
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Glycyrrhetinic acid-modified chitosan (mGA-suc-CTS) is used as liver-targeted carrier for drug delivery. In this study, nanoparticles were prepared by ionic gelation process, and glycyrrhetinic acid act as the targeting ligand. The structure of the product was confirmed by IR and NMR techniques. The main aim of this study was to deliver atorvastatin directly to the liver by using same conjugate and reduce the associated side-effects, i.e. hepatotoxicity at high dose. Characterization of the developed formulation was performed by differential scanning calorimetry, particle size measurements and cellular uptake studies. Release profile, pharmacokinetics studies and organ distribution studies showed that developed formulation shows a relative higher liver uptake. The optimized formulation showed increased plasma concentration than the CTS nanoparticles as well as plain drug and the accumulation in the liver was nearly 2.59 times more than that of obtained with the CTS nanoparticles. Pharmaceutical and pharmacological indicators suggested that the proposed strategy can be successfully utilized for liver targeting of therapeutics.
Introduction
Hepatic-targeted drug delivery systems have attracted much more attention recently for their promising roles in increasing the efficiency of pharmaceutical agents for liver, reducing drug doses and significantly decreasing the toxic side effects (Brannon-Peppas & Blanchette, Citation2012; Kaur et al., Citation2014g; Rohilla et al., Citation2014). In brief, receptor-mediated targeting system is the one that functioned with ligands via chemical coupling or physical coating ways, reaching the desired sites by the special interaction between the ligands in the system and the receptors on the surface of the desired organ (Garg et al., Citation2014b; Goyal et al., Citation2014b). There are many receptors present on the surface of hepatic cells, and receptor that is specific to hepatic parenchymal cells is asialoglycoprotein receptors, which has the ability to recognize the galactosylated ligands, lactobionic acid ligand, asialofetuin ligands, soybean-derived steryl glucoside ligands and many more (Garg et al., Citation2012; Malik et al., Citation2014). Similarly, glycyrrhetinic acid (GA) receptors are mainly found on the sinusoidal surface of mammalian hepatocytes. It has ability to recognize the GA ligands.
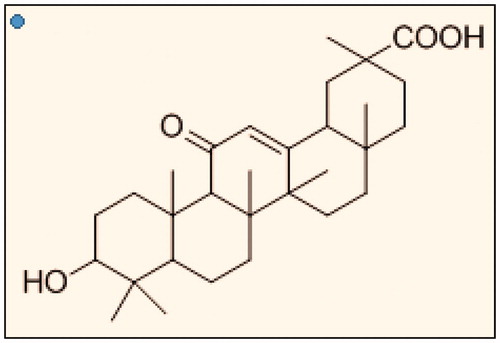
GA () is the main components of liquorice and hydrolysis product of glycyrrhizin. In 1990s, Negishi et al. confirmed that the cell membrane of rat’s liver contained specific high-binding sites for GA and reported that liposome or serum albumin modified with GA expressed a considerably high affinity to hepatocytes (Mao et al., Citation2005; Lin et al., Citation2008).
Figure 1. Structure of glycyrrhetinic acid.
Chitosan (CTS) is the naturally occurring amino-polysaccharides (Goyal et al., Citation2013) and widely used in pharmaceutics (Goyal et al., Citation2014a) due to its excellent biocompatibility and biodegradability (Chaudhary et al., Citation2014; Gagandeep et al., Citation2014). This article contains a novel hepatic drug delivery system using GA–chitosan as the carrier for delivering atorvastatin by reducing its toxic levels in blood and direct targeting to liver. Meanwhile, the drug-loaded nanoparticles were prepared by ionic gelatin method (Johal et al., Citation2014), and the release profile (Joshi et al., Citation2014) and targetability of the nanoparticles (Kataria et al., Citation2014) was evaluated by in vitro and in vivo Studies (Kaur et al., Citation2014a).
Atorvastatin, a statin is used for lowering cholesterol in coronary heart disease. At high dose, atorvastatin may result in the elevation of hepatic transaminase enzyme as well as cause hepatotoxicity (Calderon et al., Citation2010). These side effects results due to either reduced elimination of drug or due to inhibition of intestinal P-glycoprotein efflux transporter by some drugs or enhanced plasma drug level during long-term therapy (Kaur et al., Citation2014b,Citationd). By decreasing the plasma level of atorvastatin in comparison to liver, the side effects of drug may be reduced (Garg & Goyal, Citation2014; Kaur et al., Citation2014e).
GA is a ligand for liver targeting since it is recognized by the GA receptors, which are generally higher in number in hepatoma cells (Kaur et al., Citation2014f; Negishi et al., Citation1991). Conjugating GA and chitosan results in a polymer for liver targeting with sustained release of drug through it (Marwah et al., Citation2014). Nanodelivery system helps in achieving high solubility (Modgill et al., Citation2014), intracellular uptake of drug (Morie et al., Citation2014) and show high stability (Kateb et al., Citation2011; Sharma et al., Citation2014a). Liver targeting of atorvastatin will minimize the drug level in systemic circulation hence toxicity caused (Sharma et al., Citation2014b; Singh et al., Citation2014a,Citationb). GA has high efficacy against hepatotoxicity (Valan et al., Citation2010; Garg et al., Citation2014a). This project aims to formulate a liver-specific delivery of atorvastatin (to be given along with normal delivery of Ezetimibe), which may provide the high liver to plasma drug ratio leading to lower toxicity.
Materials and methods
Materials
Atorvastatin was received by Ranbaxy Laboratories Limited (Chandigarh, India) as a free gift sample. GA (HPLC > 98%), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC.HCl) and N-hydroxysuccinimide (NHS) were purchased from HIMEDIA Pvt. Ltd. (Chandigarh, India). CTS (Mw = 50 000 high molecular weight) was supplied by Sigma Aldrich (St. Louis, MO). All agents used were of analytical grade.
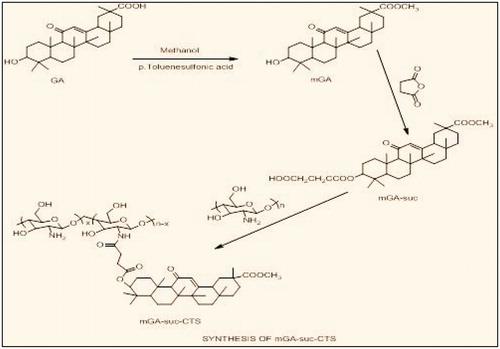
Synthesis of GA-modified chitosan (mGA-suc-CTS)
Synthesis of GA methyl ester (mGA)
The synthesis of GA methyl ester was achieved according to the literature (Tian et al., Citation2009). Briefly, to a solution of GA (7.5 g, 16.0 mmol) in 200 ml methanol, p-toluene sulfonic acid (1.0 g, 5.8 mmol) was added. The mixture was refluxed for 72 h, concentrated in vacuum, poured into water and extracted with DCM. The organic phase was washed with 5% sodium bicarbonate, dried over magnesium sulfate and concentrated to yield white solid, which was recrystallized from H2O/methanol.
Synthesis of GA methyl ester 3-O-hemisuccinate (mGA-suc)
mGA-suc was prepared according to the literature with a little modification (Kondratenko et al., Citation2001). Briefly, mGA (5.0 mmol) and succinic anhydride (20.0 mmol) were dissolved in 50 ml pyridine. The mixture was refluxed for 14–15 h under anhydrous conditions, after that the reaction mixture was poured into 300 ml water and acidified with hydrochloric acid to pH 3–4. The precipitates were filtered, washed with hot water, dried and chromatographed in a column filled with silica gel (eluted in an ethanol-ethyl acetate solvent system, 11:3, v/v).
Synthesis of mGA-suc-CTS
CTS was dissolved in 1% acetic acid (1%, w/v), followed by dilution with DMF. Different amounts of mGA-suc per NH2 group of CTS (0.2–1.0 mol/mol) were added and stirred until the solution was transparent. To activate the carboxylic acid pertaining to mGA-suc, EDC and NHS were added into the solution, and the molar ratio of EDC to NHS to mGA-suc was 1.2:1.2:1. The resulting solution was stirred at room temperature for 0.5 h and heated for 24 h. Then the final solution was poured into methanol, and the residue was filtered and dried. The structure of mGA-suc-CTS was confirmed by FTIR spectroscopy (Nicolet-380, Thermo, Waltham, MA) and 1 H NMR spectroscopy measurements (Bruker Advance II 400 NMR spectrometer, Punjab University, Punjab, India) ().
Figure 2. Synthesis of mGA-suc-CTS.
Preparation of atorvastatin-loaded mGA-suc-CTS/ tripolyphosphate nanoparticles
CTS nanoparticles are usually prepared by ionic gelation based on the interaction between the negative groups of tripolyphosphate (TPP) and the positively charged amino groups of CTS (Calvo et al., Citation1997; Janes & Alonso, Citation2003). In detail, mGA-suc-CTS was dissolved in 1% acetic acid and deacidified to pH 4.7–4.8. Then 10 ml sodium TPP (1.0 mg/mL) was added drop-wise to 10 ml of mGA-suc-CTS solution under stirring to form an opalescent suspension. Enteric coating of nanoparticles is done by using Eudragit s100 in ratio 100:3.5 (formulation: Eudragit). After complete optimization, the resulting suspension was centrifuged at 10 000 rpm for 10 min. The pelleted particles were resuspended in deionized water and lyophilized. The mean size and PDI of the mGA-suc-CTS nanoparticles were determined by photon correlation spectroscopy using a Zeta Plus particle analyzer (Brookhaven Instrument Corp., Holtsville, NY). The morphological examination of the nanoparticles was observed by transmission electron microscopy (TEM) (TECNAI, 200 Ks TEM Fei. Electron Optics, Tokyo, Japan).
In vitro studies of atorvastatin-loaded GA-suc-CTS/TPP Nanoparticles
Entrapment efficiency
The entrapment efficiency (EE) of the nanoparticles for atorvastatin was calculated by the following equation:
where A is the total amount of atorvastatin added and B is the free amount of drug in the supernatant.
In vitro drug release
In vitro drug release studies were performed by dialysis bag method at rotation speed of 100 rpm. Acidic buffer solution (pH 1.2) was used as dissolution medium. Each dialysis bag (pore size: 12 KD, Sigma Chemical Co., MO, USA) was loaded with 20 mg nanoparticles. Volume and temperature of dissolution medium were 200 ml, and 37.0 ± 0.2 °C, respectively. At predetermined time interval, samples (5 ml) were withdrawn, replaced with same volume of fresh media for duration of 2 h. The same procedure was repeated with basic buffer solution, pH 7.4, for a time period of 72 h, sample collection were at predetermined time interval, filtered and assayed for drug content at 246 nm against blank using UV-visible spectrophotometer (Shimadzu, UV-1700 Pharma Spec, Kyoto, Japan) (Kaur et al., Citation2014c).
Cellular uptake study
Cellular uptake study of the nanoparticles of GA–chitosan and chitosan nanoparticles was performed for qualitative estimation of the uptake of nanoparticles by the liver cells. Cell uptake study of the drug-loaded GA–chitosan nanoparticles was performed on Hep G2 cells (liver cell lines) by using rhodamine dye as according to the procedure reported earlier (Sayın et al., Citation2008). Formulations were incubated with the fresh culture cells using 96-well culture plate for 12 h. Images of mGA-suc-CTS nanoparticles and chitosan nanoparticles were taken using fluorescent microscope (Olympus, CK X 41, Shinjuku, Tokyo, Japan).
In vivo studies
Liver drug concentration–time profile and pharmacokinetic parameters were obtained after oral administration of atorvastatin, atorvastatin-loaded GA–chitosan nanoparticles and chitosan-atorvastatin Nanoparticles. In this study, three groups (each group had nine rats) of Wistar rats were taken for pharmacokinetic studies ().
Table 1. Experimental layout for pharmacokinetic and liver distribution studies.
For oral administration study, three group (n = 3) for each time point received a dose of 2.5 mg/kg with a cannula via the oral route. Rats were sacrificed by cervical dislocation, and liver were isolated (Hu et al., Citation2013). Liver samples were homogenized and immediately centrifuged (5000–10 000 rpm) for 10 min at an ambient temperature. After centrifugation, supernatant plasma was transferred into clean, fresh Eppendrofs and stored in freezer (at −70 °C) until analysis. Samples collected from rats were analyzed using validated bio-analytical method, and liver concentration values were determined from calibration curve (Nabavi et al., Citation2012).
Statistical analysis
Data were expressed as means of three separate experiments.
Results and discussion
Synthesis of mGA-suc-CTS
Methyl ester of GA was synthesized in dry methanol using P-TSA as an acidic catalyst. This reaction completes in very long time, and methyl ester of glycyrrhetinic (mGA) was obtained in good yield (61.14%). The hemisuccinate of mGA was achieved by reaching mGA with succinic anhydride in anhydrous condition to give mGA-suc in good yield and purity. Furthermore, the mGA-suc was competed with chitosan (CTS) by utilizing the amine of chitosan and carboxylic acid functionality of mGA-suc. The coupling agent was EDC.HCl, which activates the carboxylic acid for amide bond formation. The resultant mGA-suc-CTS were obtained in moderate yield and good purity, which was further utilized for the development of the formulation without further purification. 1H NMR spectra showed (400 MHz, DMSO, Ƨ = 0) CTS (δ, ppm): 2.0 (NHCOCH3), 3.11 (H2), 3.5–3.9 (H3, H4, H5 and H6). As compared with chitosan, new peaks at 0.6–1.6 ppm were observed in the 1H NMR spectrum of mGA-suc-CTS, which belonged to the protons of CH3, CH2 and CH groups of the steroid skeleton of mGA-suc. The results of 1H NMR were evidence of the conjugation of mGA-suc onto CTS.
In vitro characterization of the atorvastatin loaded mGA-suc-CTS nanoparticles
Optimization is performed to obtain minimum possible sized particles, as small nanoparticles size may provide a large surface area and controlled release. In this study, we have prepared Eudragit-coated GA–chitosan nanoparticles to prevent drug degradation in the stomach. Based on the above context, conjugate to TPP ratio was optimized at 5:1 for GA–chitosan nanoparticles with size of 243.5 nm and PDI of 0.117. Increase in polymer conjugate to TPP ratio increases turbidity resulting in increased size. Increase in TPP content lead to flocculation and particle aggregation due to excess polyanions (Calvo et al., Citation1997). The optimal value of ultrasonication time selected was 3.0 min. However, beyond 2.5 min, particle size increased due to increase in surface energy provided by ultrasonication, which lead to agglomeration (Saltiel et al., Citation2004) and coating of Eudragit is done in ratio of 100:3.5 (Formulation: Eudragit) with increment in particle size up to 352.5 nm.
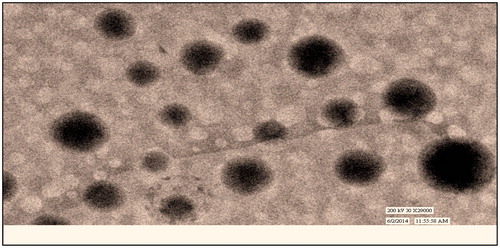
Morphology, particle size and its distribution
Optimized formulation was visualized under TEM for morphology of mGA-suc-CTS nanoparticles. shows the TEM images of nanoparticles. Electron photomicrograph results show that nanoparticles were spherically uniform and discrete. Particle size as size distribution of polymer conjugate is a measure of polymer and TPP ratio. Polymer conjugate to TPP in a ratio of 5:1 found to be optimized 352.5 nm with a PDI of 0.126. The effect of polymer conjugate concentration is found to be more critical in achieving nanoparticles of desired range. Increase in polymer conjugate concentration beyond five parts, increase viscosity of medium and leads controlled precipitation with high PDI. Whereas at polymer conjugate below four parts, lack of adequate functional group makes it unable to induce desired ionic precipitation.
Figure 3. TEM image of GA–CTS nanoparticles.
Differential scanning calorimetry
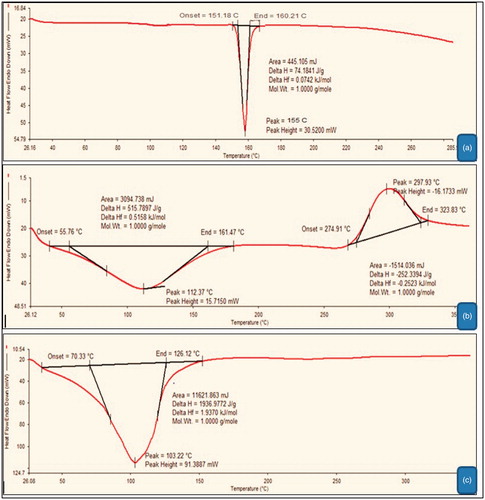
Differential scanning calorimetry thermo grams of plain atorvastatin, GA–chitosan and atorvastatin-loaded GA–chitosan nanoparticles is demonstrated in ). Prominent endothermic peaks at 155 °C, 112.37 °C and 103.22 °C represented the sharp melting point of plain atorvastatin, GA–chitosan conjugate and GA–chitosan nanoparticles, respectively. The broad endothermic peaks and absence of sharp peaks in drug-loaded nanoparticles indicates the charge in phase position of the drug in developed formulation. Furthermore absence of sharp endothermic peaks clearly resolved the loss in crystalline stage of the drug in prepared formulation. This phenomenon could be related to the instant are controlled precipitation of polymer conjugate, which inhibit the formation of stable crystal.
Figure 4. DSC of plain atorvastatin (a), glycyrrhetinic acid-chitosan (b) and atorvastatin-loaded GA–CTS nanoparticles (c).
Entrapment efficiency
EE of chitosan nanoparticles was estimated and repeated three times. All the formulations showed good reproducible results. EE of atorvastatin was found to be 79.43 ± 2.03%, which showed that nanoparticles had fairly good EE. Higher EE could be attributed poor aqueous solubility of the drug as rigid polymer matrix of nanoparticles. Below or above the optimized molar concentration of polymer conjugate and TPP leads to uncontrolled precipitation along with poor EE.
In vitro drug release studies
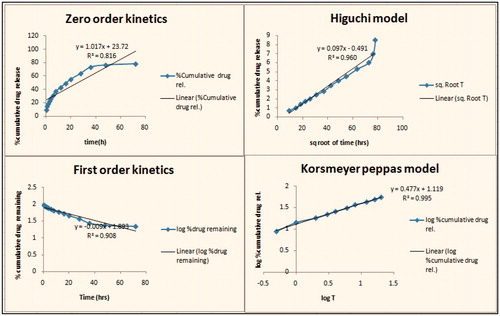
In vitro drug release was performed using dialysis bag at pH 1.2 and 7.4. ) shows the in vitro drug release from optimized GA–chitosan nanoparticles at pH 1.2 and 7.4, respectively. Results showed that the drug release was found to be 6.04 ± 0.23 after two hours at PBS (pH 1.2) and controlled release of above 78.01% at PBS (pH 7.4) after 72 h. It was concluded that a large fraction of the drug was released at pH 7.4 as compared to pH 1.2. In vitro release profiles establish the enteric coating of the prepared formulation. Polyanionic nature of the polymer remains nonprotonated in the gastric pH. Thereby it inhibits the release of encapsulated therapeutics, whereas intestinal pH induces the protonation of the coating material causes a controlled release of encapsulated material. Controlled release is further attributed to the physiochemical behavior of polymer conjugate where the conjugate remains unprotonated in intestinal pH, thus restricts the free access of dissolution medium, loads to controlled diffusion of drug. In order to completely understand the kinetics of drug release, it is essential to apply the different kinetic models like zero order, first order, Higuchi curves and Korsmeyer–Peppas model. shows kinetic analysis of drug release from nanoparticles; drug release behavior was interpreted on the basis of the regression coefficient statically obtained from different models. Results indicated that Higuchi model was the best fit model with R2 value 0.960, which describe the drug release was predominantly based on diffusion mechanism. The diffusion mechanism was further confirmed through the Peppas equation where n value was found to be less than 0.5. Diffusion behavior could be explained in the nonprotonated behavior of the polymer conjugate at the intestinal pH.
Figure 5. In vitro drug release from optimized glycyrrhetinic acid-chitosan nanoparticles at PBS pH 1.2 (a) and 7.4 (b).
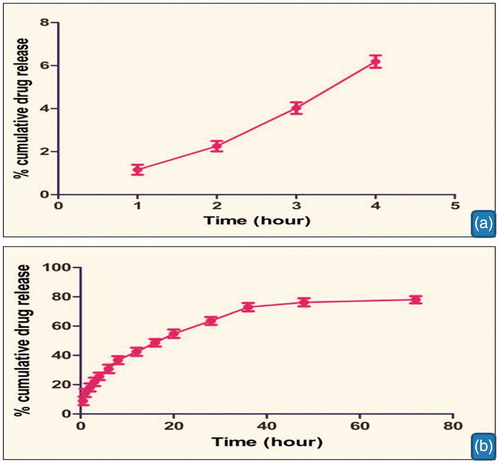
Figure 6. Drug-release kinetics of optimized mGAsuc-CTS nanoparticles of atorvastatin.
Cellular uptake study
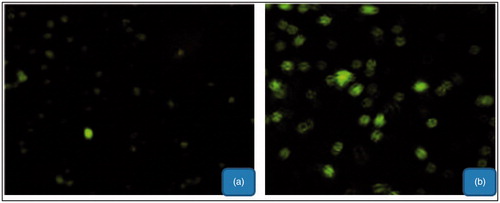
Cell uptake studies of the formulations were performed for qualitative estimation of the nanoparticle uptake by liver cells. Image of the cells taken after 12-h of incubation of drug-loaded nanoparticles is shown in . Cellular uptake studies showed that mGA-suc-CTS nanoparticles has the maximum cellular uptake as CTS nanoparticles have less cellular uptake in comparison. A high cellular uptake of polymer-conjugated nanoparticles established the proposed hypothesis. In that, the presence of GA improves the cellular efficiency of developed formulation for hepatic cells. Cell uptake study further confirms that receptor mediated endocytosis is a key mechanism for cellular uptake of developed formulation.
Figure 7. Cellular uptake of the CTS nanoparticles (a) and mGA-suc-CTS nanoparticles (b).
In vivo studies
Plasma pharmacokinetic study
From the given results (; ), it was observed that the developed optimized formulation showed increased plasma concentration (61.03 µg/ml) and T1/2 (32 h) than the CTS nanoparticles and plain drug. Furthermore, the plasma concentration takes longer time to reach its maximum value in case of mGA-suc-CTS formulation followed by CTS nanoparticles and plain drug, showing sustained release in case of mGA-suc-CTS formulation. This could be attributed to preferential localization polymer conjugate nanoparticles in the liver. Polymer conjugate 30% rise in Cmax value, which could be attributed to increased bioavailability of developed formulation attributed to nano solid dispersion, controlled release mechanism and retention of the developed formulation near the absorption window. Furthermore, a higher Tmax in developed formulation is associated with its high affinity for hepatic cell lines. Moreover, the localization of receptor along the sinusoid cell lines improves hepatic permeability and retention of the developed formulation in the liver.
Figure 8. Plasma pharmacokinetic parameters of different formulations.
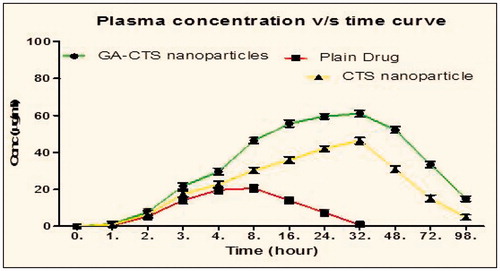
Table 2. Plasma pharmacokinetic parameters of different formulations.
Organ distribution study of atorvastatin after oral administration
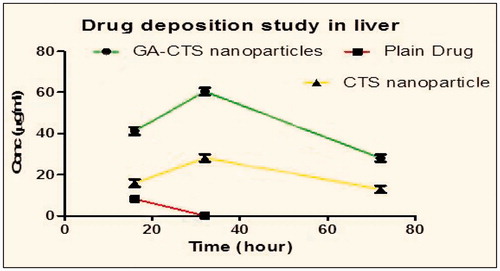
From the given data (), it is concluded that the amount of drug uptake in the liver is more in mGA-suc-CTS nanoparticles as compared to CTS nanoparticles and plain drug. mGA-suc-CTS nanoparticles increased the uptake of drug in the liver by 2.59-fold more than CTS nanoparticles and 5.15-fold more than plain drug. It can be attributed that GA has the ligand properties for the receptors presents in the liver, so can be used for the liver targeting, also it is efficient against hepatotoxicity.
Figure 9. Drug deposition study of different formulations in liver.
Conclusion
In this study, GA conjugate of chitosan was synthesized, which was used for the preparation of nanoparticles and compared with the nanoparticles of chitosan for drug release and efficacy to reduce hepatotoxicity of atorvastatin. The synthesized GA–chitosan conjugate has high affinity for liver cells, and hence the nanoparticles prepared from GA specifically target liver cells wherein they provide sustained release of the drug. GA–chitosan nanoparticles loaded with atorvastatin were successfully prepared; and by specifically delivering the drug to the liver, high reduction in hepatotoxicity was achieved compared to the chitosan nanoparticles.
Acknowledgements
Dr. Amit K. Goyal and Mr. Goutam Rath are thankful to the Department of Biotechnology (DBT), New Delhi, India.
Declaration of interest
The authors report no conflict of interest.
References
- Brannon-Peppas L, Blanchette JO. (2012). Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev 64:206–12
- Calderon RM, Cubeddu LX, Goldberg RB, Schiff ER. (2010). Statins in the treatment of dyslipidemia in the presence of elevated liver aminotransferase levels: a therapeutic dilemma. Mayo Clin Proc 2010;85:349–56
- Calvo P, Remunan-Lopez C, Vila-Jato J, Alonso M. (1997). Novel hydrophilic chitosan-polyethylene oxide nanoparticles as protein carriers. J Appl Polym Sci 63:125–32
- Chaudhary S, Garg T, Murthy RS, et al. (2014). Recent approaches of lipid-based delivery system for lymphatic targeting via oral route. J Drug Target. [Epub ahead of print]
- Gagandeep G, Garg T, Malik B, et al. (2014). Development and characterization of nano-fiber patch for the treatment of glaucoma. Eur J Pharm Sci 53:10–16
- Garg T, Goyal AK. (2014). Biomaterial-based scaffolds – current status and future directions. Expert Opin Drug Deliv 11:767–89
- Garg T, Kumar A, Rath G, Goyal AK. (2014a). Gastroretentive drug delivery systems for therapeutic management of peptic ulcer. Crit Rev Ther Drug Carrier Syst 31:531–57
- Garg T, Rath G, Goyal AK. (2014b). Ancient and advanced approaches for the treatment of an inflammatory autoimmune disease-psoriasis. Crit Rev Ther Drug Carrier Syst 31:331–64
- Garg T, Singh O, Arora S, Murthy R. (2012). Scaffold: a novel carrier for cell and drug delivery. Crit Rev Ther Drug Carrier Syst 29:1–63
- Goyal G, Garg T, Malik B, et al. (2013). Development and characterization of niosomal gel for topical delivery of benzoyl peroxide. Drug Deliv. [Epub ahead of print]
- Goyal G, Garg T, Rath G, Goyal AK. (2014a). Current nanotechnological strategies for an effective delivery of drugs in treatment of periodontal disease. Crit Rev Ther Drug Carrier Syst 31:89–119
- Goyal G, Garg T, Rath G, Goyal AK. (2014b). Current nanotechnological strategies for treating glaucoma. Crit Rev Ther Drug Carrier Syst 31:365–405
- Hu L, Song W, Zhang H, Gu D. (2013). HPLC-UV method development for atorvastatin calcium micro-emulsion determination in rat plasma and its application to elucidate pharmacokinetic behavior after oral administration to rats. Metabolism 2:3839–44
- Janes K, Alonso M. (2003). Depolymerized chitosan nanoparticles for protein delivery: preparation and characterization. J Appl Polymer Sci 88:2769–76
- Johal HS, Garg T, Rath G, Goyal AK. (2014). Advanced topical drug delivery system for the management of vaginal candidiasis. Drug Deliv. [Epub ahead of print]
- Joshi D, Garg T, Goyal AK, Rath G. (2014). Advanced drug delivery approaches against periodontitis. Drug Deliv. [Epub ahead of print]
- Kataria K, Sharma A, Garg T, et al. (2014). Novel technology to improve drug loading in polymeric nanofibers. Drug Deliv Lett 4:79–86
- Kateb B, Chiu K, Black KL, et al. (2011). Nanoplatforms for constructing new approaches to cancer treatment, imaging, and drug delivery: what should be the policy? Neuroimage 54:S106–24
- Kaur M, Garg T, Rath G, Goyal AK. (2014a). Current nanotechnological strategies for effective delivery of bioactive drug molecules in the treatment of tuberculosis. Crit Rev Ther Drug Carrier Syst 31:49–88
- Kaur M, Malik B, Garg T, et al. (2014b). Development and characterization of guar gum nanoparticles for oral immunization against tuberculosis. Drug Deliv. [Epub ahead of print]
- Kaur N, Garg T, Goyal AK, Rath G. (2014c). Formulation, optimization and evaluation of curcumin-beta-cyclodextrin-loaded sponge for effective drug delivery in thermal burns chemotherapy. Drug Deliv. [Epub ahead of print]
- Kaur P, Garg T, Rath G, et al. (2014d). Surfactant-based drug delivery systems for treating drug-resistant lung cancer. Drug Deliv. [Epub ahead of print]
- Kaur R, Garg T, Das Gupta U, et al. (2014e). Preparation and characterization of spray-dried inhalable powders containing nanoaggregates for pulmonary delivery of anti-tubercular drugs. Artif Cells Nanomed Biotechnol. [Epub ahead of print]
- Kaur R, Garg T, Malik B, et al. (2014f). Development and characterization of spray-dried porous nanoaggregates for pulmonary delivery of anti-tubercular drugs. Drug Deliv. [Epub ahead of print]
- Kaur R, Garg T, Rath G, Goyal AK. (2014g). Advanced aerosol delivery devices for potential cure of acute and chronic diseases. Crit Rev Ther Drug Carrier Syst 31:495–530
- Kondratenko R, Mustafina S, Baltina L, et al. (2001). Synthesis and antiulcer activity of 3-O-acylated glycyrrhetic acid methylates. Pharm Chem J 35:243–6
- Lin A, Liu Y, Huang Y, et al. (2008). Glycyrrhizin surface-modified chitosan nanoparticles for hepatocyte-targeted delivery. Int J Pharm 359:247–53
- Malik R, Garg T, Goyal AK, Rath G. (2014). Polymeric nanofibers: targeted gastro-retentive drug delivery systems. J Drug Target. [Epub ahead of print]
- Mao S-J, Hou S-X, He R, et al. (2005). Uptake of albumin nanoparticle surface modified with glycyrrhizin by primary cultured rat hepatocytes. World J Gastroenterol 11:3075
- Marwah H, Garg T, Goyal AK, Rath G. (2014). Permeation enhancer strategies in transdermal drug delivery. Drug Deliv. [Epub ahead of print]
- Modgill V, Garg T, Goyal AK, Rath G. (2014). Permeability study of ciprofloxacin from ultra-thin nanofibrous film through various mucosal membranes. Artif Cells Nanomed Biotechnol. [Epub ahead of print]
- Morie A, Garg T, Goyal AK, Rath G. (2014). Nanofibers as novel drug carrier – an overview. Artif Cells Nanomed Biotechnol. [Epub ahead of print]
- Nabavi SM, Nabavi SF, Eslami S, Moghaddam AH. (2012). In vivo protective effects of quercetin against sodium fluoride-induced oxidative stress in the hepatic tissue. Food Chem 132:931–5
- Negishi M, Irie A, Nagata N, Ichikawa A. (1991). Specific binding of glycyrrhetinic acid to the rat liver membrane. Biochim Biophys Acta 1066:77–82
- Rohilla R, Garg T, Goyal AK, Rath G. (2014). Herbal and polymeric approaches for liver-targeting drug delivery: novel strategies and their significance. Drug Deliv. [Epub ahead of print]
- Saltiel C, Chen Q, Manickavasagam S, et al. (2004). Identification of the dispersion behavior of surface treated nanoscale powders. J Nanopart Res 6:35–46
- Sayın B, Somavarapu S, Li X, et al. (2008). Mono-N-carboxymethyl chitosan (MCC) and N-trimethyl chitosan (TMC) nanoparticles for non-invasive vaccine delivery. Int J Pharm 363:139–48
- Sharma A, Garg T, Aman A, et al. (2014a). Nanogel-an advanced drug delivery tool: Current and future. Artif Cells Nanomed Biotechnol. [Epub ahead of print]
- Sharma R, Singh H, Joshi M, et al. (2014b). Recent advances in polymeric electrospun nanofibers for drug delivery. Crit Rev Ther Drug Carrier Syst 31:187–217
- Singh B, Garg T, Goyal AK, Rath G. (2014a). Recent advancements in the cardiovascular drug carriers. Artif Cells Nanomed Biotechnol. [Epub ahead of print]
- Singh H, Sharma R, Joshi M, et al. (2014b). Transmucosal delivery of Docetaxel by mucoadhesive polymeric nanofibers. Artif Cells Nanomed Biotechnol. [Epub ahead of print]
- Tian Q, Wang W, He X, et al. (2009). Glycyrrhetinic acid-modified nanoparticles for drug delivery: preparation and characterization. Chin Sci Bull 54:3121–6
- Valan M, Britto A, Venkataraman R. (2010). Phytoconstituents with hepatoprotective activity. Int J Chem Sci 8:1421–32