
Abstract
The present work was performed aiming to develop a new solid self-emulsifying system (SMEDDS) for poorly water-soluble drug Lornoxicam and evaluate the bioavailability in Wister rats by oral gavage. Liquid SMEDDS of Lornoxicam was formulated with Labrafil M 1944 CS as oil phase, Kolliphor HS 15 as a surfactant and Transcutol HP as a cosurfactant after screening various vehicles. The microemulsion system selected from the phase diagram and optimized by central composite design (CCD) response surface method was transformed into solid-SMEDDS (S-SMEDDS) by lyophilization using sucrose as cryoprotectant. The formulations were further characterized by the particle size, poly dispersity index (PDI), self-emulsifying time, zeta potential, transmission electron microscope (TEM), differential scanning calorimeter (DSC), in vitro drug release and in vivo pharmacokinetics. Results of DSC studies confirmed that the drug was incorporated in the S-SMEDDS. The in vitro drug release from Lornoxicam SMEDDS was found to be greatly higher in comparison with that from the commercial tablets. It was indicated that SMEDDS might be effective in reducing the effect of pH variability of Lornoxicam and improving the release performance of Lornoxicam. HPLC system was applied to study the concentration of Lornoxicam in the plasma of the Wister rats after oral administration of Lornoxicam SMEDDS and Lornoxicam commercial tablets. The pharmacokinetics parameters of the rats were Cmax 1065.91 ± 224.90 and 1855.22 ± 748.25 ngmL−1, Tmax were 2.5 ± 0.4 h and 1.8 ± 0.5 h, and AUC0∼t were 5316.35 ± 323.62 and 7758.07 ± 241.57 ngmL−1 h, respectively. Calculated by AUC0∼∞, the relative bioavailability of Lornoxicam S-SMEDDS was 151.69 ± 15.32%. It suggested that this S-SMEDDS could be used as a successful oral solid dosage form to improve the solubility and bioavailability of poorly water-soluble drug Lornoxicam as well.
Introduction
Oral administration has been the major route of drug delivery for the treatment of many diseases. However, approximately 40% of new drug candidates exhibit low solubility in water, leading to poor bioavailability, high intra- and inter-subject variability, and lack of dose proportionality (Nazzal et al., Citation2006; Ankush et al., Citation2012). Therefore, for such compounds, the bioavailability is controlled by dissolution. With the increasing number of newly developed lipophilic drug compounds, various techniques have been proposed, such as solid dispersions (Christian et al., Citation2000), co-solvents, emulsions (Jang et al., Citation2006), liposomes (Chen et al., Citation2009), and nanoparticles based on lipidic or polymer carriers. However, of various strategies reported, self-emulsifying drug delivery systems (SEDDS) are discovered to be the prominent approach to improve the solubility and the approach is prone to realize industrialization. Over recent years, much attention has been focused on lipid microemulsion formulations, with particular emphasis on liquid self-nanoemulsifying (SNEDDS), self-microemulsifying (SMEDDS), and self-emulsifying drug delivery systems (SEDDS) to make the solubility, dissolution rate, and oral absorption of water insoluble drugs ameliorated (Attama et al., Citation2003; Balakrishnan et al., Citation2009; Cui et al., Citation2009).
Self-emulsifying drug delivery system (SEDDS) is isotropic and thermodynamically stable solutions, consisting of oils, solid, or liquid surfactants, co-surfactants, and drug which could spontaneously form oil-in-water emulsions or microemulsions when mixed with water under gentle stir. Self-emulsifying formulations spread readily in gastrointestinal (GI) fluids, and the digestive motility of the stomach and the intestine provides the agitation necessary for self-emulsifying. SEDDS typically produce emulsions with a droplet size less than 5 μm, while SMEDDS form transparent microemulsion with a globule size less than 100 nm.
The SMEDDS can improve the solubility and dissolution rate of water-insoluble drugs by providing a large interfacial area for partitioning of the drugs between the oil and aqueous GI fluids (Woo et al., Citation2008; Sachan et al., Citation2010; Patel et al., Citation2007). Generally, the traditional liquid SMEDDS (L-SMEDDS) are encapsulated either in hard or in soft gelatin capsules. However, lipid formulation may interact with the capsule, resulting in either brittleness or softness of the shell. To address these limitations, more approaches were committed, such as adsorbing, lyophilization, and spray drying, which can combine the advantages of lipid-based drug delivery systems with those of solid dosage forms.
As a non-steroid anti-inflammatory drug, Lornoxicam (LX) exhibits a pH-dependent solubility and its water (pH 7.0) solubility is only 18 μg mL−1. It is a potent COX-1 and COX-2 inhibitor, providing short-term relief from mild-to-moderate pain and inflammation, notably in osteoarthritis and rheumatoid arthritis. It demonstrates a better anti-inflammatory action and abirritation than any other oxicams as well. At the same time, it plays an important part on both central and peripheral nervous. However, it is associated with some shortcomings, such as low solubility, irritation on GI tract and low bioavailability for oral administration.
In this paper, self-microemulsifying drug delivery system (SMEDDS) of LX which was not reported on this field so far was developed to increase its solubility and bioavailability in addition to reduce the side effect of Lornoxicam on GI tract. Oils and surfactants were screened out according to their solubilizing capacity. Then the pseudo ternary-phase diagram was used to determine the formulations. The optimized formulation was obtained by the central composite design (CCD) response surface method. SMEDDS of LX then was further evaluated by particle size, poly dispersity index, self-emulsifying time, zeta potential, transmission electron microscope (TEM), differential scanning calorimeter (DSC), in vitro drug release, and in vivo pharmacokinetics.
Materials and methods
Materials
Lornoxicam was a gift from Zhejiang Zhenyuan Co. (Zhejiang, China). Larbafil M 1944CS, Labrafac Lipophile WL 1349, Lauroglycol FCC, Labrasol, Transcutol HP were obtained from Gattefosse (Saint-Priest Cedex, France), Cremophor EL and Kolliphor HS 15 were purchased from BASF (Ludwigshafen, Germany), Isopropyl Myristate_(IPM, Shanghai, China), Tween 80, PEG 400,1, 2-propylene glycol, and glycerol were obtained from Bodi Chemical Co., Ltd (Tianjin, China). Other chemicals were of HPLC or analytical grade.
HPLC analysis of LX
The HPLC method was used for the determination of LX in the solubility test, dissolution test, and pharmacokinetic study in vivo. The HPLC system consisted of an LC-20AT pump, an SPD-M20A detector, and a Diamonsil® C18 column (200 mm × 4.6 mm, 5 μm, Dikma Technologies, Lake Forest, CA). The mobile phase was methanol and phosphate buffer (pH 6.0) at the ratio of 60:40. The wavelength of the UV detector, flow rate, and injection volume were set at 375 nm, 1.0 mLmin−1 and 20 μL, respectively.
Solubility determination
An excess amount of LX was added to a centrifuge tube containing 5 mL of virous oils, surfactants, and cosurfactants. The samples were vortexed for 5 min to facilitate the dissolution and then were placed in a rotary shaker for 72 h at ambient temperature under constant agitation. After equilibrium, the samples were centrifuged at 4000 rpm for 15 min and the supernatant was filtered through 0.22 μm membrane filter. The resultant solution was analyzed by HPLC.
Preparation of LX liquid SMEDDS (L-SMEDDS) formulations
Based on the previous researches, it was established that the liquid SMEDDS consisted of oils (Labrafil M 1944 CS and Labrafac Lipophile WL1349), surfactants (Tween 80, Cremophor EL, and Kolliphor HS 15), Transcutol HP, and Lornoxicam. Briefly, the calculated amount of Lornoxicam was added into the mixture of oils, surfactants, and cosurfactant. Then, the components were mixed by gentle stir at 37 °C until Lornoxicam was completely dissolved. The mixture was stored at room temperature until use.
Preparation of LX solid SMEDDS (S-SMEDDS) formulations
The LX S-SMEDDS was prepared by a lyophilization method. In brief, 1 g of the above obtained LX L-SMEDDS was dispersed into the solution of sucrose at a concentration of 3%, 10%, and 15% (w/v). The resultant microemulsion was lyophilized at −30 °C and 900 mTorr for 8 h, then at 20 °C, and 350 mTorr for 8 h, finally at 25 °C and 1000 mTorr for 8 h to obtain LX S-SMEDDS.
Construction of the pseudo ternary-phase diagram
Ternary-phase diagrams of surfactants, Tween 80, Cremophor EL, and Kolliphor HS 15, co-surfactant (Transcutol HP) and oils (Labrafac Lipophile WL 1349, Labrafil M 1944 CS) were plotted, each of them represents an apex of the triangle (Shweta et al., Citation2011). A series of LX L-SMEDDS were prepared in the formulation with varying concentrations of Labrafil M 1944 CS and Labrafac Lipophile WL 1349 (10–30% w/w), and the Km values (the weight ratio of a surfactant and a cosurfactant) were set as 2, 3, and 4. About 0.2 mL of LX L-SMEDDS was introduced into a glass beaker containing 100 mL of distilled water at 37 °C. The tendency to emulsify spontaneously and also the progress of emulsifying were observed. Dispersions with 100 nm of globule size or below were considered acceptable. Phase diagrams were constructed to identify the good self-emulsifying region using Origin 8.0 software (MA, USA).
Experimental design
The formulation factors including the amount of Labrafil M 1944 CS (X1) and Km (the weight ratio of Kolliphor HS 15 and Transcutol HP, X2) could significantly affect the mean particle size, self-emulsifying time (Gershanik et al., Citation1998), and poly dispersity index (PDI) of SMEDDS. Therefore, in our work, response surface methodology based on CCD was utilized to evaluate the formulation factors and optimize the formulations. The experimental range of each variable was selected based on the results of preliminary experiments. shows the independent factors and their design levels in the present study. The experiments were designed using Design-Expert® software (MN, USA).
Table 1. Independent variables and the correspondent values in coded and physical form.
Characterization of LX L-SMEDDS and S-SMEDDS
LX L-SMEDDS (0.2 mL) and S-SMEDDS (200 mg) were dispersed with 100 mL dispersion media (distilled water, simulated gastric fluid, and simulated intestinal fluid). The samples were vortexed for 5 min until emulsification completed, and then incubated for 30 min at 25 °C. The physicochemical characteristics of the reconstituted LX SMEDDS were further characterized in the following experiment.
Particle size, PDI, zeta-potential, and self-emulsifying time
The particle size and zeta potential of the reconstituted LX SMEDDS were accessed by Malvern Zetasizer (Nano ZS 90, Worcestershire, UK) at a wavelength of 635.0 nm and a scattering angle of 90° at 25 °C. The measurements were repeated three times and the average values were used.
The self-emulsifying time of LX S-SMEDDS was determined according to United State Pharmacopeia (USP) XXIII, dissolution apparatus II. In brief, 0.5 mL of each formulation was added dropwise to 500 mL of distilled water at 37 °C. Gentle agitation was provided by a standard stainless steel dissolution paddle rotating at the speed of 50 rpm. The self-emulsifying time was assessed visually as reported by Bachynsky et al. (Citation1997).
Physical stability after dilution
To observe if there would be precipitation in reconstituted LX SMEDDS with the increase of dilution extent. The reconstituted LX SMEDDS were diluted 50, 200, and 1000 times with distilled water, simulated gastric fluid, and simulated intestinal fluid, respectively. At the same time, the changes in mean particle size, self-emulsifying time, and PDI were evaluated.
Effect of the dispersed media on the reconstituted LX SMEDDS
The reconstituted LX SMEDDS were diluted with distilled water, simulated gastric fluid, and simulated intestinal fluid. The mean particle size, self-emulsifying time, zeta potential, and PDI of the resultant microemulsion was measured.
Transmission electron microscopy (TEM)
The TEM (JEM-1200EX, JEOL, Tokyo, Japan) was used in order to observe the morphology of LX L-SMEDDS with a negative staining method. Briefly, a drop of diluted L-SMEDDS was spread on a 200-mesh copper grid coated with carbon film and negatively stained with 1% phosphotungstic acid for 30 s. The grid was dried at room temperature and then observed by TEM whose voltage and current were 60 kV and 30 mA, respectively.
Differential scanning calorimetry
Differential scanning calorimetry (DSC) analysis was performed using a DSC 1 calorimeter (Mettler Toledo, Schwerzenbach, Switzerland). Analysis was performed under a nitrogen purge (20 mL min−1). The samples (about 3 mg) were weighed accurately, placed in aluminum pans, and then sealed with a pinhole-pierced cover. Heating curves were recorded at a scan rate of 10 min−1 from 25 °C to 250 °C, and an empty pan was used as a reference.
In vitro dissolution study
The dissolution test was carried out using the reverse dialysis method. Seven identical dialysis bags each filled with 1 mL of release medium and were placed in a dissolution tester containing 500 mL of release medium which was agitated at 50 rpm and 37 °C. LX S-SMEDDS formulations (equivalent to 8 mg of LX) and the commercial Lornoxicam tablets were added to the exterior solution. At predetermined time intervals, 1 mL samples were withdrawn from the interior of alternate dialysis bags to estimate the percentage of drug released. Meanwhile, the appearance of the reconstituted LX SMEDDS in the dissolution tester was observed.
In vivo pharmacokinetic study
All animal studies were conducted in accordance with the principles of Laboratory Animal Care and were approved by the Department of Laboratory Animal Research at Shenyang Pharmaceutical University. Male Sprague–Dawley rats weighing 200–250 g were supplied by the Animal Centre of the Shenyang Pharmaceutical University. All the SD rats were clinically healthy during the period of the experiment. SD Rats were fasted for 24 h prior to the beginning of experiments. Twelve SD rats were divided into two groups. Two types of LX formulations at a dose of 1.6 mgkg−1 were orally administrated to two groups of rats, i.e., optimized formulation LX S-SMEDDS and LX tablets were prepared by dispersing in 0.5% CMC–Na solution and sonicated for 10 min. At designated time point, blood samples were collected from the orbit. Blood samples were contained in the heparin tube, followed by centrifugation at 5000 rpm for 5 min. The obtained plasma was stored at −40 °C until analyzed.
To 0.2 mL of plasma, 0.5 mL of 1% H3PO4 and 50 μL of internal standard (100 ngmL−1 Meloxicam solution) were added and shook vigorously for 5 min and then the drug was extracted with normal hexane–dichloromethane–isopropanol (20:10:1) for 5 min and centrifuged. After centrifugation at 4000 rpm for 15 min, the supernatant was transferred to a micro tube and dried under reduced pressure. The residue was redissolved with 100 μL of the mobile phase, vortexed for 1 min, and centrifuged at 10 000 rpm for 5 min. Thereafter, 20 μL of the supernatant layer was analyzed by HPLC.
Statistical analysis
All results were expressed as mean ± SD. The data from different formulations were compared for statistical significance by a one-way analysis of variance (ANOVA).
Results and discussions
Solubility study
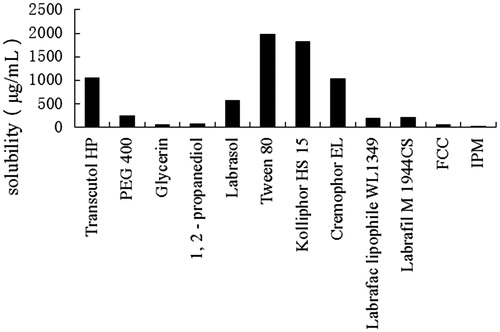
The self-emulsifying formulations composed of oils, surfactants, cosurfactants, and drug should be a clear and monophasic liquid at ambient temperature when introduced to aqueous phase. The solubility of LX in various vehicles is presented in . Among the tested vehicles, Transcutol HP showed the highest drug solubility for LX among the four cosurfactants. Therefore, Transcutol HP was chosen as the cosurfactant. Tween 80, Kolliphor HS 15, and Cremophor EL displayed better drug solubility among the surfactants, which were 1979.88 ± 111.70 μg mL−1, 1820.45 ± 102.27 μg mL−1, and 1025.45 ± 92.27 μg mL−1, respectively. Taking into account more hydrophobic nature and better drug solubility, Labrafil M 1944 CS and Labrafac Lipophile WL 1349 were chosen as oil phases. Cremophor EL, Tween 80, and Kolliphor HS 15 were preliminarily chosen as surfactants.
Figure 1. Solubility studies of Lornoxicam in various vehicles (mean ± SD, n = 3).
Preparation of LX L-SMEDDS and LX S-SMEDDS
LX SMEDDS were prepared successfully in the present study. To formulate and prepare SMEDDS, there were some basic guidelines needed to conform safety, compatibility, better drug solubility, higher self-emulsification efficiency, smaller particle size, etc. In the formulations of LX SMEDDS, Labrafil M 1944 CS was selected as the oil phase, because of its better solubility, lower toxicity, and in addition it could protect the drug from enzyme degradation in the gastrointestinal tract (Prajapati et al., Citation2007; Wei et al., Citation2012) and increases the amount of lipophilic drug transport through the intestinal lymphatic system which is an important absorption pathway of poorly water-soluble drugs (Patel et al., Citation2010). Transcutol HP as the cosurfactant exhibited the excellent solubility for LX and presented an optimal SMEDDS formulation leading to improved drug loading and form spontaneous fine emulsion. LX S-SMEDDS was prepared by lyophilization method with the sucrose at the concentration of 10% (w/v) as the protectant. This solid form of LX can provide the physical stability, accurate dosage, and good patient compliance (Gursoy et al., Citation2004).
Construction of pseudo ternary-phase diagrams
A series of LX L-SMEDDS formulations were prepared and their self-emulsifying properties were observed visually. Pseudo-ternary-phase diagrams were constructed in the absence of LX to identify the self-emulsifying regions and to optimize the percentage of oil and Km in the SMEDDS formulations. The ternary-phase diagrams of the systems containing Labrafac Lipophile WL1349 and Labrafil M 1944CS as oils, Tween 80, Kolliphor HS15, or Cremophor EL as surfactants and Transcutol HP as a cosurfactant are shown in .
Figure 2. Phase diagram prepared with the following components: oil, Labrafac Lipophile WL 1349; surfactant, Tween 80; and cosurfactant, Transcutol HP. S–Cos ratio is 2:1, 3:1, 4:1, and 5:1. The gray regions represent microemulsion phase.
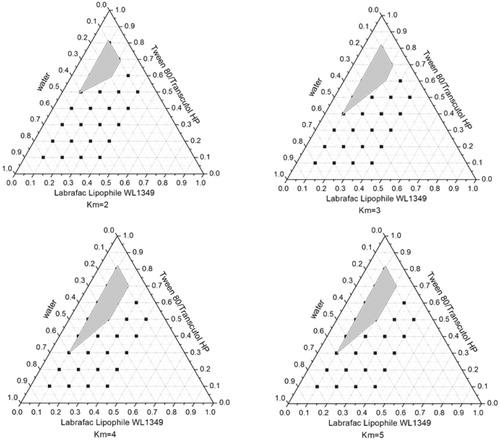
Figure 3. Phase diagram prepared with the following components: oil, Labrafil M 1944CS; surfactant, Tween 80; and cosurfactant, Transcutol HP. S–Cos ratio is 2:1, 3:1, 4:1, and 5:1. The gray regions represent microemulsion phase.
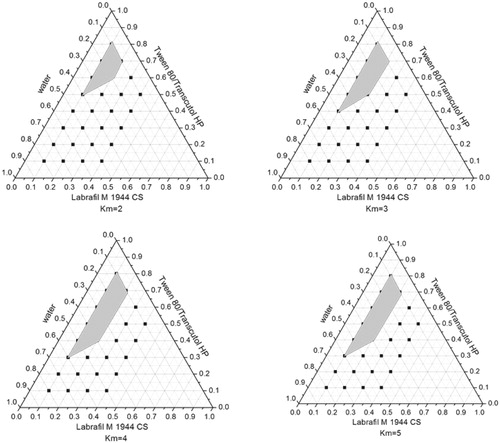
Figure 4. Phase diagram prepared with the following components: oil, Labrafil M 1944CS; surfactant, Cremophor EL; and cosurfactant, Transcutol HP. S–Cos ratio is 2:1, 3:1, 4:1, and 5:1. The gray regions represent microemulsion phase.
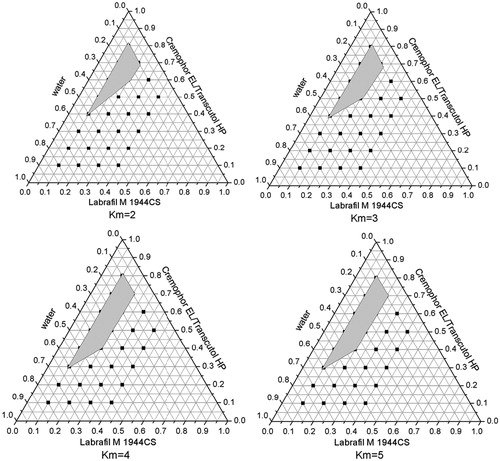
Figure 5. Phase diagram prepared with the following components: oil, Labrafil M 1944CS; surfactant, Kolliphor HS 15; and cosurfactant, Transcutol HP. S–Cos ratio is 2:1, 3:1, 4:1, and 5:1. The gray regions represent microemulsion phase.
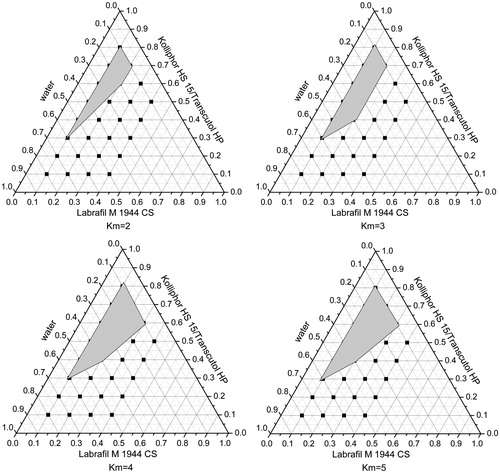
From the Pseudo ternary-phase diagrams in and , when the surfactant was fixed as Tween 80, the emulsifying area of Labrafil M 1944 CS was larger than Labrafac Lipophile WL 1349 as the oil phase indicating that the former was emulsified easier than the latter. Taking into account the better solubility of Labrafil M 1944 CS, it was chosen as the oil of LX SMEDDS formulation.
In , the oil and cosurfactant were fixed and Tween 80, Cremophor EL, and Kolliphor HS 15 were used as surfactants, respectively. The largest microemulsion region was gained when Labrafil M 1944CS was used as oil and Kolliphor HS 15 as a surfactant, which suggested that Kolliphor HS 15 had the best emulsifying ability among the three surfactants. Moreover, it was reported that Kolliphor HS 15 could enhance the intestinal absorption of drugs and exhibited higher ability of emulsifying (Seo et al., Citation2012). So it was helpful to use Kolliphor HS 15 as the surfactant in LX SMEDDS formulation.
Formulation optimization
A total of 13 experiments were carried out to study the formulation factors that affect the particle size, self-emulsifying time, and PDI of LX SMEDDS. Response data for all experimental runs of CCD are presented in .
Table 2. Experimental design table (columns 1–3) with experimentally determined values of different dependent variables (columns 4–6).
The responses were fitted into second-order polynomial models. The obtained models were validated using an ANOVA. The coefficient of determination (R2) closest to unity indicated a good model. p Values lower than 0.05 indicated that the regression equations were statistically significant.
According to the results, three second-order polynomial representing particle size, self-emulsifying time, and PDI were generated, which are shown below in terms of coded factors.
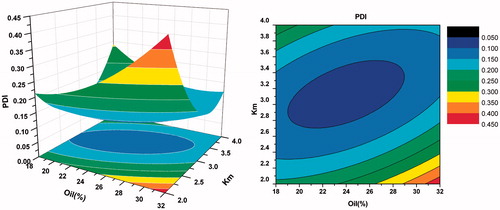
As shown in , when Km was more than three, there was no significant difference in particle size with the weight ratio of oil varying from 20% to 40%. However, when Km was less than three, with the increase of oil and decrease of surfactants, the particle size increased gradually. From the response surface of self-emulsifying time (in ), we can observe that the bigger the weight ration of oil was, the longer the self-emulsifying time was. This may be because larger amount of oil made the surfactants’ ability to reduce the surface tension weaker. As seen in , when Km was more than three and the oil was less, the value of PDI was smaller. Overall, considering the side effect of large quantity of surfactant and cosurfactant, the optimal formulation was as follows: Labrafil M 1944 CS was 25% (w/w), Kolliphor HS 15 was 56.25% (w/w), and Transcutol HP was 18.75% (w/w).
Figure 6. Response surface plot (3D) and contour plot showing the effect of the amount of oil(X1) and Km(X2) added on the response Y1, i.e., particle size.
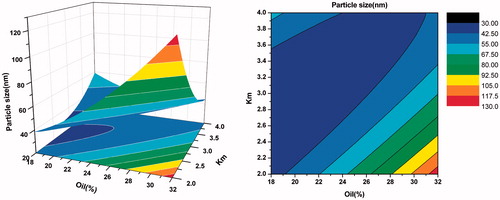
Figure 7. Response surface plot (3D) and contour plot showing the effect of the amount of oil(X1) and Km(X2) added on the response Y2, i.e., self-emulsifying time.
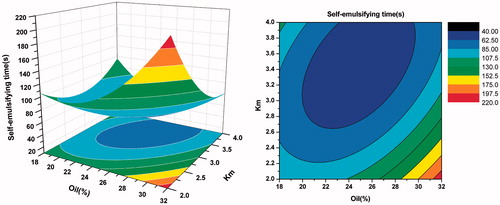
Figure 8. Response surface plot (3D) and contour plot showing the effect of the amount of oil(X1) and Km(X2) added on the response Y3, i.e., polydispersity index.
Characterization of LX L-SMEDDS and S-SMEDDS
Particle size, PDI, zeta potential, and self-emulsifying time measurement
In SMEDDS, the efficiency of self-emulsification could be estimated by determining the rate of emulsification and particle size distribution. The latter is a crucial factor in self-emulsifying performance because it determines the rate and extent of drug release, as well as absorption. The particle size of both systems was 38.42 ± 0.33 nm and 70.14 ± 1.06 nm, respectively. The particle size of the reconstituted LX S-SMEDDS was larger than that of LX L-SMEDDS, which may be due to the aggregation of particle in the process of lyophilization. The PDI of these two systems were 0.107 ± 0.013 and 0.193 ± 0.010, indicating that the particles distributed uniformly in the system (Jun et al., Citation2012). The zeta potential of the optimized LX L-SMEDDS and S-SEDDS was −18.21 ± 0.11 mV and −17.17 ± 1.13 mV. The negative charge may be possibly due to the presence of free fatty acids (Craing et al., Citation1993; Satomi et al., Citation2012). This indicated that the optimized SMEDDS generated a more stable system. It is reported that the self- emulsifying system should spread to the medium with slight stir within 2 min (Jun et al, Citation2012). The self-emulsifying time of LX L-SMEDDS and S-SMEDDS wa 55 ± 1 s and 68 ± 2 s, respectively. The above results suggested that the emulsifying efficiency of LX SMEDDS was high and it may improve the dissolution rate and extent, as well as absorption of LX.
Physical stability after dilution
LX S-SMEDDS formulations were exposed to different folds of dilution in different media in an attempt to mimic the in vivo conditions where the formulation would encounter gradual dilution. Each SMEDDS formulation was subjected to 50-, 200-, and 1000-fold dilution in water, simulated gastric fluid, and simulated intestinal fluid. As shown in , with the increase of dilution volume, the particle size, self-emulsifying time, and PDI of microemulsion did not change obviously and the formulation dispersion showed no signs of precipitation, cloudiness, or separation.
Table 3. Physicochemical characteristics of LX S-SMEDDS diluted with different media (mean ± SD, n = 3).
Effect of the dispersed media on the reconstituted LX SMEDDS
Different media (distilled water, simulated gastric fluid, and simulated intestinal fluid) were applied to simulate the in vivo environment that SMEDDS might meet after entering into body. As shown in , the mean particle size and zeta potential exhibited no obvious changes in the distilled water and simulated intestinal. However, in the simulated gastric fluid, the zeta potential of LX S-SMEDDS was higher than the other medium. It may be due to the existence of ion in the solution, which made the zeta potential change (Wei et al., Citation2005). The self-emulsifying time of the system was significantly longer in the simulated gastric fluid compared with that in the intestinal. In conclusion, the longest self-emulsifying time and the largest particle size indicated that the emulsified efficiency of LX SMEDDS was the poorest in the simulated gastric fluid. It may be because the hydrogen ions in hydrochloric acid and water combined with the ethyoxyl of surfactant competitively.
Table 4. Effect of the dispersion media on physicochemical characteristics of the reconstituted LX SMEDDS (mean ± SD, n = 3).
TEM
The morphology of the reconstituted LX L-SMEDDS is shown in . It can be seen that oil droplets were homogenous, spherical with good integrity, and did not show any precipitation of LX.
Figure 9. TEM image of L-SMEDDS of LX.

Differential scanning calorimetry (DSC)
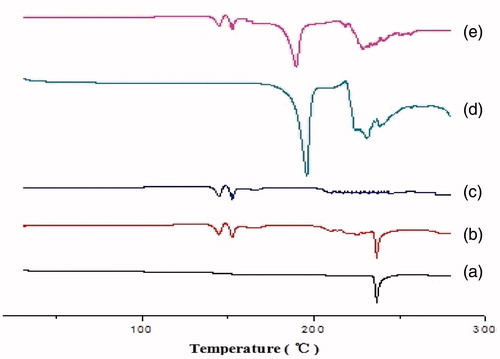
The physical state of LX in the S-SMEDDS was investigated as it would have an important influence on the in vitro and in vivo release characteristics. The DSC thermograms are shown in . The pure LX showed major endothermic peak at 236.6 °C and the physical mixture also displayed the same endothermic peak, which indicated that the crystal form did not change when LX was mixed with other excipients. In the DSC profile of S-SMEDDS, the endothermic peaks of LX disappeared completely, which suggested that LX was in the molecular or amorphous state in the S-SMEDDS. Because of more protectant in the LX S-SMEDDS formulation, there was no significant difference in the profile of and .
Figure 10. DSC thermograms (a) pure Lornoxicam powder, (b) physical mixture, (c) LX S-SMEDDS without cryoprotectant, (d) sucrose, and (e) LX-SMEDDS lyophilized powder.
Dissolution study
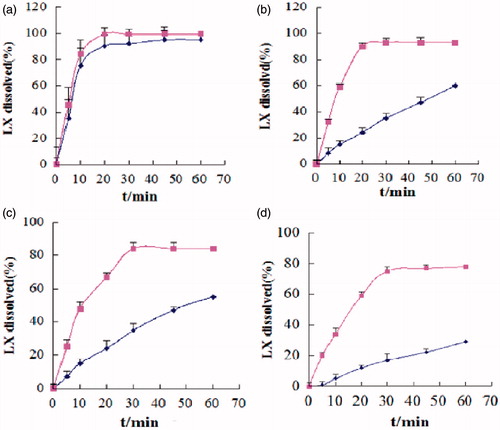
Dissolution test was carried out successfully by a reverse dialysis method. When SMEDDS encounter aqueous medium, different forms of solubilized drug are formed, including free molecular state, drug in micro-emulsion, and drug in micellar solution. Under these circumstances, it is necessary to separate free drug molecules from those entrapped in the micro-emulsion droplets or micelles to assess the real release pattern. Thereby, conventional release testing is not adequate to this system. In this field, the reverse dialysis method was recently adopted. In vitro release patterns of commercial LX tablets and S-SMEDDS after reconstitution with different buffer solutions (pH 1.2–4.0, water, and pH 6.8) are depicted in . It can be found that release rate of commercial LX tablets varied with the change of pH. More than 95% of drug released within 45 min and the complete release was obtained within 60 min in pH 6.8 buffer, while only 30% of drug was released within 60 min in pH 1.2. However, LX S-SMEDDS released 80% and 98% of drug in pH 1.2 and pH 6.8, respectively, indicating that SMEDDS might be effective in reducing the effect of pH variability of LX and improving the release performance of LX. The difference in dissolution rate between LX tablets and LX S-SMEDDS might be attributed to the difference in solubility of LX at different pH and the increased surface area. Thus, it was capable to ensure that the formulated SMEDDS improved the solubilization of LX and the dissolution rate successfully. At the same time, the appearance of the system was observed. The reconstituted LX S-SMEDDS was an uniform, transparent dispersion with blue opalescence, and there was no occurrence of precipitation.
Figure 11. Dissolution profiles of LX powder ![]()
In vivo pharmacokinetic study
The pharmacokinetic parameters of LX () were determined after oral administration of LX tablets or S-SMEDDS to Wister rats. shows the change in mean plasma concentration of LX after oral administration of preparations in rats. The total plasma concentrations of drug in S-SMEDDS were higher than those in LX powder. In particular, the initial plasma concentrations of LX in S-SMEDDS were significantly higher than those in LX tablets (p < 0.05). The S-SMEDDS gave significantly higher AUC and greater Cmax of LX than did commercial LX tablets (p < 0.05). However, the Tmax of LX S-SMEDDS was in advance apparently. Calculated by AUC0−∞, the relative bioavailability of LX S-SMEDDS was 151.69 ± 15.32%. These results indicated that LX S-SMEDDS significantly improved the bioavailability. In the present research, the superior performance of S-SMEDDS may be attributed to the following factors: (1) larger surface area provided by the fine emulsion droplets, (2) improved diffusion of the fine emulsion droplets, and (3) increased mucosal permeability due to surfactants. Our results suggested that the S--SMEDDS was as a solid form of the liquid SMEDDS kept promising for industrialization (Elnaggar et al., Citation2009; Shweta et al., Citation2011).
Figure 12. Plasma profiles of LX commercial tablets and S-SMEDDS (n = 6).
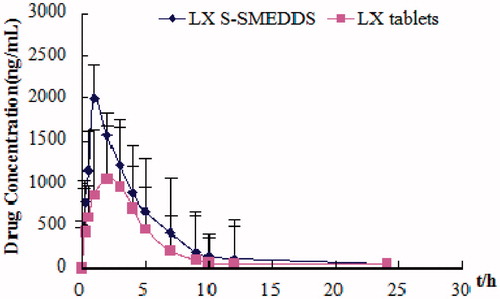
Table 5. Pharmacokinetic parameters of LX S-SMEDDS and LX tables in rats.
Conclusions
In the present study, a new solid formulation was successfully designed, employing a lyophilized self-emulsifying powder system. In vitro dissolution testing demonstrated a marked improvement in dissolution behavior of LX S-SMEDDS through self-emulsification in aqueous media. The DSC analysis suggested that LX in the S-SMEDDS may be in the amorphous state. Also the LX S-SMEDDS showed a significant increase in the bioavailability of LX compared with the LX commercial tablets. Above all, this solid self-emulsifying system may provide a worthy oral solid dosage form for poorly water-soluble drug LX.
Declaration of interest
This work was supported by the Joint Specialized Research Fund for the Doctoral Program of Higher Education, MOE & Department of Education of Liaoning Province (20102134120002), State Key Laboratory of Long-acting and Targeting Drug Delivery System, and the special construction projects fund which belongs to “Taishan Scholar-Pharmacy Specially Recruited Experts”.
Reference
- Ankush C, Avtar CR, Geeta A, et al. (2012). Development and characterization of anatorvastatin solid dispersion formulation using skimmed milk for improved oral bioavailability. Acta Pharmaceutica Sinica B 2:421–8
- Attama AA, Nzekwe IT, Nnamani PO, et al. (2003). The use of solid self-emulsifying systems in the delivery of diclofenac. Int J Pharm 262:23–8
- Bachynsky MO, Shah NH, Patel CI, et al. (1997). Factors affecting the efficiency of self-emulsifying oral delivery system. Drug Dev Ind Pharm 23:809–16
- Balakrishnan P, Lee BJ, Oh DH, et al. (2009). Enhanced oral bioavailability of coenzyme Q10 by self-emulsifying drug delivery systems. Int J Pharm 374:66–72
- Christian L, Jennifer D. (2000). Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm 50:47–60
- Chen YP, Lu Y, Chen JM, et al. (2009). Enhanced bioavailability of the poorly water-soluble drug fenofibrate by using liposomes containing a bile salt. Int J Pharm 376:153–60
- Cui SX, Nie SF, Li L, et al. (2009). Preparation and evaluation of self-microemulsifying drug delivery system containing vinpocetine. Drug Dev Ind Pharm 35:603–11
- Craing DQM, Lievens HSR, Pitt KG, et al. (1993). An investigation into the physico-chemical properties of self- emulsifying measurements and particle size analysis. Int J Pharm 96:1–12
- Elnaggar YSR, Massik MA, Abdalla OY. (2009). Self-nanoemulsifying drug delivery systems of tamoxifen citrate: design and optimization. Int J Pharm 380:133–41
- Gershanik T, Benzen OS, Beni ta S. (1998). Interaction of a self-emulsifying lipid drug delivery system with the reverted rat intestinal mucosa as a function of droplet size and surface charge. Pharm Res 15:863–9
- Gursoy RN, Benita S. (2004). Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother 58:173–82
- Jang DJ, Jeong EJ, Lee HM, et al. (2006). Improvement of bioavailability and photostability of amlodipine using redispersible dry emulsion. Eur J Pharm Sci 28:405–11
- Jun HK, Dong HO, Chul SY, et al. (2012). Effects of solid carriers on the crystalline properties, dissolution and bioavailability of flurbiprofen in solid self-nanoemulsifying drug delivery system (solid SNEDDS). Eur J Pharm Biopharm 80:289–97
- Nazzal S, Khan MA. (2006). Controlled release of a self-emulsifying formulation from a tablet dosage form: stability assessment and optimization of some processing parameters. Int J Pharm 315:110–21
- Patel D, Sawant KK. (2007). Bioavailability enhancement of acyclovir by self-microemulsifying drug delivery systems (SMEDDS). Drug Dev Ind Pharm 33:1318–26
- Prajapati BG, Patel M. (2007). Conventional and alternative methods to improve oral bioavailability of lipophilic drugs. Asian J Pharm 1:1–8
- Patel MJ, Patel SS, Patel NM, et al. (2010). A self-microemulsifying drug delivery system (SMEDDS). Int J Pharm Res 4:29–35
- Sachan R, Khatri K, Kasture SB. (2010). Self-emulsifying drug delivery system a novel approach for enhancement of bioavailability. Int J Pharm Tech Res 2:1738–45
- Shweta G, Sandip C, Krutika KS. (2011). Self-nanoemulsifying drug delivery system for adefovir dipivoxil: design, characterization, in vitro and ex vivo evaluation. Colloids Surf A: Physicochem Eng Aspects 392:145–55
- Satomi O, Atushi U, Kazuki K. (2012). Novel solid self-emulsifying drug delivery system of coenzyme Q10 with improved photochemical and pharmacokinetic behaviors. Eur J Pharm Sci 46:492–9
- Seo SW, Han HK, Chun MK, et al. (2012). Preparation and pharmacokinetic evaluation of curcumin solid dispersion using Solutol HS15 as a carrier. Int J Pharm 424:18–25
- Woo JS, Song YK, Hon JY, et al. (2008). Reduced food-effect and enhanced bioavailability of a self-microemulsifying formulation of itraconazole in healthy volunteers. Eur J Pharm 33:159–16
- Wei YH, Ye XL, Shang XG. (2012). Enhanced oral bioavailability of silybin by a supersaturatable self-emulsifying drug delivery system (S-SEDDS). Colloids Surf A: Physicochem Eng Aspects 396:22–8
- Wei L, Sun P, Nie S. (2005). Preparation and evaluation of SEDDS and SMEDDS containing carvedilol. Drug Dev Ind Pharm 31:785–94