
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Sirolimus is recognized as a P-glycoprotein (P-gp) substrate with poor water-solubility. To improve its solubility and bioabsorption, self-microemulsifying drug delivery systems (SMEDDS) containing a novel P-gp inhibitor, honokiol, were prepared. The aim of this work was to evaluate the enhanced transport of sirolimus SMEDDS as well as the roles of honokiol. In situ single-pass intestinal perfusion and in vitro human colon adenocarcinoma (Caco-2) cell models were applied to study the effects of honokiol within SMEDDS on the transport of sirolimus. The results indicated that a combination of honokiol with sirolimus in SMEDDS did not significantly alter the particle size, polydispersity index and release of drugs. In addition, the absorption rate constant (Ka) as well as the effective permeability coefficients (Peff) of sirolimus in situ intestinal absorption, and the apparent permeability coefficients (Papp) of sirolimus in caco-2 cells were significantly enhanced by cremophor EL-based SMEDDS with honokiol as compared with those of SMEDDS without honokiol. Rhodamine123 uptake rate in caco-2 cells and in vitro cytotoxicity of sirolimus were enhanced by honokiol in SMEDDS indicating a substantial P-gp inhibition of honokiol. In conclusion, coadministration of honokiol with poor soluble P-gp substrate in SMEDDS, could serve as a favorable approach for oral delivery.
Introduction
Self-microemulsifying drug delivery system (SMEDDS) (Dokania & Joshi, Citation2014) is one of the most effective and favorite routes of drug administration usually adopted for active components with low bioavailability and a narrow therapeutic index. The advances in these avant-garde drug delivery systems are increasing solubility of hydrophobic drugs, ameliorating permeability or transport of poorly pervious drugs, adjusting biodistribution and metabolism of drugs, avoiding acid hydrolysis and enzymatic degradation of drugs in physiological milieu, and improving targeted delivery of the drugs to the onset position, which makes SMEDDS ideal carriers for the purpose of parenteral administration (Sun et al., Citation2011).
However, oral delivery of SMEDDS is a great challenge for several drugs with special physicochemical properties attributed to not only low water solubility and/or poor intestinal permeability, but also high level of P-glycoprotein (P-gp) efflux. P-gp is a large plasma membrane protein of the ATP-binding cassette (ABC) family of proteins. As an energy-dependent transporter or efflux pump, P-gp can decrease the intracellular accumulation of drugs by extruding exogenous substance from the cell, which causes lower oral bioavailability of drugs (Di et al., Citation2008). Therefore, transmembrane efflux of drugs may be tackled by appropriate P-gp modulators or inhibitors coadministrated with the drugs.
Honokiol (3′,5-di-(2-propenyl)-1,1′-biphenyl-2,2′-diol) is a bioactive natural compound obtained from Magnolia spp. (Esumi et al., Citation2004) and a small bi-phenolic lignan () with molecular formula C18H18O2, which has multifunctional pharmacological effects, such as in vitro anti-arrhythmic, anti-angiogenesis, anti-tumor and anti-oxidative activities (Tsai et al., Citation1999; Esumi et al., Citation2004; Ma et al., Citation2011; Zhao & Liu, Citation2011; Avtanski et al., Citation2014; Li et al., Citation2014) in preclinical models. Besides, it has been reported that honokiol can down-regulate the expression of P-gp at protein and mRNA levels in a human breast multi-drug resistant (MDR) cancer cell line (MCF-7/ADR), leading to a partial recovery of the intracellular drug accumulation and of the sensitivity toward antineoplastic drug doxorubicin (Xu et al., Citation2006). However, there has been no investigation to evaluate whether or not honokiol can enhance the intestinal absorption of drugs.
Figure 1. Structure formula of honokiol.
The aim of this study was to investigate coadministration of honokiol with sirolimus (also known as rapamycin), a P-glycoprotein substrate as model, in SMEDDS which would be more convenient than the intravenous dosage form, in an attempt to facilitate the transport of sirolimus in human colon adenocarcinoma (caco-2) cells and to promote the absorption of the drug across the intestinal tract.
Materials and methods
Materials
Sirolimus (≥99.3%) was purchased from Guangzhou eastbang Pharmaceutical Technology Co., Ltd. (Guangzhou, China). Honokiol was obtained from Xi'an Tian Ben Bio-Engineering Co., Ltd (Xi'an, China). Cremophor EL was supplied by Yunhong Chemical Co., Ltd. (Shanghai, China) and medium chain triglycerides (MCT) were purchased from Tieling Beiya Pharmaceutical Oil Co., Ltd (Tieling, China). Minimum essential medium (MEM) was obtained from Gibco, Invitrogen (Ontario, CA). Non-essential amino acid solution, penicillin-streptomycin (10 000 IU/ml penicillin-G and 10 mg/ml streptomycin), Fetal bovine serum (FBS), PBS and Hank's balance salt solution (HBSS, pH 7.4) were obtained from M&C Gene Technology (Beijing, China). 96-well culture plates and Transwell 12-well plate (12 mm, 3.0 μm pore size inserts, Costar3401) were purchased from Costar (Corning Incorporated, Tewksbury, MA). SulforhodamineB (SRB) was obtained from Sigma–Aldrich (St. Louis, MO). Rhodamine123 (R123) was purchased from J&K Scientific Ltd. (Beijing, China).
Animals
Healthy male Sprague–Dawley rats, weighing 230–250 g, were provided by Experimental Animal Center of Peking University. All animal procedures were approved by the Institutional Authority for Laboratory Animal Care of Peking University. Rats, housed under normal laboratory conditions at 24 °C, were fed a standard chow diet and tap water ad libitum for at least one week before the experiments.
Formulation and preparation of sirolimus SMEDDS
Several formulations of SMEDDS () were prepared containing a fixed proportion of sirolimus (2 mg/g) and different proportion of honokiol (0, 0.4, 1.1 and 2.2 mg/g) dissolved in a mixture of vehicles composed of cremophor EL (emulsifier), propylene glycol (PG, co-emulsifier) and MCT as oil phase (9:2:9, w/w/w) (Li et al., Citation2010). These components were accurately weighed and mixed using a magnetic stirrer until a clear solution was obtained. All of the liquid formulations were stored in air-tight glass containers at 4 °C until required for use as below.
Table 1. Composition (mg/g) of SMEDDS formulations.
Particle size and polydispersity index
The measurement of particle size/distribution, determined using Zetasizer (ZEN 3600, Malvern Instrument, Malvern, UK) was performed with a scattering angle of 90 ° at 25 °C after diluting the SMEDDS solution to an appropriate volume with water. Each sample was analyzed in triplicate.
In vitro release
To compare the release behaviors, sirolimus SMEDDS with different amount of honokiol and honokiol SMEDDS were tested using a USP30 Dissolution apparatus II with paddle (TIANDA TIANFA-pharmaceutical testing instrument manufacturer, Tianjin, China). The samples were filled in size No. 0 hard gelatin capsules. A total of 900 ml of simulated gastric fluid (SGF, pH 1.2) and simulated intestinal fluid (SIF, pH 6.8) were used as the media and were controlled at 37 ± 0.5 °C. During the release studies, the paddles were rotated at 50 rpm. A total of 1 ml of sample of release medium was withdrawn and replaced with fresh media after 5, 10, 15, 20, 25, 30, 45, 60, 90 and 120 min. Samples were filtered using a 0.45 μm filter and analyzed using an HPLC assay as described in the following section.
HPLC analysis and analytical method validation
The high-pressure liquid chromatography (HPLC) system used for this study was a Waters Series System, which consisted of a E2695 pump, a 2998 DAD detector, a column heater set at 40 °C, a model auto-sampler with injection volume at 20 μl, and a model Empower 3 Personal Single System software (Waters Corporation, Milford, MA). The mobile phase (acetonitrile/water, 4:1, v/v) was online mixed and pumped by a pump at a flow rate of 1.0 ml/min. Sirolimus and honokiol were separated by a ODS-C18 column (Diamonsil, 4.6 mm × 250 mm, 5 μm; Dikma, Beijing, China) and detected at 272 nm.
To validate the assay specificity, the chromatogram of a blank SMEDDS, perfusion medium and transport buffer were compared with that of sirolimus and honokiol. Linearity was assessed using a calibration curve for sirolimus and honokiol over the concentration range from 2 μg/ml to 200 μg/ml. Moreover, precision, sensitivity of detection and accuracy (% bias) were evaluated.
In vitro permeability study by using caco-2 cells
Caco-2 cells were cultured in MEM culture medium supplemented with 10% fetal bovine serum, 1% non-essential amino acid, 1% penicillin-streptomycin, 1% l-glutamine and 1% of HBSS at 37 ± 2 °C in a humidified atmosphere of 95% O2/5% CO2. The cells were passaged using trypsin–EDTA after reaching 80% confluency, and plated in a 75 cm2 tissue culture flask.
At first, caco-2 cells (passage number 27-35) were seeded on the apical site of Transwell tissue culture plate at a density of 1 × 105 cells/plate for 0.5 ml. The medium was changed within 24 h to remove any non-adherent cells. Cell monolayer with trans-epithelial electrical resistance (TEER) values greater than 500 Ω/cm2, as measured by an electrical resistance system and a MERSSTX01 electrode (Millicell ERS-2, Millipore, Billerica, MA), was used for the transport study 21–28 days after seeding (Zhang et al., Citation2010).
For the apical to basolateral (A to B) transport study, the cell monolayer was pre-incubated at both the apical (0.5 ml) and basolateral (1.5 ml) sites with Hank's balanced salts solution (HBSS) at 37 ± 2 °C for 30 min. At the zero time point, HBSS was replaced by the same volume of sirolimus microemulsion (20 μg/ml) with honokiol (0, 4, 11 and 22 μg/ml), atenolol (1 mmol/l) and propranolol (0.1 mmol/l) dissolved or diluted in HBSS, respectively. At 0, 30, 60, 90, 120 and 150 min, the sirolimus content in medium from the basolateral site was analyzed by HPLC. Similarly, for basolateral to apical (B to A) transport study, HBSS was added to pre-treat the cell monolayer for 30 min at 37 °C. After rinsing the monolayer with HBSS, Transwell inserts with 0.5 ml of HBSS at the apical side were inserted into the basolateral side containing 1.5 ml of above-mentioned sirolimus microemulsion. 200 μl of medium in the apical side was taken and substituted with the fresh medium at predetermined time intervals. Finally, it was determined by HPLC analysis as above.
In situ perfusion
In situ single-pass intestinal perfusion was carried out as previously reported [21] with some modifications. The sample groups received sirolimus SMEDDS with or without honokiol that was diluted with Krebs–Ringer buffer to a final sirolimus concentration of 20 μg/ml just before use. Rats, segregated into four groups (3/group), were anesthetized by an intraperitoneal injection of 1.5 g/kg of urethane. Cannulation was made at jejunum with polyethylene tubing at the proximal end for infusion of superfusion fluid, and tail end for collection of effluent liquid. Polyethylene tubing was then connected to the inlet cannula, and a perfusion pump (BT100-1L, Baoding Longer Precision Pump Co., Ltd, Baoding, China) was placed between the perfusate reservoir and the inlet cannula. The experiment started by delivering the perfusion solution containing sirolimus microemulsion dilution at a flow rate of 0.20 ml/min to the tubes and the intestinal segment. After a stabilization period of 30 min to remove the intestinal content until the outlet solution appeared clear, intestinal effluent was collected in pre-weighed 1.5-ml vials at 10 min intervals for 120 min. Samples were weighed and centrifuged (10 000 rpm for 10 min), and the supernatant was stored at −20 °C until analysis by HPLC. The length of the intestinal segment was measured at the end of the experiment, and finally mercy killing of the animals was exercised.
In vitro cytotoxicity
The cytotoxic activity was assessed in vitro using the SRB colorimetric assay. Caco-2 cells were inoculated in 96-well culture plates (2 × 104 cells/well) for 48 h allowing close attachment of the cell to the wall of the plate prior to treatment with the sample groups. Free sirolimus and free honokiol were dissolved in ethanol and diluted with PBS to the appropriate volume, respectively. The sirolimus SMEDDS, with or without honokiol, was diluted with PBS to form microemulsion with graded concentrations of sirolimus that were added into the 96-well plates containing the caco-2 cells for 4 and 24 h, respectively. The cells were fixed with ice-cold trichloroacetic acid (TCA) for 1 h at 4 °C. Then, the culture plates were washed by distilled water five times and dried by airing. Fifty microliter of 0.4% (w/v) SRB solution was added into each well allowed staining for 30 min at ambient temperature, and afterwards removed by washing the plates rapidly with 1% v/v acetic acid, five times, in order to get rid of unreactive dye. After drying in the air, the bound SRB in each well was solubilized by adding 200 μl of 10 mM unbuffered Tris base (pH 10.5), and then the plates were shook for 5 min on a shaker platform. Finally, the plates were read in a 96-well plate reader at 540 nm (Houghton et al., Citation2007). Cells devoid of any sample were used as control.
Evaluation of cellular uptake by flow cytometry
Caco-2 cells were seeded into six-well plates at a density of 4.0 × 105 cells/well in 4 ml of MEM containing 10% FBS, and then cultured in 5% CO2 atmosphere at 37 °C for one day. After that, the cells were incubated with honokiol microemulsion at a final concentration of 2.5 μg/ml of R123 in MEM for 4 h at 37 °C. After the culture medium was removed, the cells were washed three times with PBS to remove microemulsions that were not internalized by the cells. The cells were detached using trypsin for 5 min, and then terminated by MEM. About 1 ml of PBS was added to each well and the cell suspensions were centrifuged at 1500 rpm for 5 min. After removal of the supernatants, the cells were resuspended in 1 ml of PBS. The suspensions were centrifuged again, and then the cells were suspended in 400 μl of PBS, followed by filtration through a 40 μm nylon mesh to remove cell aggregates before measurements were made. The mean fluorescence intensity of R123 in cells was analyzed by FCM (Epics XL, Beckman Coulter, Inc., Kraemer Boulevard Brea, CA) at an excitation wavelength of 485 nm and an emission wavelength of 525 nm. Results were expressed as the ratio of the control value for R123 accumulation.
Data analysis
The absorption rate constants (Ka) and effective permeability coefficients (Peff) of sirolimus across rat intestine were calculated based on the disappearance of the drug in perfusate using the following equations (Cummins et al., Citation2003; Zhang et al., Citation2014).
where Q is the flow rate of the drug through the intestine (0.2 ml/min), r is the radius of the rat intestine, and L is the length of intestinal segment perfused after completion of the perfusion experiment. Cin, Cout, Vin and Vout are the drug concentration (μg/ml) and volume (ml) in the inlet of the perfusate entering the intestinal segment and the exiting solution, respectively.
Apparent permeability coefficients (Papp, cm/s) of sirolimus across caco-2 monolayer were calculated according to the following equation:
where dQ/dt (μg/s) is the drug permeation rate, A is the surface area of the membrane filter (1.13 cm2), and C0 (μg/ml) is the initial sirolimus concentration in the donor compartment at t = 0 min.
Cell viability of each group was expressed as % cell viability using the following formula:
where Atested is the absorbance of cells treated with various samples, and Acontrol is the absorbance of control at 540 nm.
All results were expressed as mean ± SD. Student's t-test was employed to evaluate statistically significant difference between two groups. Values of p < 0.05 was considered statistically significant for all tests.
Results
HPLC method validation
Sirolimus and honokiol were eluted at the desirable retention time of 8.0 and 4.4 min, respectively, and well separated from interfering components in blank SMEDDS, perfusion medium and transport buffer as shown in supplementary materials. Besides, over the concentration range of 2–200 μg/ml, a good linearity was achieved with a correlation coefficient of 0.9997 and 0.9998 for sirolimus and honokiol separately. There were no clear decrease in chromatographic area of sirolimus and honokiol in perfusion medium and transport buffer at 37 °C for more than 12 h. The intra- and inter-day precision, as well as accuracy values for the detection of drugs were all below 2%. The limit of quantitation (LOQ) value for sirolimus and honokiol were 0.8 ng and 1.0 ng, respectively. The accuracy of the method was verified with recovery values of 98–102%. Therefore, this analytical method was valid in terms of specificity, linearity, precision and accuracy.
Particle size analysis
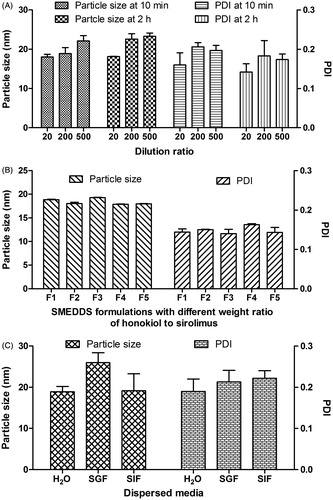
In our study, several influencing factors of particle size, such as dilution ratio and storage time of diluted solution (microemulsions), drug weight ratio of honokiol to sirolimus, and different dispersed media, were investigated. At first, the effect of dilution ratio and storage time on particle size and PDI of sirolimus SMEDDS with honokiol (formulation F3 as listed in ) in distilled water is shown in . When the dilution ratio was ranged from 1:20 to 1:500, the particle size seemed to be increased slightly from ∼18 nm to ∼23 nm, and the range of PDI was 0.14-0.20. Moreover, there was no remarkable increase in particle size and PDI of reconstituted microemulsions diluted by distilled water at 10 min and 2 h. The results demonstrated that dilution volume and period took no effect on particle size and PDI of reconstituted microemulsions. According to the result, reconstituted microemulsions diluted by 200-fold volume of medium were investigated as below. Second, the effect of drug weight ratio on particle size is shown in . As the honokiol loading increased from 0 to 2.2 mg/g in SMEDDS (formulation F1-F5), the particle size of reconstituted microemulsions diluted by distilled water remained almost unchanged indicating that the amount of honokiol in formulation had no obvious effect on particle size of SMEDDS. Third, the effect of medium on particle size was also investigated in our study as shown in . When the SMEDDS dispersed in distilled water, SIF and SGF for 2 h, the resulted particle size was 18.9 ± 1.3 nm, 26.0 ± 2.4 nm and 19.2 ± 4.1 nm, respectively. Additionally, the results as shown in demonstrated that dilution methods had little influence on particle size for all of the SMEDDS. Sirolimus microemulsions in the absence or presence of honokiol were thermodynamically stable dispersions with lower particle size below 30 nm and narrow distribution. Therefore, the optimal formulation of SMEDDS without drug, i.e. PG of 45% (v/v), MCT of 10% (v/v) and cremophor EL of 45% (v/v), was selected for the following study.
Figure 2. The particle size and polydispersity index (PDI) of different SMEDDS formulation diluted by dispersed media (n = 3). SGF: simulated gastric fluid; SIF: simulated intestinal fluid.
In vitro release
An important observation on in vitro release was made as shown in , when cremophor EL, MCT and PG were used in combination, this system was able to effectively provide quick release of sirolimus, while little sirolimus was released (below 5%) from drug powder due to a lack of solubilizer in the release medium indicating unsatisfied sink condition. Further, the SMEDDS significantly increased the release of sirolimus providing constant release up to 120 min in different media, when compared to unformulated sirolimus.
Figure 3. The release profiles of sirolimus and honokiol SMEDDS formulation (F1-F5) in distilled water, simulated gastric fluid (SGF, pH 1.2) and/or simulated intestinal fluid (SIF, pH 6.8) as compared with those of sirolimus powder. (n = 3).
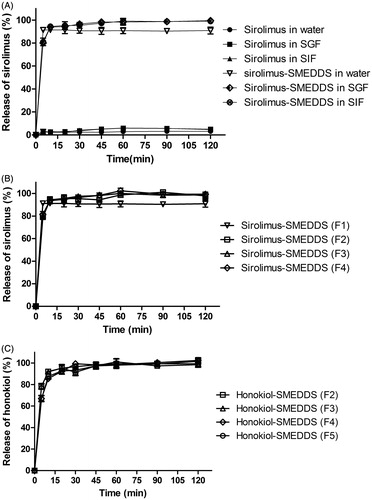
The release profiles of sirolimus and honokiol from SMEDDS in water were compared among the tested formulations (formulation F1-F5) described in . Also, the release of honokiol from microemulsions was investigated. As shown in , there were no significant differences in release of sirolimus among the sirolimus SMEDDS formulations indicating that the amount of honokiol in SMEDDS had little impact on the release of sirolimus from reconstituted microemulsions. Similarly, the release percentage of honokiol for microemulsions (formulation F2-F5) was almost 90% within 10 min demonstrating that the rapid release of honokiol was independent of the amount of drug itself in the formulation.
In vitro permeability study
The caco-2 cell permeability of sirolimus SMEDDS is shown in . Atenolol and propranolol, used as low and high permeability controls, had Papp values of 0.48 ± 0.02 and 13.05 ± 0.30 (×10−6 cm/s), respectively. Importantly, it was shown that the Papp of the microemulsions with different amount of honokiol were significantly higher than that of microemulsion without honokiol in the A to B transport (p < 0.001). In addition, the A to B transport of sirolimus from each formulation was in the following order: sirolimus (20 μg/ml) microemulsion with honokiol (22 μg/ml) > sirolimus (20 μg/ml) microemulsion with honokiol (11 μg/ml) > sirolimus (20 μg/ml) microemulsion with honokiol (4 μg/ml) indicating a clear dose-dependent manner. Similarly, the efflux ratio, calculated with Papp(B-A)/Papp(A-B), remarkably decreased with the increased amount of honokiol in the microemulsion. In particular, the efflux ratio of sirolimus from microemulsion containing the highest amount of honokiol (22 μg/ml) was around 35 times lower than that from microemulsion without honokiol (0.074 versus 2.62). The results demonstrated that the transport of sirolimus through caco-2 cell monolayer was improved by introduction of honokiol into the microemulsion formulation.
Table 2. Apparent permeability coefficients (Papp) of sirolimus across caco-2 cell monolayer from each formulation.
In situ perfusion
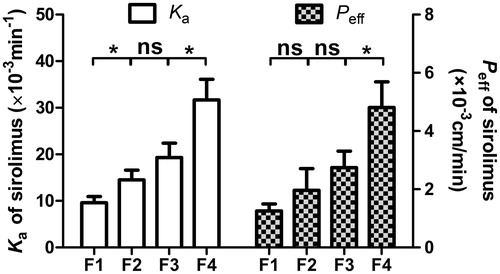
In the intestinal absorption studies, the absorption characteristics of the reconstituted microemulsion containing sirolimus and honokiol were assessed in jejunum segment. The absorption rate constants (Ka) and effective permeability coefficients (Peff) obtained in the single-pass intestinal perfusion (SPIP) model are presented in . The results indicated that the Ka and Peff of sirolimus appeared to be high, predicting clear intestinal absorption in the jejunum perfusion. In addition, when the honokiol content in formulation increased, the Ka and Peff values presented an incremental increase although there was not always a significant difference (p < 0.05) in above values between any of the two adjacent groups. The results that the absorption increased gradually with the enhancement of honokiol content in formulation, well correlated with the results of Papp in caco-2 cells transport experiments as shown in , indicated that the transport of sirolimus via intestine could be stimulated by honokiol in a dose-dependent manner. It has been reported that P-gp protein expression showed an overall increase in rat and human small intestine (Dahan & Amidon, Citation2009). So the results might attribute the significant change of P-gp inhibition by honokiol causing the remarkable increase in sirolimus permeability. Although the mechanisms need to be further elucidated, the results still indicated that honokiol played an indelible role in the improved absorption of sirolimus formulated in SMEDDS in intestine.
Figure 4. The Ka and Peff obtained for in situ perfusion in the single-pass intestinal perfusion model in jejunum segment. The data are presented as the mean ± SD (n = 3).
In vitro cytotoxicity
The dose−response studies against sirolimus and honokiol as a single agent and in combination on caco-2 cells for 24 h are shown in and . The IC50 for free sirolimus and blank SMEDDS was 35.2 and 49.5 μg/ml (the concentration of cremophor EL), respectively. A 6.7-fold reduction in IC50 was noted when the sirolimus formulated in microemulsion compared to free sirolimus in caco-2 cells. Although free honokiol exhibited relatively high IC50 versus free sirolimus, the sirolimus delivered in microemulsions with honokiol to caco-2 cells showed a 9.8-fold lowered concentration in IC50 as compared with those without honokiol. The results demonstrated that the enhancement of cytotoxicity of caco-2 cells was primarily induced by blank SMEDDS or honokiol within SMEDDS.
Figure 5. Toxicity results of various concentration of free sirolimus, free honokiol, blank SMEDDS or sirolimus-SMEDDS with or without honokiol on caco-2 cells after incubation for 24 h. Results were represented as cell viability % ± SD (n = 6).
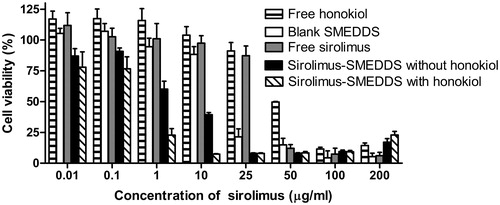
Table 3. IC50 of various sirolimus formulations against caco-2 cells (n = 6).
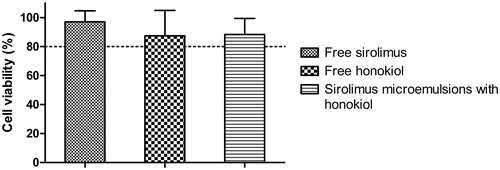
In addition, the viabilities of caco-2 cells incubated with several samples for 4 h were also evaluated as shown in . It was noteworthy that all the viabilities were more than 80% after incubation for 4 h, indicating hardly any toxicity on caco-2 cells for free sirolimus, free honokiol, and microemulsions loading sirolimus and honokiol, whose concentration was below 25 μg/ml. Therefore, the low cytotoxicity of sirolimus formulations ensured reliable results of the transport and the uptake study.
Figure 6. Toxicity results of free sirolimus, sirolimus-SMEDDS with honokiol or free honokiol on caco-2 cells after incubation for 3 h. The final concentrations of sirolimus or honokiol in all dilutions of SMEDDS were 25 μg/ml. Results were represented as cell viability % ± SD (n = 6).
Cellular uptake
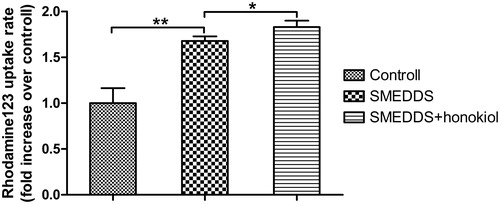
Uptake results of R123 on caco-2 cells are shown in . R123 SMEDDS incubation resulted in higher levels of accumulation of R123 in caco-2 cells than free R123 solution incubation (p < 0.01), suggesting that the SMEDDS were capable of enhancing the uptake rate of R123. In addition, the uptake rates of R123 SMEDDS on caco-2 cells without honokiol were observed to be lower than that of R123 SMEDDS with honokiol (p < 0.05). From the results, it could be inferred that the uptake rates of R123 were increased by the introduction of honokiol in SMEDDS.
Figure 7. Uptake rates of R123 on caco-2 cells incubated with cremophor EL-based SMEDDS without honokiol and SMEDDS with honokiol for 4 h. R123 solution was incubated as a control. Results were represented as fold increase of uptake rate over the corresponding control (n = 3). **p < 0.01; *p < 0.05.
Discussion
Efforts are being put forth to construct and estimate a suitable oral delivery system that obtains several advantages of SMEDDS, such as simple preparation technique, ease of drug dispersion, synchronous solubilization of drugs with varied levels of lipophilicity, improved absorption and bioavailability (Zeng et al., Citation2010), in order to enhance the solubility and absorption for poorly water-soluble sirolimus. Non-ionic surfactants (emulsifier), used in most of SMEDDS, were low-toxic and unaffected by pH and ionic strength of solution. Among them, polyoxyethylene castor oil including cremophor RH and cremophor EL, had relatively larger self-microemulsion region, and the ability to solubilize effectively lipophilic drugs. Therefore, the desirable emulsifier cremophor EL, one of the components of the cyclosporin microemulsion, the ritonavir oral solution, and the ritonavir and lopinavir soft gelatin capsules (Gaylord, Citation1999; Ershov et al., Citation2002; Lei et al.,, Citation2012), was selected in our study. Another, it is considered that oil in the formulation would distinctly dominate the spontaneity of the microemulsified process, droplet size of the microemulsion, drug solubility and biological fate of microemulsions (Bouchemal et al.,, Citation2004; Anton et al., Citation2008; Pouton &Porter, Citation2008). Medium chain triglycerides (MCT) retained greater solubilization capability for hydrophobic drugs and permeation-enhancing properties as compared to long-chain triglycerides (Lundin et al., Citation1997; Pouton & Porter, Citation2008), and thereby MCT was chosen in SMEDDS for sirolimus. Next, the incorporation of the coemulsifiers in SMEDDS was able to expand self-microemulsification region in the phase diagrams. In case of our study, propylene glycol (PG), as a amphiphilic solubilizer often used in the SMEDDS to improve drug loading (Basalious et al., Citation2010; Yoo et al., Citation2010; Mekjaruskul et al., Citation2013; Hintzen et al., Citation2014), was used.
Particle size is a critical factor to evaluate SMEDDS. The smaller the particle size, the larger the superficial area, which surely influenced drug transport accompanied by delivering them to specific sites (Gershanik & Benita, Citation2000; Kang et al., Citation2004). In our study, the results shown in revealed that the formed microemulsions were capable of keeping sirolimus and honokiol solubilized, even if the dilution ratio was as large as 500. Besides, there was no significant difference (p > 0.05) in particle size of microemulsions between SGF and SIF indicating that microemulsion systems might not be affected by alteration of pH or the ionic strength of digestive juice. Importantly, loading of honokiol had no effect on particle size significantly, possibly due to the fact that lipophilic drugs located in the hydrophobic core space of the microemulsions (Mu et al., Citation2005), demonstrating that the optimized SMEDDS kept relatively stable particle size improving release and transport of sirolimus.
Although there were limitations on the traditional in vitro release method, it was still a plain and quick method to carry out, and to measure the concentration of drug dissolved by the SMEDDS formulation. Usually, the release medium contained a percentage of solubilizing agent, such as ethanol, sodium lauryl sulfate or tween to enhance very poorly soluble drug solubility. In case of our study, none of them was added to both media simulating in vivo condition. As shown in , sirolimus SMEDDS rapidly achieved more than 90% drug release within 10 min in water, SGF and SIF, respectively. It could be proposed that all SMEDDS resulted in spontaneous formation of nanosized microemulsions dissolving in the gastrointestinal fluids, which gave faster release of drug into aqueous media compared to plain sirolimus powder (Juliano et al., Citation2005). This solublised form of sirolimus could lead to higher absorption and higher bioavailability. In addition, similarly quick release of honokiol as shown in was a guarantee of fast and efficient action, such as P-gp inhibition of honokiol.
Non-toxicity was a basic principle to study the effects of SMEDDS on drug absorption using cell models due to the widely known fact that several surfactant-comprising delivery carriers had potential toxicity on caco-2 cells (Gursoy et al., Citation2003). Therefore, the toxicities of sirolimus or honokiol SMEDDS on caco-2 cells need to be investigated before the cellular uptake experiments. From the results as shown in , the viabilities of caco-2 cells incubated with free drug or sirolimus microemulsions were higher than 80% after incubation for 4 h, which was generally considered to be non-toxic to the cells (Wahlang et al., Citation2011). It could be attributed to the short time of incubation to caco-2 cells. Therefore, during the transport and uptake experiments, sirolimus SMEDDS did not induce clear toxic effect on cells and the results could be considered reliable.
In order to investigate the ability to deliver sirolimus across the intestinal mucosa of the developed SMEDDS, a caco-2 cell permeability study, commonly used as an in vitro approach to predict in vivo absorption (Yee, Citation1997), was conducted. Meaningfully, about 35-fold decrease in the transport (Efflux ratio) of sirolimus through caco-2 cell monolayer was observed upon co-administration with honokiol in SMEDDS. Similarly, the transport (Ka and Peff) obtained in the SPIP model was remarkably improved by SMEDDS with honokiol. The results demonstrated that honokiol provided the exact and effective promotion for sirolimus in SMEDDS.
Sirolimus is a known substrate for the P-gp highly expressed by caco-2 cells, and the large dose required to exhibit cytotoxicity is attributed to P-gp transporter mediated drug efflux in caco-2 cells. Therefore, cytotoxicity on cells by adequate incubation (24 h), together with uptake of R123, another P-gp substrate, reflected the inhibitory effect for P-gp on caco-2 cells. IC50 (0.54 μg/ml) of sirolimus SMEDDS with honokiol was remarkably lower than that of SMEDDS without honokiol (5.28 μg/ml), which were lower as compared with other control groups as listed in . The enhanced cytotoxicity caused by honokiol in SMEDDS was consistent with the uptake result as shown in . On the other hand, the increase in uptake rates of R123 for honokiol within SMEDDS corresponded to high amount of drug into caco-2 cells indicating more pronounced transport capacity or bioabsorption.
There were several possible explanations about the improvement of transport and absorption of sirolimus by SMEDDS with honokiol. (1) the augmentation of drug solubility and dissolution rate due to nano-size (below 30 nm) microemulsions and a large superficial area for more absorption; (2) oil phase (MCT) in SMEDDS might keep the drug from enzyme degradation in the digestive tract (Gursoy & Benita, Citation2004) and improved transport through the intestinal lymphatic system which was an important absorption pathway for highly lipophilic drugs (Patel et al., Citation2010); (3) the enhancement of the fluidity of the membrane due to permeation by surfactant and/or coemulsifier (Sachan & Kasture, Citation2010). The last but not the least is the inhibition of P-gp expressed in small intestine.
P-glycoprotein, a drug efflux inducing transporter, localized in normal tissues and operated physiologically in the excretion of endogenous metabolites and xenobiotics (Ambudkar et al., Citation1999). It might function as an “absorptive barrier” in the intestine (Lin & Yamazaki, Citation2003), and therefore, it was expected that the bioavailability of P-gp substrate sirolimus could be improved by inhibition of P-gp. In our study, there were two distinct mechanisms of inhibition involved emulsifier (cremophor EL) and honokiol, respectively. First of all, emulsifiers have been widely used in formulations with the intention of enhanced drug absorption. Besides wetting and solubilization, P-gp inhibition capability for emulsifier including fatty acid ester surfactants, such as hydroxyl stearates, e.g. solutol HS 15, cremophor EL, etc., has recently gained extensive support (Rege et al., Citation2002; Collnot et al., Citation2006). Among them, cremophor EL, cremophor RH, etc., gave rise to a significant increase in the uptake of [3H]mitoxantrone in P-gp-expressing cells (Yamagata et al., 2007), acting even at very low concentration (Friche et al., Citation1990). Mechanistically, they altered the membrane fluidity and bound with hydrophobic domain of P-gp thereby transforming its conformation resulting in weaken functionality (Yin et al., Citation2009). Secondly, it is reported that several compounds previously have potency to inhibit P-gp's efflux pump, including some natural products, such as curcuminoids (Anuchapreeda et al., Citation2002; Limtrakul et al., Citation2004), and the calcium channel blockers, such as verapamil (Muller et al., Citation1994; Sonneveld &Wiemer, Citation1997). In case of our study, a newcomer of compounds down-regulating P-gp over-expressed in cells, honokiol, was introduced. Differed from verapamil and curcuminoids (Sonneveld & Wiemer, Citation1997; Anuchapreeda et al., Citation2002), honokiol only functioned as a down-regulator of the expression of P-gp, although the effectiveness of honokiol to inhibit the over-expression of P-gp seemed to be similar to curcuminoids and verapamil (Muller et al., Citation1994; Anuchapreeda et al., Citation2002). The ideal nature of honokiol as a mono-modulator rather than a P-gp substrate might have advantage over verapamil, a substrate of this protein, avoiding the induction of P-gp low-expressed in cell, due to a possible fact that the substrate of P-gp also served as the inducer of this protein, in the event of continuous exposure to cells (Chaudhary &Roninson, Citation1993; Herzog et al., Citation1993). To sum up, honokiol within microemulsion was highly efficient in delivering sirolimus into caco-2 cells and transport through caco-2 cell monolayer. More importantly, combination of honokiol with cremophor EL-based SMEDDS might present synergistic effects on inhibition of exocytosis of caco-2 cells, indicating that a combination of two different inhibition mechanism became effective and significative ways to reverse excretion from caco-2 cells. The detailed mechanism about the inhibition of honokiol in caco-2 cells will be explored in the future studies.
Conclusion
Honokiol was effective at improving the oral absorption and transport of sirolimus, a P-gp substrate, possibly by down-regulating the expression of P-gp presented in the epithelial cells of the intestine. At the same time, the SMEDDS would be a very useful strategy for loading drug as well as functional compound, such as P-gp inhibitor providing nano-size and rapid release for oral delivery of the poor soluble drug.
Supplementary material available online
Supplementary Figures S1--S5
SUPPLEMENTAL
Download PDF (219.3 KB)Declaration of interest
We would like to acknowledge the support of this work by the National Major Science and Technology Project of China (Pharmaceutical Research for Microbial Medicine and Biopharmaceutics, 2012ZX09301002-001-019) and the National Basic Research Program of China (973 program, 2015CB932100).
References
- Ambudkar SV, Dey S, Hrycyna CA, et al. (1999). Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol 39:361–98
- Anton N, Benoit JP, Saulnier P. (2008). Design and production of nanoparticles formulated from nano-emulsion templates-a review. J Control Release 128:185–99
- Anuchapreeda S, Leechanachai P, Smith MM, et al. (2002). Modulation of P-glycoprotein expression and function by curcumin in multidrug-resistant human KB cells. Biochem Pharmacol 64:573–82
- Avtanski DB, Nagalingam A, Bonner MY, et al. (2014). Honokiol inhibits epithelial-mesenchymal transition in breast cancer cells by targeting signal transducer and activator of transcription 3/Zeb1/E-cadherin axis. Mol Oncol 8:565–80
- Basalious EB, Shawky N, Badr-Eldin SM. (2010). SNEDDS containing bioenhancers for improvement of dissolution and oral absorption of lacidipine. I: development and optimization. Int J Pharm 391:203–11
- Bouchemal K, Briancon S, Perrier E, Fessi H. (2004). Nano-emulsion formulation using spontaneous emulsification: solvent, oil and surfactant optimisation. Int J Pharm 280:241–51
- Chaudhary PM, Roninson IB. (1993). Induction of multidrug resistance in human cells by transient exposure to different chemotherapeutic drugs. J Natl Cancer Inst 85:632–9
- Collnot EM, Baldes C, Wempe MF, et al. (2006). Influence of vitamin E TPGS poly(ethylene glycol) chain length on apical efflux transporters in Caco-2 cell monolayers. J Control Release 111:35–40
- Cummins CL, Salphati L, Reid MJ, Benet LZ. (2003). In vivo modulation of intestinal CYP3A metabolism by P-glycoprotein: studies using the rat single-pass intestinal perfusion model. J Pharmacol Exp Ther 305:306–14
- Dahan A, Amidon GL. (2009). Segmental dependent transport of low permeability compounds along the small intestine due to P-glycoprotein: the role of efflux transport in the oral absorption of BCS class III drugs. 6:19–28
- Di YM, Li CG, Xue CC, Zhou SF. (2008). Clinical drugs that interact with St. John's wort and implication in drug development. Curr Pharm Design 14:1723–42
- Dokania S, Joshi AK. (2014). Self-microemulsifying drug delivery system (SMEDDS) - challenges and road ahead. Drug Deliv. Early Online: 1-16. DOI: 10.3109/10717544.2014.896058
- Ershov FI, Kas'ianova NV, Vasil'ev AN. (2002). Kaletra (lopinavir/ritonavir) -- a new HIV protease inhibitor. Antibiot Khimioter 47:27–9
- Esumi T, Makado G, Zhai H, et al. (2004). Efficient synthesis and structure-activity relationship of honokiol, a neurotrophic biphenyl-type neolignan. Bioorg Med Chem Lett 14:2621–5
- Friche E, Jensen PB, Sehested M, et al. (1990). The solvents cremophor EL and Tween 80 modulate daunorubicin resistance in the multidrug resistant Ehrlich ascites tumor. Cancer Commun 2:297–303
- Gaylord G. (1999). New Norvir soft-gel capsule formulation approved by FDA. Posit Living 8:11
- Gershanik T, Benita S. (2000). Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur J Pharm Biopharm 50:179–88
- Gursoy N, Garrigue JS, Razafindratsita A, et al. (2003). Excipient effects on in vitro cytotoxicity of a novel paclitaxel self-emulsifying drug delivery system. J Pharm Sci 92:2411–18
- Gursoy RN, Benita S. (2004). Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother 58:173–82
- Herzog CE, Tsokos M, Bates SE, Fojo AT. (1993). Increased mdr-1/P-glycoprotein expression after treatment of human colon carcinoma cells with P-glycoprotein antagonists. J Biol Chem 268:2946–52
- Hintzen F, Perera G, Hauptstein S, et al. (2014). In vivo evaluation of an oral self-microemulsifying drug delivery system (SMEDDS) for leuprorelin. Int J Pharm 472:20–6
- Houghton P, Fang R, Techatanawat I, et al. (2007). The sulphorhodamine (SRB) assay and other approaches to testing plant extracts and derived compounds for activities related to reputed anticancer activity. Methods 42:377–87
- Juliano C, Cossu M, Alamanni MC, Piu L. (2005). Antioxidant activity of gamma-oryzanol: mechanism of action and its effect on oxidative stability of pharmaceutical oils. Int J Pharm 299:146–54
- Kang BK, Lee JS, Chon SK, et al. (2004). Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int J Pharm 274:65–73
- Lei Y, Qi J, Nie S, et al. (2012). Solid self-nanoemulsifying cyclosporine A pellets prepared by fluid-bed coating: stability and bioavailability study. J Biomed Nanotechnol 8:515–21
- Li W, Wang Q, Su Q, et al. (2014). Honokiol suppresses renal cancer cells' metastasis via dual-blocking epithelial-mesenchymal transition and cancer stem cell properties through modulating miR-141/ZEB2 signaling. Mol Cells 37:383–8
- Li X, Yuan Q, Huang Y, et al. (2010). Development of silymarin self-microemulsifying drug delivery system with enhanced oral bioavailability. AAPS PharmSciTech 11:672–8
- Limtrakul P, Anuchapreeda S, Buddhasukh D. (2004). Modulation of human multidrug-resistance MDR-1 gene by natural curcuminoids. BMC Cancer 4:13
- Lin JH, Yamazaki M. (2003). Clinical relevance of P-glycoprotein in drug therapy. Drug Metab Rev 35:417–54
- Lundin PD, Bojrup M, Ljusberg-Wahren H, et al. (1997). Enhancing effects of monohexanoin and two other medium-chain glyceride vehicles on intestinal absorption of desmopressin (dDAVP). J Pharmacol Exp Ther 282:585–90
- Patel SSP, Patel NM, Patel MM. (2010). A self-microemulsifying drug delivery system (SMEDDS). Int J Pharm Sci Res 4:29–35
- Ma L, Chen J, Wang X, et al. (2011). Structural modification of honokiol, a biphenyl occurring in Magnolia officinalis: the evaluation of honokiol analogues as inhibitors of angiogenesis and for their cytotoxicity and structure-activity relationship. J Med Chem 54:6469–81
- Mekjaruskul C, Yang YT, Leed MG, et al. (2013). Novel formulation strategies for enhancing oral delivery of methoxyflavones in Kaempferia parviflora by SMEDDS or complexation with 2-hydroxypropyl-beta-cyclodextrin. Int J Pharm 445:1–11
- Mu L, Elbayoumi TA, Torchilin VP. (2005). Mixed micelles made of poly(ethylene glycol)-phosphatidylethanolamine conjugate and d-alpha-tocopheryl polyethylene glycol 1000 succinate as pharmaceutical nanocarriers for camptothecin. Int J Pharm 306:142–9
- Muller C, Bailly JD, Goubin F, et al. (1994). Verapamil decreases P-glycoprotein expression in multidrug-resistant human leukemic cell lines. Int J Cancer 56:749–54
- Pouton CW, Porter CJ. (2008). Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev 60:625–37
- Rege BD, Kao JP, Polli JE. (2002). Effects of nonionic surfactants on membrane transporters in Caco-2 cell monolayers. Eur J Pharm Sci 16:237–46
- Sachan KK, Kasture SB. (2010). Self-emulsifying drug delivery system a novel approach for enhancement of bioavailability. Int J Pharm Tech Res 2:1738–45
- Sonneveld P, Wiemer E. (1997). Inhibitors of multidrug resistance. Curr Opin Oncol 9:543–8
- Sun M, Zhai X, Xue K, et al. (2011). Intestinal absorption and intestinal lymphatic transport of sirolimus from self-microemulsifying drug delivery systems assessed using the single-pass intestinal perfusion (SPIP) technique and a chylomicron flow blocking approach: linear correlation with oral bioavailabilities in rats. Eur J Pharm Sci 43:132–40
- Tsai SK, Huang CH, Huang SS, et al. (1999). Antiarrhythmic effect of magnolol and honokiol during acute phase of coronary occlusion in anesthetized rats: influence of L-NAME and aspirin. Pharmacology 59:227–33
- Wahlang B, Pawar YB, Bansal AK. (2011). Identification of permeability-related hurdles in oral delivery of curcumin using the Caco-2 cell model. Eur J Pharm Biopharm 77:275–82
- Xu D, Lu Q, Hu X. (2006). Down-regulation of P-glycoprotein expression in MDR breast cancer cell MCF-7/ADR by honokiol. Cancer Lett 243:274–80
- Yamagata T, Kusuhara H, Morishita M, et al. (2007). Effect of excipients on breast cancer resistance protein substrate uptake activity. J Control Release 124:1–5
- Yee S. (1997). In vitro permeability across Caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man- fact or myth. Pharm Res 14:763–6
- Yin YM, Cui FD, Mu CF, et al. (2009). Docetaxel microemulsion for enhanced oral bioavailability: preparation and in vitro and in vivo evaluation. J Control Release 140:86–94
- Yoo JH, Shanmugam S, Thapa P, et al. (2010). Novel self-nanoemulsifying drug delivery system for enhanced solubility and dissolution of lutein. Arch Pharmacal Res 33:417–26
- Zeng Z, Zhou G, Wang X, et al. (2010). Preparation, characterization and relative bioavailability of oral elemene o/w microemulsion. Int J Nanomed 5:567–72
- Zhang Y, Li X, Zhou Y, et al. (2010). Cyclosporin A-loaded poly(ethylene glycol)-b-poly(d,l-lactic acid) micelles: preparation, in vitro and in vivo characterization and transport mechanism across the intestinal barrier. Mol Pharm 7:1169–82
- Zhang Z, Chen Y, Deng J, et al. (2014). Solid dispersion of berberine-phospholipid complex/TPGS 1000/SiO(2): preparation, characterization and in vivo studies. Int J Pharm 465:306–16
- Zhao C, Liu ZQ. (2011). Comparison of antioxidant abilities of magnolol and honokiol to scavenge radicals and to protect DNA. Biochimie 93:1755–60