
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Poly(d,l-lactic-co-glycolic acid) (PLGA) nanoparticles (NP) of Val-Val dipeptide monoester prodrugs of ganciclovir (GCV) including L-Val-L-Val-GCV (LLGCV), L-Val-D-Val-GCV (LDGCV) and D-Val-L-Val-GCV (DLGCV) were formulated and dispersed in thermosensitive PLGA-PEG-PLGA polymer gel for the treatment of herpes simplex virus type 1 (HSV-1)-induced viral corneal keratitis. Nanoparticles containing prodrugs of GCV were prepared by a double-emulsion solvent evaporation technique using various PLGA polymers with different drug/polymer ratios. Nanoparticles were characterized with respect to particle size, entrapment efficiency, polydispersity, drug loading, surface morphology, zeta potential and crystallinity. Prodrugs-loaded NP were incorporated into in situ gelling system. These formulations were examined for in vitro release and cytotoxicity. The results of optimized entrapment efficiencies of LLGCV-, LDGCV- and DLGCV-loaded NP are of 38.7 ± 2.0%, 41.8 ± 1.9%, and 45.3 ± 2.2%; drug loadings 3.87 ± 0.20%, 2.79 ± 0.13% and 3.02 ± 0.15%; yield 85.2 ± 3.0%, 86.9 ± 4.6% and 76.9 ± 2.1%; particle sizes 116.6 ± 4.5, 143.0 ± 3.8 and 134.1 ± 5.2 nm; and zeta potential −15.0 ± 4.96, −13.8 ± 5.26 and −13.9 ± 5.14 mV, respectively. Cytotoxicity studies suggested that all the formulations are non-toxic. In vitro release of prodrugs from NP showed a biphasic release pattern with an initial burst phase followed by a sustained phase. Such burst effect was completely eliminated when NP were suspended in thermosensitive gels with near zero-order release kinetics. Prodrugs-loaded PLGA NP dispersed in thermosensitive gels can thus serve as a promising drug delivery system for the treatment of anterior eye diseases.
Introduction
Infection with herpes simplex virus type 1 (HSV-1) transmitted by non-sexual contact is a most common cause of corneal epithelial and stromal keratitis. Herpetic keratitis is one of the most common causes of ocular morbidity and corneal blindness (Wilhelmus, Citation2000). Recently, it was estimated on the basis of seroprevalence that HSV-1 affects 58% of general population in the US (Xu et al., Citation2006). Approximately, 400 000 patients are annually infected by HSV-1 with a 50% recurrence rate within two years (Liesegang, Citation1988). After the primary infection, HSV-1 remains life-long in latent form within sensory neurons of trigeminal ganglia as a reservoir (Perng & Jones, Citation2010; Kennedy et al., Citation2011). Subsequent recurrence of keratitis often causes corneal scarring, thinning, stromal opacity and eventually vision loss (Suresh & Tullo, Citation1999; Ohara et al., Citation2000; Perng & Jones, Citation2010).
Ganciclovir (GCV) exhibits antiviral activity against all herpes virus strains and demonstrates significant therapeutic efficiency against HSV-1 infection (Matthews & Boehme, Citation1988). Ganciclovir ophthalmic gel (0.15%) has been recently approved and is available in the US market (Zirgan®, Bausch & Lomb Inc, Rochester, NY, USA) for the treatment of acute ocular herpetic keratitis. However, ocular bioavailability of GCV is poor due to limited aqueous solubility (4.3 mg/mL in water), low partition coefficient (log P = −1.55) and low corneal penetration (Majumdar et al., Citation2003). To achieve therapeutic concentrations at deeper corneal layers, i.e. stroma, a high-concentration gradient across the tight corneal epithelium and/or frequent topical instillation is required.
Many strategies have been investigated to improve aqueous solubility and corneal permeation of GCV (El-Samaligy et al., Citation1996). Novel transporter-targeted prodrug strategy has been proven to be an effective approach. Dipeptide monoester prodrugs of GCV, such as valine-valine-GCV (VVGCV) was selected due to its high aqueous solubility and chemical stability, excellent affinity for oligopeptide transporter type 1 (PEPT-1), an influx transporter present on the corneal epithelium, resulting in appreciable corneal permeability (Plourde et al., Citation1995; Majumdar et al., Citation2005; Kansara et al., Citation2007). Valine-valine-GCV can produce deeper corneal penetration, which further contributes to greater therapeutic efficacy against stromal keratitis and uveal infections. Moreover, VVGCV is susceptible to precorneal esterase and peptidases. The prodrug generates parent GCV in ocular tissues. However, a major drawback of VVGCV ophthalmic solution is its very short residence on the corneal surface in the precorneal area requiring frequent administration.
Many novel formulations, such as hydrogels and colloidal nanocarriers made of biocompatible and biodegradable polymers have been employed to improve ocular bioavailability (Sood et al., Citation2014). However, nanocarrier delivery can enhance concentration and precorneal residence time due to their non-mucoadhesive property (Ali et al., Citation2014). It can overcome physiological mechanisms, such as blinking and nasolacrimal drainage (Baeyens & Gurny, Citation1997). Thermosensitive PLGA-PEG-PLGA gel solution of concentrations ranging between 20 and 25% w/w can transition into polymeric gel at 32 °C and remain in gel state till 60 °C (Duvvuri et al., Citation2005). Hence, another interesting approach is to combine polymeric NP and in situ thermosensitive gel to achieve ocular retention (Gupta et al., Citation2013; Ramyadevi & Sandhya, Citation2014).
Therefore, a new ophthalmic delivery systems has been designed which aims at (1) enhancing corneal permeability and ocular bioavailability; (2) prolonging precorneal residence time (more than 2 min); (3) providing constant and sustained release of entrapped drugs; (4) minimizing precorneal drug loss and side effects; and (5) reducing the recurrence of keratitis (Diebold & Calonge, Citation2010). In this study, various alternative strategies are employed to achieve overall therapeutic goals for GCV prodrugs after topical instillation. Stereoisomeric dipeptide GCV prodrugs including L-Val-L-Val-GCV (LLGCV), L-Val-D-Val-GCV (LDGCV) and D-Val-L-Val-GCV (DLGCV) are synthesized and encapsulated in PLGA NP. Effect of lactide/glycolide ratio on prodrug entrapment efficiency, drug loading, yield and particle size is investigated. Nanoparticles with optimum entrapment efficiency and particle size have been further characterized for surface morphology, zeta potential, in vitro release and cytotoxicity. Mechanism of drug release from the NP was delineated. Optimized NP is then suspended in thermogelling vehicle (23% w/w PLGA-PEG-PLGA aqueous solution) and in vitro release properties are studied. The results can provide useful and essential information for further in vitro cell experiments and in vivo animal studies.
Materials
Ganciclovir was purchased from Hubei Gedian Humanwell Pharmaceutical Co. Ltd (Gedian, Hubei, China). All the stereoisomeric dipeptide monoester GCV prodrugs were synthesized and purified in our laboratory. Poly(D,L-lactic-co-glycolic acid) (PLGA) polymers 65:35 with Mw 45 000–75 000 Da; PLGA 75:25 with Mw 66 000–107 000 Da; PLGA 85:15 with Mw 50 000–75 000 Da; and polyvinyl alcohol (PVA) with Mw 31 000–50 000 Da were procured from Sigma Chemicals (Balcatta, WA, USA). All these PLGA polymers are end capped with ester group. Lactide and glycolide monomers for synthesis of thermosensitive gel PLGA-polyethylene glycol (PEG)-PLGA (PLGA-PEG-PLGA) were donated by Purac America (Lincolnshire, IL) and PLGA-PEG-PLGA with Mw 4759 Da was further synthesized, purified and characterized in our laboratory (Duvvuri et al., Citation2005). Dichloromethane (DCM) was purchased from Sigma-Aldrich Co (St. Louis, MO). HCEC cells were a generous gift from Dr Araki-Sasaki (Kinki Central Hospital, Japan). Culture flasks (75 cm2 growth area), 12-well Costar® plates, all buffer components and solvents were purchased from Fisher Scientific Co (Pittsburgh, PA, USA). Distilled deionized (DDI) water was added in the preparation of all buffers and mobile phases. All other required chemicals were obtained from Sigma-Aldrich Co.
Methods
Synthesis
Stereoisomeric monoester dipeptide prodrugs of GCV (LLGCV, LDGCV and DLGCV) were synthesized from parent drug GCV according to a previously published procedure with slight modifications (Majumdar et al., Citation2005). Intermediate compound O-tert-butyldimethylsilyl-GCV-OH (OTBDMS-GCV-OH) was obtained following previous methods (Majumdar et al., Citation2004). Fmoc protected amino acids, Fmoc-L-Val-OH and Fmoc-D-Val-OH, were used in the initial conjugation. The product Fmoc-Val-GCV-OTBDMS was purified by silica column chromatography and deprotected from Fmoc group under basic condition twenty percent piperidine/DMF. Boc protected amino acids, N-Boc-L-Val-OH and N-Boc-D-Val-OH, were added for further conjugation. Final products were obtained after deprotection of the N-Boc and tert-butyldimethylsilyl (TBDMS) groups with 80% TFA/DCM followed by recrystallization from cold diethyl ether. The reaction was monitored by thin layer chromatography (TLC) and liquid chromatography/mass spectrometry (LC/MS). The structures of all the compounds were confirmed with high-resolution nuclear magnetic resonance (1H-NMR) spectroscopy and LC/MS. 1H-NMR spectra was carried out on Varian Mercury 400 Plus spectrometer (Varian, Inc. Palo Alto, CA, USA) with tetramethylsilane (TMS) as an internal standard. Chemical shifts (δ) are reported in parts per million (ppm) relative to the NMR solvent signal (CD3OD, 3.31 ppm for proton). Mass analysis was carried out with a hybrid triple quadrupole linear ion trap mass spectrometer (Q-Trap LC/MS/MS spectrometer, Applied Biosystems-2000, Waltham, MA, USA) under enhanced mass (EMS) mode. Electrospray ionization (ESI) was selected as an ion source and was operated in positive ion mode.
Preparation of NP by water in oil in water (W/O/W) method
Since prodrugs (LLGCV, LDGCV and DLGCV) are hydrophilic, NP was prepared by W/O/W emulsion solvent evaporation method. Briefly, prodrug solution (0.5 mL, 10 mg/mL) was emulsified in dichloromethane (5 mL) containing various amounts of PLGA by sonication over an ice-bath with a Fisher Scientific 100 ultrasonic probe for 2 min. The primary emulsion was slowly added to PVA solution (20 mL, 2.0% w/v) and sonicated at a constant output of 55 W for 5 min to form a W/O/W type double emulsion. Organic solvent residues were removed by stirring at room temperature for 2 h and evaporating under vacuum for 30 min to ensure complete removal of organic solvents. Nanoparticle suspension was washed with distilled deionized water followed by centrifugation at 5000 g (22 000 rpm) for 1 h. This procedure was repeated three times to remove free prodrug and PVA residue. The NP suspension was frozen and lyophilized for 48 h under a freeze-drier system to obtain the final NP.
Entrapment efficiency, drug loading and yield
Entrapment efficiency and prodrug loading in NP were also determined according to the following method. Freeze-dried NP (1 mg) was taken in triplicate, dissolved in 2 mL of dichloromethane and vortexed for 1 min to completely destruct the NP. Subsequently, the mixture was dried under inert atmosphere and dissolved in 500 µL acetonitrile:water (65:35). After vortexing and centrifugation at 12 000 rpm for 10 min, the supernatant was collected to determine the prodrug concentration by high-performance liquid chromatography (HPLC). Entrapment efficiency, drug loading and yield were calculated according to Equations (Equation1(1)
(1) ), (Equation2
(2)
(2) ) and (Equation3
(3)
(3) ), respectively. The measurements were made in triplicate.
(1)
(1)
(2)
(2)
(3)
(3)
Particle size and polydispersity by dynamic light scattering
Dynamic light scattering analyzer (Brookhaven Zeta Plus instrument, Holsville, NY) was employed for the measurement of particle size and polydispersity index (PDI) of 27 different NP formulations (n = 3). It was considered monodispersed when PI ≤ 0.05.
Zeta potential
To determine zeta potential, the NP suspension was diluted and measured using Nano ZS zetasizer (Malvern Instruments Ltd, Worcestershire, UK). All measurements were performed in triplicate.
Surface morphology analysis by scanning electron microscope
The morphological examination of NP with optimal entrapment efficiency was performed with a FEG ESEM XL 30 SEM (FEI, Hillsboro, OR). To obtain the specimen, freeze-dried NPs were attached to a double-sided tape, spray-coated with gold-palladium at 0.6 kV. Nanoparticles were viewed under the electron microscope and digital images were recorded.
Differential scanning calorimetry
Glass transition temperatures (Tg) of the polymers and prodrug-loaded NP were measured with DSC Thermal Analysis (DSC-60, Shimadzu Corporation, Kyoto, Japan) with a heating cycle of 30 °C to 350 °C at a heating rate of 5 °C/min. A steady stream of nitrogen at 20 mL/min was supplied to control the heating and cooling rates. Samples (5 mg) were sealed in aluminum pans for analysis with an empty pan as reference and thermograms were finally recorded.
X-ray diffraction
X-ray diffraction measurements of NP were performed on a high resolution MiniFlex automated X-ray prototype diffractometer (Rigaku, The Woodlands, TX) based on Ni-filtered Cu-Kα radiation under 30 kV and 15 mA. Recordings were made at ambient temperature with the diffraction angle from 2θ = 5° to 40° using steps of 0.05° and a count time of 3 s/step (effectively 1°/min). The diffraction patterns were processed using Jade 8+ (Materials Data, Inc., Livermore, CA).
Cell culture
A human corneal epithelial cell line, HCEC cells were cultured in a humidified incubator at 37 °C with 5% carbon dioxide in air atmosphere. Cells were grown in the culture medium containing minimum essential medium supplemented with 10% calf serum (non-heat inactivated), 100 U/L of penicillin, 100 U/L of streptomycin, 1.76 g/L lactalbumin enzymatic dehydrolysate and 1.3 g/L HEPES. The medium was changed every alternate day and cells were passaged weekly at a 1/3 split ratio.
Cytotoxicity studies
In vitro cytotoxicity of all stereoisomeric dipeptide monoester prodrugs and prodrug-loaded NP were evaluated with the dye [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] (MTS) with electron coupling reagent phenazine methosulfate (PMS). Phenazine methosulfate has enhanced chemical stability, which allows it to be combined with MTS to form a stable solution eliminating the need to solubilize formazan crystals with an external source. Briefly, cells were grown in 96-well plates at 10 000 cells per well for 24 h prior to sample addition. Cells were incubated with 100 µL medium containing prodrugs and prodrug-loaded NP at equivalent drug concentrations ranging from 100 to 500 µg/mL for 24 and 48 h. The culture medium without any drug or formulations was used as the control. At specified time intervals, the medium was aspirated and rinsed with Dulbecco’s phosphate-buffered saline (DPBS) twice. Twenty microliters of MTS stock solution was then added to each well. After the addition of the MTS dye, cells were incubated for 4 h at 37 °C. Cell viability was then measured by absorbance at 485 nm (reference at 590 nm) on an automated plate reader (BioRad, Hercules, CA). The absorbance is directly proportional to the number of living cells in the culture and the relative cell viability (%) compared to control cells was calculated as given in Equation (Equation4(4)
(4) ):
(4)
(4)
Absdrug is the absorbance of cells incubated with samples; Abscontrol is the absorbance of cells incubated with the culture medium only; Absblank is the absorbance of culture medium devoid of cells.
In vitro drug release from NP suspended in buffer and thermosensitive gel
Prodrug-loaded NP (10 mg) were dispersed in 1 mL of isotonic phosphate-buffered saline (IPBS, pH 7.4) and subsequently filled into dialysis bags (Mw CO 6275 g/mol). Poly(d,l-lactic-co-glycolic acid) NP were suspended in 1 mL of 23% w/w PLGA-PEG-PLGA aqueous polymer solution and added to dialysis bags. The bags were placed into vials containing IPBS (10 mL) and sodium azide (0.025% w/v) was added to prevent microbial growth. The polymer solution inside the bags gelled at 37 °C over 30–60 s entrapping the suspended NP (Duvvuri et al., Citation2005). The vials were kept in a shaker bath at 37 ± 0.5 °C with a constant agitation of 60 oscillations/min. At predetermined time intervals, 200 µL of sample was withdrawn and replaced with equal volumes of fresh buffer. Samples were analyzed by HPLC. All experiments were carried out in triplicate.
HPLC analytical method
A high-performance liquid chromatography system (Waters 600 pump, Waters, Milford, MA), equipped with a fluorescence detector (HP1100, Hewlett Packard, Waldbronn, Germany) and a reversed-phase C8 column (4 µm, 250 mm × 4.6 mm, Phenomenex, Torrance, CA) were employed for analysis. The detector was used at 16 pmt, at the excitation and emission wavelengths of 265 and 380 nm, respectively. The mobile phase consisted of a mixture of 15 mM phosphate buffer (pH 2.5) and 2.5% acetonitrile pumped at a flow rate of 1 mL/min. Sample injection volume was kept to be 50 µL. The limits of quantification were found to be 50 ng/mL and 1 µg/mL for GCV and prodrugs, respectively.
Drug release mechanism
Drug release parameters were calculated by several mathematical models: zero-order [Equation (Equation5(5)
(5) )], first-order [Equation (Equation6
(6)
(6) )-, Higuchi [Equation (Equation7
(7)
(7) )] and Korsmeyer-Peppas [Equation (Equation8
(8)
(8) )] models. Release data were fitted into the model equations in order to delineate drug release mechanism from formulations.
(5)
(5)
(6)
(6)
(7)
(7)
(8)
(8)
Qt, Q0 and Q∞ represent the cumulative amount of drug released at time t, initial amount of drug and total amount of drug in dosage form, respectively. K0 is the zero-order release rate constant obtained by plotting Qt against time. K1 represents the first-order release rate constant determined by plotting log(Qt/Q∞) against time. Kh is the Higuchi release rate constant obtained by plotting Qt against the square root of time. Kp denotes the release rate constant of the Korsmeyer-Peppas model and the constant n is the release exponent which characterizes different release mechanisms. The parameters Kp and n can be calculated by plotting log(Qt/Q∞) against logarithm of time.
Statistical analysis of data
All measurements were taken at least in triplicate. Results were expressed as mean ± standard deviation (SD). The significance of results was assessed by the Student’s t-test. The level of significance was taken at p value <0.05.
Results and discussion
Properties of prodrugs-loaded NP
The structure of LLGCV has been confirmed with LC/MS and 1H-NMR as suggested by Majumdar et al. (Citation2005) from our laboratory. The yield, mass and NMR data for LDGCV and DLGCV are as following: L-Val-D-Val-GCV-OH: White solid; Yield 78%; LC/MS (m/z): 454.4; 1H-NMR (CD3OD): δ 0.83–1.05 (m, 12H), 2.01–2.16 (m, 2H), 3.40–3.61 (m, 2H), 3.96–4.38 (m, 5H), 5.50–5.54 (m, 2H), 8.04 (s, 1H); D-Val-L-Val-GCV-OH: White solid; Yield 82%; LC/MS (m/z): 454.4; 1H-NMR (CD3OD): δ 0.83–1.12 (m, 12H),1.93–2.13 (m, 2H), 3.52–3.63 (m, 2H), 3.75–3.77 (m, 1H), 3.95–4.26 (m, 4H), 5.54–5.59 (m, 2H), 8.11 (s, 1H).
A series of hydrophobic PLGA polymers with various lactide/glycolide ratios were incorporated in the preparation of NP. Valine-valine-GCV is highly hydrophilic, and many partition into the aqueous phase during PLGA NP preparation. Nanoparticle entrapment efficiencies of LLGCV, LDGCV and DLGCV prepared by W/O/W double emulsion solvent evaporation were determined with various PLGA polymers (PLGA 65:35, PLGA 75:25 and PLGA 85:15). represents the experimental values of entrapment efficiencies, drug loading, size and PDI of prodrug-loaded PLGA NP with different drug/polymer ratio (1:10, 1:15 and 1:20). The results show good reproducibility of W/O/W double emulsion solvent evaporation method with relative standard deviation (RSD) less than 6%. There was a variation in particle size distribution, where PDI value was greater than 0.005, but a large fraction of particles had a diameter below 280 nm. Entrapment efficiency of VVGCV was significantly higher with PLGA 65:35 and PLGA 75:25 polymers than PLGA 85:15 (). Due to the higher hydrophobicity of PLGA 85:15, PLGA NPs with particle size smaller than 200 nm are considered to be optimal to achieve higher ocular penetration (Baba et al., Citation2011). Nanoparticles with smaller particle size possess higher specific surface area, which may lead to lower encapsulation efficiency. The higher polymer/drug ratio also contributes to higher entrapment efficiency and larger particle size. L-Val-L-Val-GCV loading in all three polymers and DLGCV loading in PLGA 75:25 diminished with increasing polymer percentage. However, LDGCV loading in PLGA 65:35 and PLGA 75:25 and DLGCV loading in PLGA 65:35 and PLGA 85:15 were maximum at drug/polymer ratio 1:15; LDGCV loading in PLGA 85:15 with drug/polymer ratio 1:20 was the highest. LLGCV NP with drug/polymer ratio 1:10 exhibited highest yield, whereas LDGCV NP and DLGCV NP generated optimized yield when drug/polymer ratio was 1:15. Prodrugs LLGCV-, LDGCV- and DLGCV-loaded NP were optimized based on favorable entrapment efficiency, high drug loading, optimal yield, small particle size and low PDI (). The optimized NP for LLGCV, LDGCV and DLGCV display entrapment efficiencies of 38.7, 41.8 and 45.3%, respectively, drug loadings 3.87 ± 0.20%, 2.79 ± 0.13% and 3.02 ± 0.15%; yield 85.2 ± 3.0, 86.9 ± 4.6 and 76.9 ± 2.1, particle sizes 116.6 ± 4.5, 143.0 ± 3.8 and 134.1 ± 5.2 nm (). PLGA 75:25 was related for LLGCV and DLGCV, and PLGA 65:35 was considered to be most suitable for LDGCV on the basis of optimization.
Figure 1. Particle size distribution curves and scanning electron photographs of NP. (A–B) LLGCV-loaded PLGA 75:25 NP, (C–D) LDGCV-loaded PLGA 65:35 NP, and (E–F) DLGCV-loaded PLGA 75:25 NP.
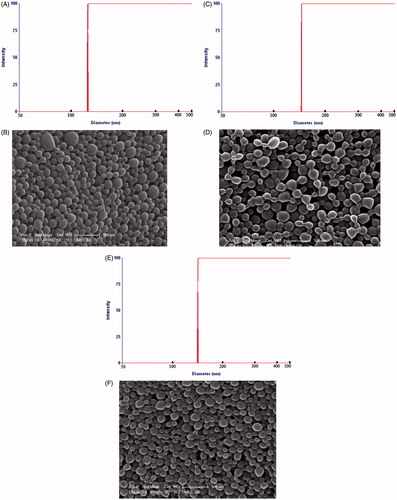
Table 1. Physicochemical characteristics of drug-loaded nanoparticles using various grades of PLGA.
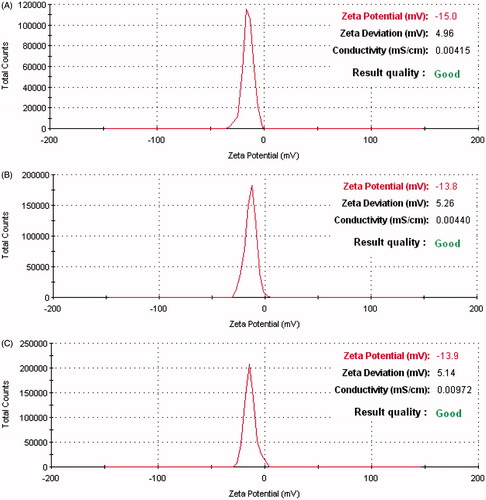
Scanning electron microscope (SEM) revealed the optimized NP to be of spherical shape with uniform, smooth surface (). All the optimized NP exhibited uniform size distribution with low polydispersity and negative zeta potential around −15.0 ± 4.96, −13.8 ± 5.26 and −13.9 ± 5.14 mV, respectively (). Such surface negative charge may be attributed to the presence of ionized carboxyl groups of PLGA polymer. It is known that topical administration of PLGA NP with a negative charge range of −10 to −25 mV indicates incipient instability of formulation. Incorporation of thermosensitive gel in this formulation may reduce the probability of particle aggregation due to its semisolid state (Zhao et al., Citation2012).
Figure 2. Zeta potential distribution of NP. (A) LLGCV-loaded PLGA 75:25 NP, (B) LDGCV-loaded PLGA 65:35 NP, and (C) DLGCV-loaded PLGA 75:25 NP.
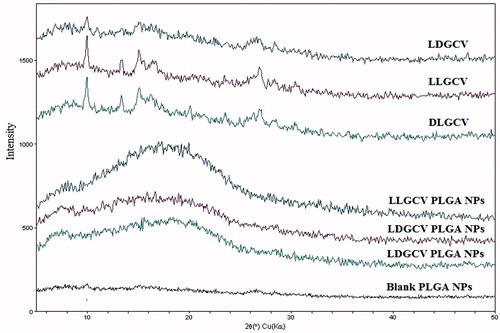
Glass transition temperatures (Tg) by DSC thermograms for VVGCV-loaded NP was considerably higher compared to blank NP and polymers (). An increase in Tg may be a result of rapid solidification of the polymer and strong binding between PLGA and drug molecules in NP. PLGA 65:35 and PLGA 75:25 polymers display the glass transition temperature (Tg) values of 43.8 and 44.5 °C, respectively (Lee et al., Citation2007). An increase in the Tg values of blank NP (48.06 °C for PLGA 65:35 NP; 50.72 °C for PLGA 75:25 NP) was observed relative to the PLGA polymers (). It may be due to rapid solidification of polymer during the evaporation step of preparation process. However, DSC thermogram of prodrugs did not show any endothermic peak because of the amorphous nature of the prodrugs. It may also be one of the reasons for the higher aqueous solubility of prodrugs and higher efficiency in treating viral infections. Furthermore, DSC thermogram of prodrug-loaded NP exhibited slight Tg shift to higher values (LDGCV-PLGA 65:35 NP: 46.17 °C; LLGCV-PLGA 75:25 NP: 51.46 °C; DLGCV-NP: 49.02 °C). Similar glass transitions of blank NP and loaded NP suggest that the PLGA polymer remained unaffected during encapsulation. No prodrug endothermic peak was observed in prodrug-loaded NP, which indicates that prodrugs may be in amorphous or disordered crystalline form. XRD studies of prodrug-loaded NP displayed no diffraction peaks (). These studies were conducted to delineate the crystalline or amorphous nature of prodrugs in the polymeric NP. Prodrugs exhibited a non-characteristic peak on the X-ray diffractogram. X-ray of blank PLGA NP illustrated no diffraction peaks, indicating non-crystalline polymeric NP. Similarly, XRD diffractogram of prodrug-loaded NP suggested a lack of crystallinity, indicating existence of prodrugs in amorphous form or molecular dispersion. Both DSC and X-ray studies confirmed the amorphous form of prodrug in the formulation.
Figure 3. DCS thermograms of (A) blank PLGA 65:35 NP, (B) LDGCV-loaded PLGA 65:35 NP, (C) blank PLGA 75:25 NP, (D) LLGCV-loaded PLGA 75:25 NP, and (E) DLGCV-loaded PLGA 75:25 NP.
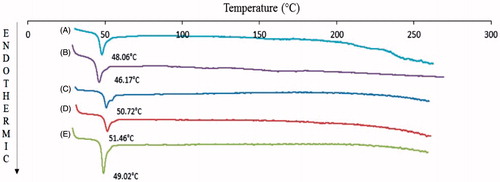
Figure 4. X-ray diffractograms of prodrugs, blank PLGA NP and prodrugs-loaded PLGA NP.
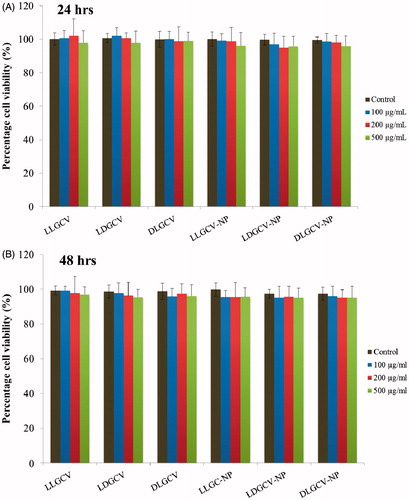
Prodrugs and prodrug-loaded NP did not show any significant cytotoxicity at 100, 200 and 500 µg/mL after 24 and 48 h, suggesting that compounds and formulations are safe and non-toxic in HCEC cell lines ().
Figure 5. MTS assay of HCEC cells after treated with prodrugs of GCV and prodrug-loaded NP with various concentrations. (A) HCEC cells at 24 h, and (B) HCEC cells at 48 h. Each data point is expressed as mean ± SD (n = 3).
In vitro drug release from NP suspended in buffer and thermosensitive gel
In vitro drug release from the pure prodrug occurs within 2 h (), but it is sustained for up to 14–17 days from prodrug-loaded NP (). A biphasic release pattern is observed in all release profiles. An initial rapid phase (burst release) with about 69.1% LLGCV, 72.5% LDGCV and 66.4% DLGCV release in this duration is noted after three days. One of the reasons for the initial burst release phase from NP may be due to the presence of hydrophilic active ingredients in amorphous state. Water can penetrate more easily through the amorphous matrix consisting of polymer and prodrugs. The second phase was a sustained release phase with the diffusion of prodrugs from the matrix or the polymer degradation. A cumulative percentage release of prodrugs from the PLGA NP was over 97% for 14–17 days. Moreover, raising lactide content of PLGA polymers leads to an increase in lipophilicity of polymer and a slower drug release rate (Mu & Feng, Citation2003). Higher polymer hydrophobicity in turn diminishes the rate of water penetration into polymer matrices. The release rate was higher in the case of formulation prepared from PLGA 65:35 relative to PLGA 75:25.
Figure 6. Percent cumulative release profiles of steroid from dialysis bags only and thermosensitive gel in dialysis bags. Each data point is expressed as mean ± SD (n = 3).
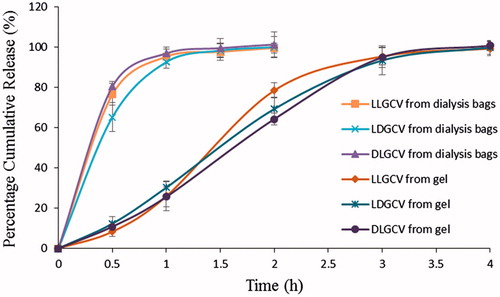
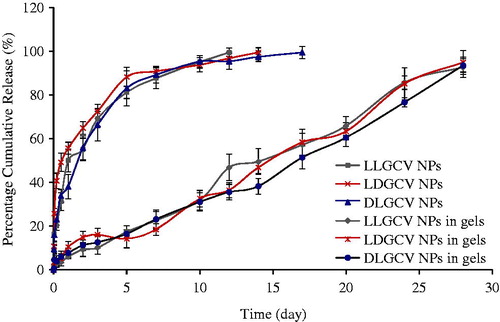
Figure 7. Percent cumulative release profiles of steroids from NP and NP suspended in thermosensitive gels. Each data point is expressed as mean ± SD (n = 3).
In vitro release data from NP were fitted to first order and Higuchi model to describe the pattern of drug release. The first-order release rate is dependent on concentration. The Higuchi model describes the release of drugs from an insoluble matrix based on Fickian diffusion. The release rate constants along with regression values (R2) are shown in . The regression coefficients with p value <0.05 are highly significant. The optimum model was selected based on the correlation value of various models. The Higuchi model showed higher correlation and linearity than first-order model. The Higuchi equation indicated the release rate constants of LLGCV, LDGCV and DLGCV from NP as 5.60, 4.47 and 5.18 h−1/2, respectively. To determine the mechanism of drug release, the data were fitted into the Korsmeyer-Peppas equation and high linearity was observed. The diffusion exponent (n value) was found to be 0.366, 0.290 and 0.356, respectively, for LLGCV, LDGCV and DLGCV. The exponent indicated that the release mechanism from PLGA NP was Fickian in nature, as the n value was less than 0.43 for spherical shape (Peppas, Citation1985; Lopez-Gasco et al., Citation2011). It can be concluded that the release patterns of LLGCV, LDGCV and DLGCV from PLGA NP followed the Higuchi model and Fickian diffusion mechanism.
Table 2. Kinetic parameters of dissolution profile of nanoparticles and formulations.
Dispersion of NP in thermosensitive gel can be an effective alternative to retard prodrug diffusion into release buffer. Profiles of three prodrug release from the formulations prepared by dispersing PLGA NP in thermosensitive gel also followed similar kinetics. Synthesis, characterization and role of triblock copolymer PLGA-PEG-PLGA was investigated in our laboratory. Since the temperature inside the eye ranges from 34 °C to 37 °C, a 23% w/w solution was employed as a vehicle for suspending PLGA NP. Ocular administration of such nanoparticulate formulations can result in spontaneous gelation at ocular surface temperature. Prodrug release studies from the gel revealed that the gel structure may not control prodrug release in vitro. The gel alone is not effective for sustained delivery of prodrugs. When prodrug (500 µg) alone was dispersed in the thermosensitive gel, it is completely released in about 4 h suggesting negligible resistance of gel structure on prodrug diffusion (). However, NP dispersed in gels produce none or minimal burst release of prodrugs. An elimination of such initial burst and slower long-term release from PLGA NP suspended in gels may be due to diffusional resistance of the gel in semisolid nature, and/or polymeric adhesion of gel. The thickness of diffusion boundary layer arises before the parent molecules are released into the aqueous buffer. After the first week, a fraction of NP, within the gel, is released into the medium due to a gradual breakdown of polymeric structure over time. The formulation of NP in thermosensitive gels indicates a linear zero-order release behavior over 28 days relative to biphasic release in the absence of gel (). The release rates are independent of drug concentration. Release rate constants from NP suspended in gels are observed to be 0.538, 0.559 and 0.570 μg/h for LLGCV, LDGCV and DLGCV, respectively (). The regression coefficients with p value <0.05 are highly significant. The zero-order model exhibited very high regression values (R2 > 0.95). An association of PLGA blocks in the tri-block gel with PLGA NP may exert some effect to prevent burst release. Therefore, dispersion of NP inside thermosensitive gels may be an effective alternative as controlled release ocular prodrug/drug delivery systems.
The Korsmeyer–Peppas equation was also applied to determine the mechanism of drug release from the formulations of NP suspended in gels. The n values were calculated to be 0.631, 0.522 and 0.565, which indicated the non-Fickian (anomalous) release for all three formulations due to 0.43 < n < 0.89 (Duvvuri et al., Citation2005). Non-Fickian release refers to the combination of both diffusion and erosion mechanisms of controlled release. The Korsmeyer-Peppas model also showed high correlation for all formulations (). From these results, we concluded that drug release from NP suspended in gels appears to be governed by two phenomena: erosion based degradation and drug diffusion (non-Fickian diffusion) compared to the drug release from NP following Fickian drug diffusion mechanism. Nanoparticles suspended in PLGA-PEG-PLGA thermosensitive gels may result in longer residence time at the site administration.
Conclusions
The prepared NPs possess small particle size, good entrapment and reasonable loading of prodrugs causing sustained release. The application of thermosensitive gel may eliminate the burst release phase and extend the release rate of prodrug from NP. Moreover, these formulations may form a depot in the cul-de-sac to provide a robust concentration gradient for controlled release and efficient permeation of prodrugs across the cornea following topical administration. Further research is required to elucidate the exact mechanism involved in retarding prodrug release from PLGA NP suspended in gels. In vivo pharmacokinetic studies of aqueous humor concentrations of these formulations following topical administration will be reported in the future.
Acknowledgements
We sincerely thank Dr James B. Murowchick from the Department of Geosciences at University of Missouri-Kansas City for his assistance with X-ray pictures of compounds and NP. Authors would also like to thank Dr Piyush Jain for useful discussion.
Declaration of interest
The authors report no declarations of interest.
References
- Ali J, Fazil M, Qumbar M, et al. (2014). Colloidal drug delivery system: amplify the ocular delivery. Drug Deliv 3:1–17
- Baba K, Tanaka Y, Kubota A, et al. (2011). A method for enhancing the ocular penetration of eye drops using nanoparticles of hydrolyzable dye. J Control Release 153:278–87
- Baeyens V, Gurny R. (1997). Chemical and physical parameters of tears relevant for the design of ocular drug delivery formulations. Pharm Acta Helv 72:191–202
- Diebold Y, Calonge M. (2010). Applications of nanoparticles in ophthalmology. Prog Retin Eye Res 29:596–609
- Duvvuri S, Janoria KG, Mitra AK. (2005). Development of a novel formulation containing poly(d,l-lactide-co-glycolide) microspheres dispersed in PLGA-PEG-PLGA gel for sustained delivery of ganciclovir. J Control Release 108:282–93
- El-Samaligy M, Rojanasakul Y, Charlton JF, et al. (1996). Ocular disposition of nanoencapsulated acyclovir and ganciclovir via intravitreal injection in rabbit's eye. Drug Deliv 2:93–7
- Gupta H, Aqil M, Khar RK, et al. (2013). Nanoparticles laden in situ gel of levofloxacin for enhanced ocular retention. Drug Deliv 20:306–9
- Kansara V, Hao Y, Mitra AK. (2007). Dipeptide monoester ganciclovir prodrugs for transscleral drug delivery: targeting the oligopeptide transporter on rabbit retina. J Ocul Pharmacol Ther 23:321–34
- Kennedy DP, Clement C, Arceneaux RL, et al. (2011). Ocular herpes simplex virus type 1: is the cornea a reservoir for viral latency or a fast pit stop? Cornea 30:251–9
- Lee LY, Wang CH, Smith KA. (2007). Micro-porous paclitaxel-loaded PLGA foams – a new implant material for controlled release of chemotapeutic agents. Cambridge, MA: Massachusetts Institute of Technology
- Liesegang TJ. (1988). Ocular herpes simplex infection: pathogenesis and current therapy. Mayo Clin Proc 63:1092–105
- Lopez-Gasco P, Iglesias I, Benedi J, et al. (2011). Paclitaxel-loaded polyester nanoparticles prepared by spray-drying technology: in vitro bioactivity evaluation. J Microencapsul 28:417–29
- Majumdar S, Macha S, Nashed Y, et al. (2004). Expression of peptide transporters on rabbit retina: a strategy to improve retinal delivery of ganciclovir. Lett Drug Des Discov 1:73–7
- Majumdar S, Nashed YE, Patel K, et al. (2005). Dipeptide monoester ganciclovir prodrugs for treating HSV-1-induced corneal epithelial and stromal keratitis: in vitro and in vivo evaluations. J Ocul Pharmacol Ther 21:463–74
- Majumdar S, Tirucherai GS, Pal D, et al. (2003). Functional differences in nucleoside and nucleobase transporters expressed on the rabbit corneal epithelial cell line (SIRC) and isolated rabbit cornea. AAPS PharmSci 5:72–85
- Matthews T, Boehme R. (1988). Antiviral activity and mechanism of action of ganciclovir. Rev Infect Dis 10:S490–4
- Mu L, Feng SS. (2003). A novel controlled release formulation for the anticancer drug paclitaxel (Taxol): PLGA nanoparticles containing vitamin E TPGS. J Control Release 86:33–48
- Ohara PT, Chin MS, LaVail JH. (2000). The spread of herpes simplex virus type 1 from trigeminal neurons to the murine cornea: an immunoelectron microscopy study. J Virol 74:4776–86
- Peppas NA. (1985). Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv 60:110–11
- Perng GC, Jones C. (2010). Towards an understanding of the herpes simplex virus type 1 latency-reactivation cycle. Interdiscip Perspect Infect Dis, 2010:1–18. Article ID 262415
- Plourde R, Merwin J, Ernst MF, et al. (1995). Acyclovir–glycoprotein conjugates are potent inhibitors of hepatitis B virus replication. Drug Deliv 2:136–43
- Ramyadevi D, Sandhya P. (2014). Dual sustained release delivery system for multiple route therapy of an antiviral drug. Drug Deliv 21:276–92
- Sood N, Bhardwaj A, Mehta S, et al. (2014). Stimuli-responsive hydrogels in drug delivery and tissue engineering. Drug Deliv 21:1–23
- Suresh PS, Tullo AB. (1999). Herpes simplex keratitis. Indian J Ophthalmol 47:155–65
- Wilhelmus KR. (2000). The treatment of herpes simplex virus epithelial keratitis. Trans Am Ophthalmol Soc 98:505–32
- Xu F, Sternberg MR, Kottiri BJ, et al. (2006). Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA 296:964–73
- Zhao C, Yuan G, Han CC. (2012). Stabilization, aggregation, and gelation of microsphere induced by thermosensitive microgel. Macromolecules 45:9468–74