, likely due to selective degradation by colonic microflora (Basit et al., Citation2009; Shah et al., Citation2011). Polysaccharides that have been well established for use in colon-specific drug delivery include pectin, guar gum, chitosan, amylose, inulin, dextran and alginate (Liu et al., Citation2007; Prabaharan, Citation2011; Shah et al., Citation2011; Gulbake & Jain, Citation2012). Polysaccharide-based matrix and coated systems based on polysaccharide have been developed for colon-specific delivery of a variety of active ingredients (Basit, Citation2005; Prabaharan, Citation2011). Compression-coated systems have attracted a lot of attention recently due to their versatility and ability to deliver a wide range of drugs; however, preparation of these systems requires a special compression machine and highly skilled personnel (Shah et al., Citation2011).
Pectin is one of the most extensively studied natural polysaccharides due to its suitability for colon-specific drug delivery (Liu et al., Citation2007; Sriamornsak, Citation2011; Wong et al., Citation2011; Mamani Crispin et al., Citation2012). However, pectin’s colon-specificity is compromised by its high solubility. A previous study in which pectin alone was used as the compression coating layer required a minimum of 700 mg of pectin to achieve reliable colon-specificity (Ashford et al., Citation1993). Therefore, methods that reduce pectin solubility and/or strengthen the pectin gel are often used to take full advantage of the selective degradation of pectin in the colon. Insoluble salt like calcium pectinate has been reported to provide a better shielding effect, via the formation of an “egg-box” configuration, leading to a more controlled drug release (Rubinstein et al., Citation1993; Hiorth et al., Citation2006; Jain et al., Citation2007). However, poor compressibility and difficulties with quality control, particularly with respect to the amount of calcium in the formulation, has impeded the use of calcium pectinate. Stronger polymers, such as hydroxypropyl methylcellulose, have been employed to strengthen pectin gels (Ugurlu et al., Citation2007; Hodges et al., Citation2009); however, the presence of these polymers often compromises the sensitivity of the compression coat to colonic microflora.
In previous study, we reported that fortification of pectin gels could be achieved via calcium cation-induced in situ crosslinking (Wei et al., Citation2006, Citation2009; Wu et al., Citation2007a; Wu et al., Citation2008). Consequently, significant slowing of the initial drug release and a sigmoid release pattern were observed with use of these pectin/calcium chloride (P/Ca) matrix systems. However, pre-colonic release is unavoidable when using these systems since drugs are dispersed in the matrix and will be released as the matrix erodes. Nevertheless, the in situ crosslinking P/Ca system might be suitable for use as a compression coating material in colon-specific drug delivery. In the previous study, complete erosion of a 75/75 (mg/mg) P/Ca matrix occurred in 4 h (Wei et al., Citation2006). Therefore, it is reasonable to assume that a P/Ca compression-coated tablet, with a heavier coating and the proper P/Ca ratio, may prevent premature release of a drug. In addition, adjusting the degree of gelation in the in situ crosslinking pectin system can be done by simply altering the amount of calcium salt and is much easier than in calcium pectinate-based systems. Furthermore, calcium ions can activate or stimulate pectinase activity (Miller & Macmilla, Citation1970). As a result, the calcium in situ crosslinking not only slows the release of drug but the calcium chloride may also enhance the susceptibility of pectin to pectinase, thus achieving quick release at the proximal colon.
In this study, a compression-coated tablet, containing the non-steroidal, anti-inflammatory drug, indomethacin, was developed using pectin and calcium chloride as coating materials. The P/Ca ratio, coating weight and enzymatic effects on release characteristics were studied. The pharmacokinetic characteristics and gastrointestinal transportation of the P/Ca compression-coated tablet were also analyzed to verify colon-specific delivery.
Materials and methods
Materials
Micronized indomethacin (<5 µm) was purchased from Sine Pharmaceuticals (Shanghai, China). Pectin HM (high methoxylated) 70, crospovidone and PVP K30 were kindly provided by International Specialty Products (Kowloon, Hong Kong). Lactose was purchased from Lactose Company of New Zealand Ltd. (Kaponga, New Zealand) Microcrystalline cellulose was provided by J. Rettenmaer & Sohne Gmbh & Co. (Rosenberg, Germany). Pectinex Ultra SP-L was provided by the Shanghai Division of Novo Nordisk Ferment Ltd. (Shanghai, China) Technetium 99 m [99mTc] was provided by the Department of Nuclear Medicine, Zhongshan Hospital, Fudan University, Shanghai, China. The calcium chloride (CaCl2) was purchased from Shanghai Chemical Regent Corp. (Shanghai, China). All chemicals were of analytical grade.
Preparation of the core tablets containing indomethacin
The core tablets of indomethacin were prepared using the direct powder compression technique. Each tablet contained 25 mg indomethacin, 40 mg microcrystalline cellulose, 10 mg crospovidone, 20 mg lactose and 1% magnesium stearate (w/w) as a lubricant. The powders were mixed homogeneously and compressed into 5.5 mm flat tablets using a single-punch compressor (Shanghai FarEast Pharmaceutical Machinery Co., Shanghai, China).
Preparation of P/Ca compression-coated tablets
The P/Ca compression-coated tablets were prepared as previously reported (Wu et al., Citation2007b). The P/Ca coating material was prepared via wet granulation. First, the pectin and calcium chloride were mixed homogeneously. PVP K30 ethanol solution (10%, w/v) was then gradually added, and the mixture was milled into a paste. The wet mass was then forced through a 20-mesh sieve to make wet granules, which were then dried at 70 °C for 2 h. After mixing with 1% magnesium stearate (w/w), about 50% of the coating granules were poured into the die cavity. The core tablet was then carefully placed at the center of the die cavity, and the remainder of the coating granules were poured into the cavity. The core tablet and coating granules were then compressed into 10 mm round, flat, plain tablet using the single-punch compressor, which was used for preparation of the core tablet. The compression-coated indomethacin tablets were evaluated for drug content, hardness, friability and thickness. The formulations and their properties are listed in .
Table 1. The composition and physical properties of the compression-coated tablets.
Preparation of 99mTc-labeled compression-coated tablets
99mTc-labeled compression-coated tablets were prepared by gradually adding 100 µl of 99mTc, via micro-syringe, to both sides (50 µl on each side) of the core tablet. After the liquid was absorbed, the core tablet was dried using an air blower. The core tablet was then compression-coated into a 10 mm tablet with a coating material composed of 250 mg pectin and 250 mg calcium chloride.
Determination of indomethacin in tablets, release media and dog plasma
Indomethacin in tablets and release media were determined using a HPLC method (Wei et al., Citation2006). The Agilent 1100 series HPLC system consisted of a quaternary pump, a degasser, an autosampler, a column heater and a tunable wavelength UV detector. The separation was performed at 40 °C using a C18 column (Diamonsil®, 5 µm, 4.6 mm × 150 mm, Dikma, Beijing, China) and a refillable precolumn that was used as a guard column (C18, 1.0 mm × 20 mm, Alltech, Deerfield, IL). The mobile phase consisted of a mixture of acetonitrile and 0.3% acetic acid (ratio of 58/42) and pumped at a rate of 1.0 mL/min. The detection wavelength was set at 320 nm.
The above HPLC method was also adapted for the determination of indomethacin in dog plasma (Ji et al., Citation2007). Samples were prepared by liquid–liquid extraction. Briefly, 0.1 mL naproxen aqueous solution (internal standard, 10 µg/mL), 0.5 mL citric acid (2%, v/v) and 5 mL diethyl ether were added to 0.5 mL plasma. The mixture was then vortexed for 5 min and centrifuged at 1600 × g for 15 min. The upper organic layer was collected and evaporated at 40 °C under a stream of nitrogen. The dried residue was reconstituted with 0.1 mL mobile phase and centrifuged at 10 000 × g for 10 min. The supernatant was analyzed by HPLC for indomethacin content. This method has been validated for linearity, accuracy, precision, extraction recovery and limit of quantification (Ji et al., Citation2007).
Release study
Release tests were performed in ZRS-8 G dissolution tester (Tianjin, China) and were based on the Chinese Pharmacopoeia Method I (Wei et al., Citation2006). Basket rotation speed was set to 100 rpm. The temperature of dissolution medium was thermostatically maintained at 37 ± 0.5 °C. At predetermined time intervals, 5 mL samples were withdrawn and filtered (Millex® AP, Millipore, 0.4 µm, Billerica, MA). The filtrate was analyzed by HPLC for indomethacin content as described above. In the meantime, an equal volume of the same medium was added in order to keep the volume constant. If not specified otherwise, the release test was initially done in 900 mL of 0.1 M hydrochloride solution and thereafter was transferred to 900 mL PBS (pH 6.8) to mimic the gastrointestinal pH gradient.
Enzyme-triggered drug release was evaluated by spiking the solution with rat cecal content. Rat cecal content was collected using a reported method (Prasad et al., Citation1998). Wistar rats, weighing 200–300 g, were fed 2 mL of pectin aqueous solution (2%, w/v) daily, for seven days, to induce pectinase activity in the colon. Thirty minutes prior to the experiment, the rats were euthanized via urethane overdose. The abdomens were opened, and the ceca were ligated at both ends and immediately transferred to N2-saturated PBS (pH 6.8). The cecal bags were then opened, and the contents were collected and weighed. The contents were dispersed into the dissolution medium at predetermined times during the release test to concentration of 2% or 4%. Anaerobic conditions were maintained in the presence of rat cecal contents by continuous bubbling of N2.
Pharmacokinetic study in beagle dogs
The pharmacokinetics of indomethacin following administration of compression-coated tablets was evaluated in beagle dogs and compared with that of the core tablet counterparts in a two-period crossover study. There was a washout period of one week between the two treatments. Six male beagle dogs, weighing 8–10 kg, were kept in an environmentally controlled breeding room for one week prior to the experiments. The dogs were fasted overnight before the experiments but had access to water ad libitum. Guidelines issued by the Ethical Committee of Fudan University regarding experiments involving the use of animals were strictly followed.
A single dose, equivalent to 25 mg indomethacin, was placed at the pharyngeal site and spontaneously swallowed by the animals. Two milliliter blood samples were withdrawn from the dogs and placed into heparinized test tubes at predetermined intervals of 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 14, 24 and 30 h post-drug administration. Samples were centrifuged at 3000 × g for 10 min, and the plasma was collected and stored at −20 °C for future assay. Pharmacokinetic analysis was performed using a 3p97 computer program (issued by the State Food and Drug Administration of China for pharmacokinetic study). Maximum concentration (Cmax) and peak time (tmax) were observed as raw data. The lag time (tlag) was defined as the time elapsed between the administration of the preparation and the appearance of the drug in plasma. Area under the curve to the last measurable concentration (AUC0–t) was calculated using the linear trapezoidal rule.
Human scintigraphic evaluation
Preliminary scintigraphic evaluation was performed in two healthy male volunteers aged 27 and 23 years. Both abstained from medicine and alcohol for 24 h prior to dosing and throughout the study period. The study was approved by the Ethics Committee of Zhongshan Hospital Fudan University, Shanghai, China. Radioactivity assays were done for each volunteer immediately prior to dosing. Observed values were 0.96 mCi and 0.92 mCi, respectively. The purpose of the study was explained, and each volunteer provided written consent prior to the beginning of the study.
After fasting overnight, a 99mTc-labeled compression-coated tablet was given to each volunteer along with 250 mL water. Anterior and posterior scintigraphic images, lasting 60 s each, were recorded every hour. 99mTc-stained filter was used as an external marker and placed at the middle of the left clavicle, ensisternum and navel. Volunteers remained moderately active during the study. Images were acquired with volunteers lying down between the anterior and posterior cameras. Gamma radiation was detected using a Triple Head Gamma Camera (Philips IRIX, Andover, MA). All subjects received a standard lunch 4 h post-dose.
Results and discussion
Preparation of the core tablets and compression-coated tablets
The core tablets were produced to a crushing strength of over 10 kg in order to withstand the compression force during the second compression. The core tablets were designed to quickly disintegrate and dissolve in order to evaluate the retardant effects of P/Ca coating layer. As a consequence, the core tablets disintegrated within 1 min, and approximately 90% of the indomethacin was released from the core tablet within 15 min. The fast disintegration and dissolution prevented the core tablet from being the rate-limiting factor.
Physical properties such as weight, crushing strength, thickness and friability of P/Ca compression-coated tablets are listed in . However, since pectin lacks compressibility, it was difficult to obtain P/Ca compression-coated tablets that were extremely hard. Thus, the crushing strength of the final P/Ca compression-coated tablets was adjusted to 6–8 kg.
Effect of the P/Ca ratio and coating weight on release profiles
A variety of variables affect the release characteristics of compression-coated tablets. In this study, effects of the P/Ca ratio and the coating weight, on both lag time and release patterns, were analyzed. The effect of P/Ca ratio on indomethacin release is shown in . The release pattern, which is initially slow and later accelerates, was observed in all preparations. Pectin alone had some retardant effect on indomethacin release with a lag time of about 4 h and a release of approximately 90% at 12 h. The release lag time was longer as the amount of calcium chloride in the pectin matrix increased, with the longest release lag time reaching approximately 6 h. Interestingly, there was little difference in the release lag times of preparations with different P/Ca ratios. However, the overall release rate obviously decreased as the amount of calcium in the coating layer increased. The time it takes for 50% of the drug to be released (T50) for compression-coated tablets with P/Ca ratios of 200/0, 200/50, 200/100 and 200/200 was 7.7, 9.0, 9.4 and 10.6 h, respectively. Coating weight, at a fixed P/Ca ratio of 1/1, also had significant effects on release patterns (). The lag time was prolonged, and the release rate was decreased as coating weight gain increased. The lag time was prolonged from 3.0 h to 7.0 h, and the T50 increased from 5.2 h to 11.6 h when the P/Ca coating weight increased from 100/100 to 250/250 (mg/mg).
Figure 1. Release profile of indomethacin from compression-coated tablet prepared with constant pectin amount and varying amounts of calcium chloride. The hardness (crushing strength) of the tablets was around 7.0 kg. Release test was initially done in 900 mL of 0.1 M hydrochloride solution and thereafter was transferred at 2 h to 900 mL pH 6.8 PBS. Both the release media was thermostatically maintained at 37 ± 0.5 °C. Experiments were done in triplicate.
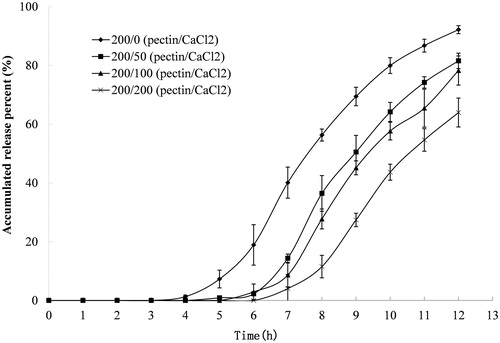
Figure 2. Release profile of indomethacin from compression-coated tablet prepared with different coating weights and a fixed pectin/calcium chloride ratio of 1/1. The hardness (crushing strength) of the tablets was around 7.0 kg. Release test was initially done in 900 mL of 0.1 M hydrochloride solution and was thereafter transferred at 2 h to 900 mL pH 6.8 PBS. Both the release media was thermostatically maintained at 37 ± 0.5 °C. Experiments were done in triplicate.
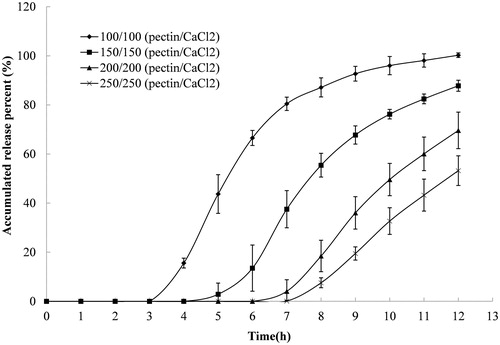
The results verified the assumption that colon-specific delivery could be achieved using a P/Ca compression coating. A lag time over 3 h can easily be attained using the compression-coated system, whereas a matrix system that is composed of the same amounts of pectin and calcium chloride cannot achieve this lag time due to the matrix erosion (Wei et al., Citation2006). The lag time appeared to be related to gel thickness rather than calcium chloride content, since similar lag times were observed for tablets with different ratios of calcium chloride in the coating layer (). Consequently, simply adjusting the coating weight can prolong the lag time.
Effect of rat cecal contents and enzymes on release profiles
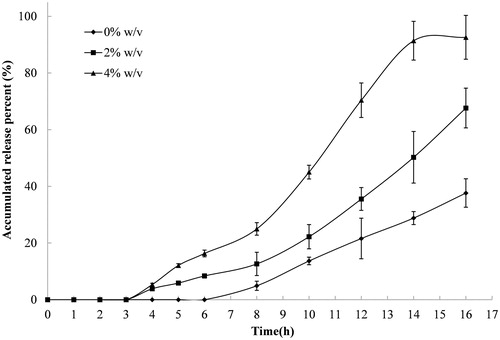
The release results of compression-coated tablets (P/Ca coating weight of 250/250 (mg/mg)) placed in PBS containing rat cecal contents at 0%, 2% and 4% are shown in . The release of indomethacin from these tablets increased as the concentration of rat cecal contents increased. The accumulated percent released over 16 h from tablets placed in solutions containing 2% and 4% rat cecal contents was 70% and 90%, respectively, while tablets placed in medium that had no rat cecal content only released 40%. Furthermore, the lag time decreased from 6 h to 3 h when the concentration of cecal content increased in the dissolution medium. The decreased lag time is likely due to pectin degradation by pectinase in the rat cecal contents. Pectinase-induced degradation of pectin accelerates the erosion of the coating layer, which shortens the release lag and promotes the release rate.
Figure 3. Release profiles of indomethacin from compression-coated tablet at pectin/calcium chloride coating weight of 250/250 (mg/mg) in pH 6.8 PBS containing different concentrations of rat cecal contents, thermostatically maintained at 37 ± 0.5 °C. The hardness (crushing strength) of the tablets was around 7.0 kg. Experiments were done in triplicate.
Effect of calcium content on enzyme-induced degradation
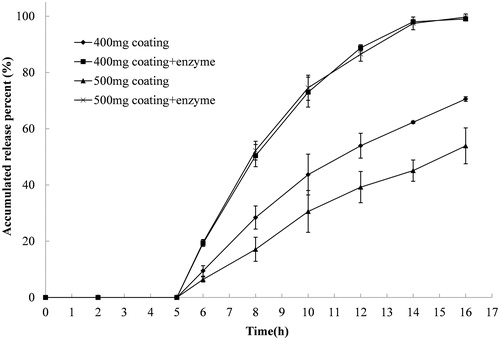
The release rate of compression-coated tablets with P/Ca coating weights of 200/200 (mg/mg) and 250/250 (mg/mg) in medium with and without pectinase were compared (). The release was initially done in 900 mL of 0.1 M hydrochloride solution and was transferred at 2 h to 900 mL PBS with and without Pectinex Ultra SP-L (2 mL) added to the dissolution medium 5 h after the initiation of the release. Drug release was reduced in medium without pectinase when the coating weight was increased from 400 mg to 500 mg. The gel layer became thicker as the amount of coating layer increased. In this case, a longer time was needed for erosion of the gel layer, which contributed to the reduced drug release. However, when pectinase was added to the release medium after 5 h, the release increased and was similar in the two tablets regardless of the coating weight (). This increased release was mainly ascribed to the enzymolysis of pectinase to the pectin in the coating layer. Although it was thought that tablets with higher coating weights would have a longer degradation time, the increased amount of calcium in the coating layer increased the activity of pectinase and accelerated the degradation of pectin. Therefore, similar release behavior was observed between the tablet with a coating weight of 400 mg and the tablet with a coating weight of 500 mg.
Figure 4. Release profiles of indomethacin from compression-coated tablet at pectin/calcium chloride coating weights of 200/200 and 250/250 (mg/mg). The hardness (crushing strength) of the tablets was around 7.0 kg. The release test was initially done in 900 mL of 0.1 Mhydrochloride solution and was thereafter transferred at 2 h to 900 mL PBS with and without Pectinex Ultra SP-L added to the dissolution media 5 h after the initiation of the release. Experiments were done in triplicate.
Pharmacokinetics in beagle dogs
The plasma indomethacin concentration versus time after oral administration of the core tablet and the compression-coated tablet is shown in . A rapid absorption phase was observed with the core tablet. Furthermore, indomethacin could not be detected in the plasma within 24 h after administration of the core tablet. However, since the crosslinking of pectin and calcium in the coating layer can minimize pre-colonic release of indomethacin, an obvious lag time along with slow absorption and elimination phases was observed with the compression-coated tablet. Plasma indomethacin could not be detected until 30 h. The main pharmacokinetic parameters for these two preparations are shown in . The lag time varied from 1.8 h to 5.8 h with an average value of 3.52 ± 1.73 h and was mainly attributed to individual differences among dogs. Nevertheless, the value was coincident with the average time it takes to reach the colon, which has been estimated by colon-specific delivery capsules to be 3.3 h (Niwa et al., Citation1995; Takaya et al., Citation1995). The quick disintegration of the core tablet in the stomach and subsequent dissolution resulted in a comparatively higher concentration of indomethacin in upper gastrointestinal tract. Since indomethacin can be absorbed throughout the gastrointestinal tract, a high Cmax of 5137.2 ± 614.0 ng/mL and a quick tmax of 1.4 ± 0.4 h were observed for the core tablet. However, for the compression-coated tablet, although the gel layer of pectin and calcium can be degraded by pectinase in the colon contents, the tablet remained integrated and the incorporated indomethacin was released slowly, and the release was sustained throughout the diffusion and erosion of the coating layer. The compression-coated tablet had a slow absorption phase with a tmax of 5.4 ± 1.8 h and a prolonged apparent t1/2 of 6.79 ± 2.14 h. Furthermore, since metabolism was concurrent with the absorption, the slow absorption of indomethacin from the compression-coated tablet led to a low Cmax of 1099.6 ± 309.3 ng/mL. The compression-coated tablet ultimately had an AUC0–t of 5615.6 ± 1302.7 ng h/mL, which was lower than the AUC0–t of 8091.1 ± 3889.1 ng h/mL associated with the core tablet.
Figure 5. Plasma indomethacin concentration versus time plot after a single oral dose of core tablet and compression-coated tablet at Pectin/calcium chloride coating weight of 250/250 (n = 6).
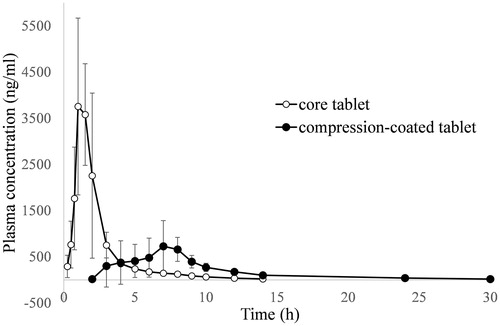
Table 2. Main pharmacokinetic parameters of indomethacin following a single oral dose of two formulations in Beagle dogs (n = 6).
Scintigraphic evaluation in humans
γ-scintigraphy based on 99m-Tc was employed in order to evaluate the gastrointestinal transportation of the compression-coated tablet. The scintigraphic images that were obtained from two volunteers after oral administration of compression-coated tablets are shown in . The formulation presents as a circular shape, before disintegration, under ECT observation, while scattered plots are observed if 99m-Tc was released. The approximate location of the preparation in the gastrointestinal tract can be tracked with the aid of external markers in body surface.
Figure 6. Scintigraphic images of 99m-Tc labeled compression-coated tablet at pectin/calcium chloride coating weight of 250/250 (mg/mg) in two volunteers (A and B) after oral administration.
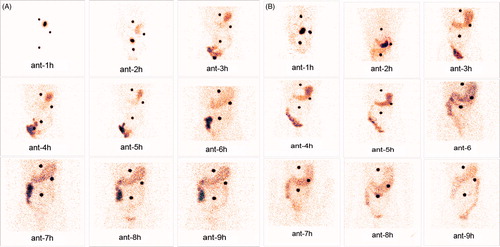
The scintigraphic images of volunteer A are shown in . The tablet (circular shape) reached the upper part of the stomach, the upper site of small intestine, the terminal ileum and the ileo-cecal junction at 1, 2, 3 and 4 h, respectively. Furthermore, the tablet remained intact during this period. However, the tablet loses integrity when it was transported to the ascending colon at 5 h since pectin in the coating layer might have been degraded by the secreted pectinase from the colonic microflora. As a consequence, the incorporated 99m-Tc started to be released. From then on, the image of the tablet became irregular, and the tablet continued to move upward in the ascending colon between 5 h and 9 h. A faster gastrointestinal transit rate was observed for volunteer B as compared with volunteer A (). The tablet reached the ileo-cecal junction at 3 h and started to disintegrate. Then the imaging band kept moving upward along the ascending colon and entered the transverse colon after 6 h. The radionuclide could be detected at descending and sigmoid colons at 9 h. Although individual variation in gastrointestinal transportation was observed, the compression-coated tablet started to disintegrate in the cecum in both volunteers. These results indicate that the P/Ca compression-coated tablet achieved excellent colon specificity.
Conclusions
Pectin and calcium chloride can be compressed onto a core tablet. The in situ crosslinking of pectin with the calcium cations in the coating layer can slow the release of incorporated indomethacin. The release lag time was mainly dependent on the coating weight. In addition, the release of indomethacin from coated tablets can be triggered by pectinase; this release was accelerated by the calcium chloride incorporated in the coating layer. Slow and sustained pharmacokinetic behavior was observed in compression-coated tablets compared to core tablets. Colon-specific delivery of the P/Ca compression-coated tablet was verified by scintigraphic evaluation in humans despite individual variations in gastrointestinal transportation.
Acknowledgements
We thank Professor Jianhua Zhu for his assistance in scintigraphic evaluation. Dr. Wu would like to thank the Shanghai Commission of Education (10SG05) and Ministry of Education (NCET-11-0114) for personnel fostering funding.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
This work was partly supported by Shanghai Municipal Commission of Science and Technology (11DZ1920200 and 11DZ1920906).
References
- Ashford M, Fell J, Attwood D, et al. (1993). An evaluation of pectin as a carrier for drug targeting to the colon. J Control Release 26:213–20
- Basit AW. (2005). Advances in colonic drug delivery. Drugs 65:1991–2007
- Basit AW, Short MD, McConnell EL. (2009). Microbiota-triggered colonic delivery: robustness of the polysaccharide approach in the fed state in man. J Drug Target 17:64–71
- Gulbake A, Jain SK. (2012). Chitosan: a potential polymer for colonspecific drug delivery system. Expert Opin Drug Deliv 9:713–29
- Hiorth M, Versland T, Heikkila J, et al. (2006). Immersion coating of pellets with calcium pectinate and chitosan. Int J Pharm 308:25–32
- Hodges LA, Connolly SM, Band J, et al. (2009). Scintigraphic evaluation of colon targeting pectin-HPMC tablets in healthy volunteers. Int J Pharm 370:144–50
- Jain A, Gupta Y, Jain SK. (2007). Potential of calcium pectinate beads for target specific drug release to colon. J Drug Target 15:285–94
- Ji C, Xu H, Wu W. (2007). In vitro evaluation and pharmacokinetics in dogs of guar gum and Eudragit FS30D-coated colon-targeted pellets of indomethacin. J Drug Target 15:123–31
- Liu L, Fishman ML, Hicks KB. (2007). Pectin in controlled drug delivery – a review. Cellulose 14:15–24
- Mamani Crispin PL, Ruiz Caro R, Dolores Veiga M. (2012). Pectin: pharmaceutical and therapeutic uses. Anales De La Real Academia Nacional De Farmacia 78:82–97
- Miller L, Macmilla JD. (1970). Mode of action of pectic enzymes. II. Further purification of exopolygalacturonate lyase and pectinesterase from Clostridium multifermentans. J Bacteriol 102:72–8
- Niwa K, Takaya T, Morimoto T, et al. (1995). Preparation and evaluation of a time-controlled release capsule made of ethylcellulose for colon delivery of drugs. J Drug Target 3:83–9
- Prabaharan M. (2011). Prospective of guar gum and its derivatives as controlled drug delivery systems. Int J Biol Macromol 49:117–24
- Prasad YV, Krishnaiah YS, Satyanarayana S. (1998). In vitro evaluation of guar gum as a carrier for colon-specific drug delivery. J Control Release 51:281–7
- Rubinstein A, Radai R, Ezra M, et al. (1993). In vitro evaluation of calcium pectinate: a potential colon-specific drug delivery carrier. Pharm Res 10:258–63
- Shah N, Shah T, Amin A. (2011). Polysaccharides: a targeting strategy for colonic drug delivery. Expert Opin Drug Deliv 8:779–96
- Sriamornsak P. (2011). Application of pectin in oral drug delivery. Expert Opin Drug Deliv 8:1009–23
- Takaya T, Ikeda C, Imagawa N, et al. (1995). Development of a colon delivery capsule and the pharmacological activity of recombinant human granulocyte colony-stimulating factor (rhG-CSF) in beagle dogs. J Pharm Pharmacol 47:474–8
- Ugurlu T, Turkoglu M, Gurer US, et al. (2007). Colonic delivery of compression coated nisin tablets using pectin/HPMC polymer mixture. Eur J Pharm Biopharm 67:202–10
- Wei X, Chen Z, Lu Y, et al. (2009). Physicochemical characterization of a pectin/calcium matrix containing a large fraction of calcium chloride: implications for sigmoidal release characteristics. J Appl Polym Sci 113:2418–28
- Wei X, Sun N, Wu B, et al. (2006). Sigmoidal release of indomethacin from pectin matrix tablets: effect of in situ crosslinking by calcium cations. Int J Pharm 318:132–8
- Wong TW, Colombo G, Sonvico F. (2011). Pectin matrix as oral drug delivery vehicle for colon cancer treatment. AAPS Pharmscitech 12:201–14
- Wu B, Chen Z, Wei X, et al. (2007a). Biphasic release of indomethacin from HPMC/pectin/calcium matrix tablet: I. Characterization and mechanistic study. Eur J Pharm Biopharm 67:707–14
- Wu B, Deng D, Lu Y, et al. (2008). Biphasic release of indomethacin from HPMC/pectin/calcium matrix tablet: II. Influencing variables, stability and pharmacokinetics in dogs. Eur J Pharm Biopharm 69:294–302
- Wu B, Shun N, Wei X, et al. (2007b). Characterization of 5-fluorouracil release from hydroxypropylmethylcellulose compression-coated tablets. Pharm Dev Technol 12:203–10