
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Purpose: The objective of this study is to investigate cellular uptake of prodrug-loaded nanoparticle (NP). Another objective is to study bioconversion of stereoisomeric dipeptide prodrugs of ganciclovir (GCV) including L-Val-L-Val-GCV (LLGCV), L-Val-D-Val-GCV (LDGCV) and d-Val-l-Val-GCV (DLGCV) in human corneal epithelial cell (HCEC) model.
Methods: Poly(D,L-lactic-co-glycolic acid) (PLGA) NP encapsulating prodrugs of GCV were formulated under a double emulsion method. Fluorescein isothiocyanate isomer–PLGA conjugates were synthesized to fabricate biocompatible fluorescent PLGA NP. Intracellular uptake of FITC-labeled NP was visualized by a fluorescent microscope in HCEC cells.
Results: Fluorescent PLGA NP and non-fluorescent NP display similar hydrodynamic diameter in the range of 115–145 nm with a narrow particle size distribution and zeta potentials around −13 mV. Both NP types showed identical intracellular accumulation in HCEC cells. Maximum uptake (around 60%) was noted at 3 h for NP. Cellular uptake and intracellular accumulation of prodrugs are significantly different among three stereoisomeric dipeptide prodrugs. The microscopic images show that NPs are avidly internalized by HCEC cells and distributed throughout the cytoplasm instead of being localized on the cell surface. Following cellular uptake, prodrugs released from NP gradually bioreversed into parent drug GCV. LLGCV showed the highest degradation rate, followed by LDGCV and DLGCV.
Conclusion: LLGCV, LDGCV and DLGCV released from NP exhibited superior uptake and bioreversion in corneal cells.
Introduction
Corneal epithelial keratitis caused by autoinoculation of ocular tissues following infection by herpes simplex virus type 1 (HSV-1) is a common opportunistic infection, often leading to corneal opacity and blindness if left untreated (Remeijer et al., Citation2002; Duan et al., Citation2009; Refai & Tag, Citation2011). Primary herpetic keratitis can heal without scarring. However, HSV-1 itself remains latent in the trigeminal ganglion. The virus particles can be trapped in the corneal lamellae due to its large size, which limits its diffusion out of the cornea. The cornea may thus present a sanctuary site for the virus (Perng & Jones, Citation2010; Kennedy et al., Citation2011). Subsequent recurrence of keratitis with a 50% rate within two years often causes corneal scarring, thinning and stromal opacity and eventually vision loss (Xu et al., Citation2006; Perng & Jones, Citation2010).
Ganciclovir (GCV), a guanosine nucleoside, has demonstrated dramatic anti-herpes virus activity in rabbits and humans (Shiota et al., Citation1987; Taylor et al., Citation1991; Tomicic et al., Citation2002; Billaud et al., Citation2009). However, poor ocular uptake, owing to the relatively low lipophilicity, significantly limits topical administration of GCV in solution form, thus leading to the failure of the treatment of deep corneal infections (Biron, Citation2006). Many studies from our laboratory have demonstrated that Val-Val-GCV, a dipeptide monoester prodrug of GCV possesses higher aqueous solubility, stability and favorable cell uptake through oligopeptide transporter type 1 (PEPT-1), an influx transporter present on the corneal epithelium (Majumdar et al., Citation2005; Kansara et al., Citation2007). However, VVGCV is susceptible to enzymes such as esterases and peptidases in the corneal epithelial cell (Tirucherai et al., Citation2002; Tirucherai & Mitra, Citation2003; Majumdar et al., Citation2005; Gunda et al., Citation2006). Hence, a fraction of prodrug can bioconvert back into the parent drug prior to reaching the site of action such as deeper layers of corneal epithelium and the stroma (Macha & Mitra, Citation2002; Macha et al., Citation2004). Premature prodrug bioconversion may reduce corneal and stromal permeability, resulting in suboptimal therapeutic efficacy. The peptidases and esterases are stereospecific and have high affinity for l-isomers. Therefore, incorporation of d-isomer in the prodrug can improve enzymatic stability of the prodrug in tissue (Talluri et al., Citation2009; Jwala et al., Citation2011). It was reported that l-valine in the terminal position increases the affinity of prodrugs to the peptide transporter protein in rabbit primary corneal epithelial cells (rPCEC) (Jwala et al., Citation2011). l-Val-l-Val-acyclovir and l-Val-d-Val-acyclovir exhibited optimal properties in terms of enzymatic stability, uptake and cytotoxicity. l-Val-d-Val demonstrated maximal bioavailability following intravenous and oral administration in rats, because the position of d-isomer in Val-Val-acyclovir impacted the enzymatic stability of acyclovir prodrug (Talluri et al., Citation2009). However, a stereoisomeric dipeptide VVGCV ophthalmic solution, like other topical solutions, possesses short residence time in the precorneal area. As a result, frequent administration is necessary to achieve minimum inhibitory concentration of the parent drug in deep ocular tissues. Polymeric nanoparticle (NP) prepared from biodegradable poly(D,L-lactic-co-glycolic acid) (PLGA) can be engineered to provide sustained release for an encapsulated prodrug following topical administration.
The present study is aimed at investigating the cellular uptake of prodrug-loaded NP and bioconversion of stereoisomeric dipeptide monoester prodrugs of GCV in corneal cells. The extent of intracellular delivery was confirmed by fluorescent PLGA NP. The results of this study demonstrate that a stereoisomeric dipeptide prodrugs-loaded PLGA NP may provide improved bioavailability and sustained release of GCV in the corneal layers for the treatment of HSV-1 keratitis.
Materials
Stereoisomeric monoester dipeptide prodrugs of GCV (LLGCV, LDGCV and DLGCV) were synthesized and purified in our laboratory according to a previously published procedure (Majumdar et al., Citation2005; Yang et al., Citation2015). GCV was obtained from Hubei Gedian Humanwell Pharmaceutical Co. Ltd (Wuhan, China) (USP31, batch no. 090920). PLGA polymers (PLGA 65:35 with Mw 45 000–75 000 Da; PLGA 75:25 with Mw 66 000–107 000 Da) and polyvinyl alcohol (PVA) (Mw 31 000–50 000 Da) were purchased from Sigma Chemicals (St. Louis, MO). Fluorescein isothiocyanate isomer I (FITC), triethylamine, 1,4-diaminobutane, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), 4-dimethylaminopyridine (DMAP), dichloromethane (DCM) and dimethylformamide (DMF) were supplied from Sigma-Aldrich Co. Human corneal epithelial cells (HCECs) were the generous gift from Dr. Araki-Sasaki (Kinki Central Hospital, Osaka, Japan). Culture flasks (75 cm2 growth area), 12-well Costar@ plates, all buffer components and solvents were purchased from Fisher Scientific Co (Hampton, NH). All other chemicals used in this study were of analytical reagent grade and used without further purification.
Methods
Preparation of FITC–PLGA conjugates
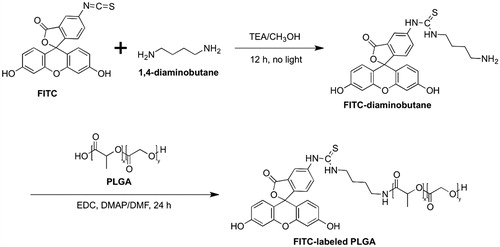
To fabricate FITC–PLGA NP, FITC was conjugated to the terminal carboxyl group of PLGA. FITC (100 mg) dissolved in methanol containing triethylamine (200 µL) was mixed with 1,4-diaminobutane (300 µL) for 12 h. The precipitate was filtered and washed with ether, then dried in a vacuum oven overnight at room temperature. The product fluorescein thiocarbamyl butanediamine (FTB) was added for further coupling to the PLGA polymer. FITC conjugated PLGA was produced by reacting PLGA (570 mg, 7.2 µmol) dissolved in DMF (5) with the primary amine terminated FTB (10 mg, 21.7 µmol) in anhydrous DMF (5 mL) along with the addition of EDC and DMAP. The sample was dialyzed against deionized water to remove unreacted FITC and then lyophilized. There was no detectable change in molecular weight of PLGA after FITC conjugation. The synthetic scheme of FITC conjugated PLGA is shown in .
Figure 1. Synthetic scheme of FITC conjugated PLGA polymer.
Preparation of NP and FITC–PLGA NP by water in oil in water (W/O/W) emulsion method
Hydrophilic prodrugs (LLGCV, LDGCV and DLGCV) loaded NP were prepared at room temperature using a previously described W/O/W emulsion solvent evaporation method (Yang et al., Citation2015). Briefly, the prodrug solution (0.5 mL, 10 mg/mL) was emulsified in DCM (5 mL) containing various amounts of polymer (50 mg of PLGA75:25, 75 mg of PLGA75:25, and 75 mg of PLGA 75:25, for LLGCV, LDGCV and DLGCV, respectively) with sonication over an ice-bath with a Fisher Scientific 100 ultrasonic probe for 2 min. The primary emulsion was added drop wise to 20 mL of aqueous phase, containing 2.0% (w/v) PVA under sonication at a constant output of 55 W for 5 min to form a W/O/W type double emulsion. Organic solvent residues were removed by stirring at room temperature for 2 h and evaporated under a vacuum for 30 min to ensure complete removal of organic solvents. NP suspension was further ultracentrifuged at 5000g (22 000 rpm) for 1 h to collect NP and then washed three times with distilled water to remove the surfactant and PVA residues in the system. Finally, the NP suspension was frozen under −80 °C then lyophilized for 48 h using a lab-scale freeze-drier system and stored at 4 °C until further use. FITC-PLGA NPs were prepared by blending PLGA and FITC conjugated PLGA at a 90:10 weight ratio according to the previously described double emulsion solvent evaporation method.
NP characterization
NP (1 mg) was dissolved in 2 mL DCM by sonication for 1 min. Prodrugs were then extracted from the organic phase by three portions of 500 µL of acetonitrile: water (65:35). Samples were subsequently analyzed by high-performance liquid chromatography (HPLC). All studies were conducted from three batches (n = 3/batch). Entrapment efficiency, drug loading and yield were calculated according to equations (1), (2), and (3), respectively. The measurements were made in triplicate.
(1)
(1)
(2)
(2)
(3)
(3)
The average particle size and size distribution of the various samples (nanosuspension of lyophilized NP powder) were measured at 25 °C by a dynamic light scattering method (Brookhaven Zeta Plus instrument, Holsville, NY). The zeta potential of NP suspension was measured using a Nano ZS zetasizer (Malvern Instruments Ltd, Worcestershire, UK). The morphological examination of NP was performed with a FEG ESEM XL 30 SEM (FEI, Hillsboro, OR).
Cell culture
HCEC cells were grown and routinely maintained at 37 °C in the culture medium containing minimum essential medium supplemented with 10% calf serum (non-heat inactivated), 100 U/L of penicillin, 100 U/L of streptomycin, 1.76 g/L lactalbumin enzymatic dehydrolysate and 1.3 g/L HEPES.
NP uptake studies in HCECs
For uptake studies, cells were cultured in 12-well plates at a density of 250 000 cells/well and grown for 5 d until confluent. Cells were then exposed to 50 μg/mL of NP in DPBS and incubated in the same medium. Uptake of NP was carried out for 1–4 h at 37 °C. After each uptake period, cells were washed three times with DPBS to ensure removal of all free NPs. Cells were lysed by adding 1 mL DDI water and then frozen at −80 °C. Cells were then thawed and scraped into 1 mL DDI. Samples were centrifuged at 1000g for 10 min at 4 °C, and then supernatant was collected. Cell pellets were sonicated in 200 µL of methanol and centrifuged at 10 000g for 15 min at 4 °C. The methanol extract was stored at −80 °C until HPLC analysis was performed. The extent of intracellular uptake of FITC-PLGA NP by HCEC cells was also examined by a Leica DMI3000 fluorescence microscope using a 494 nm excitation wavelength.
HPLC analytical method
Samples were analyzed by a HPLC procedure capable of simultaneous analyses of the parent drug and prodrugs. A Waters's HPLC equipped with a Waters 600 pump (Milford, MA) was used to analyze samples. The column selected was a reversed-phase C8 column 250 mm × 4.6 mm with 4 µm particles (Phenomenex, Torrance, CA). The mobile phase consisted of a mixture of 15 mM phosphate buffer (pH 2.5) and 2.5% acetonitrile pumped at a flow rate of 1 mL/min. The samples were detected with a fluorescence detector (HP1100) set at excitation wavelength of 265 nm and emission wavelengths 380 nm.
Statistical analysis of data
After triplicate experiments, results were expressed as means ± standard deviation (SD). Compared to controls, the significance of results was determined using the Student's t-test. A p value < 0.05 was considered statistically significant.
Results
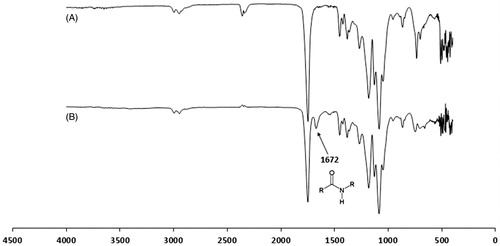
In our attempt to evaluate the uptake of PLGA NP, the first step was the development of a fluorescent derivative of PLGA that provides a stable fluorescence signal without causing significant changes in the characteristics of the NP. Association of fluorescein to the polymer was first identified by spectrofluorimetry. The emission and excitation wavelengths of the conjugate were 520 nm and 490 nm, respectively, whereas those of fluorescein were 519 nm and 495 nm, respectively. Moreover, the structure of conjugate was confirmed by Fourier transform infrared spectroscopy (FTIR) with an amide peak at 1672 cm−1 ().
Figure 2. FTIR spectra of (A) PLGA 75:25 and (B) FITC-labeled PLGA 75:25.
Properties of prodrugs-loaded NP
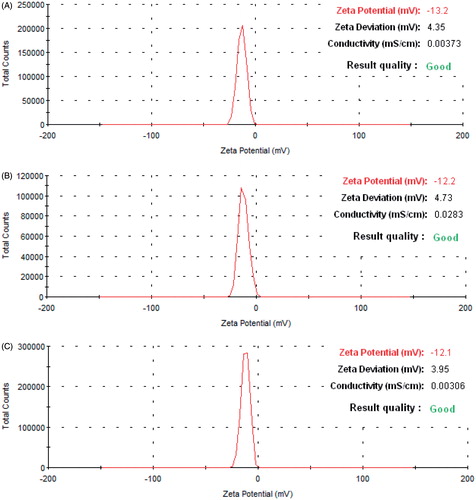
As previously published from our laboratory, LLGCV-, LDGCV- and DLGCV-loaded NP generated 38.7 ± 2.0%, 41.8 ± 1.9% and 45.3 ± 2.2% of entrapment efficiencies; 3.87 ± 0.20%, 2.79 ± 0.13% and 3.02 ± 0.15% of drug loadings; 85.2 ± 3.0%, 86.9 ± 4.6% and 76.9 ± 2.1% of yield; 116.6 ± 4.5, 143.0 ± 3 and 134.1 ± 5.2 nm of size; and −15.0 ± 4.96, −13.8 ± 5.26 and −13.9 ± 5.14 mV of zeta potential, respectively (Yang et al., Citation2015). FTIC-labeled NP in this study exhibited 122.4 ± 8.7, 145.9 ± 4.2 and 130.5 ± 5.0 nm in particle size (, and ). These FTIC-labeled NPs were further characterized for morphology and in vitro uptake study. SEM revealed the spherical shape and uniform, smooth surface for NP samples (, and ). The zeta potential values −13.2 ± 4.57, −12.2 ± 3.70, and −12.1 ± 3.48 mV of the FTIC-labeled NP samples are shown in .
Figure 3. Particle size distribution curves and scanning electron photographs of NP. (A and B) FITC-labeled LLGCV-NP, (C and D) FITC-labeled LDGCV-NP and (E and F) FITC-labeled DLGCV-NP.
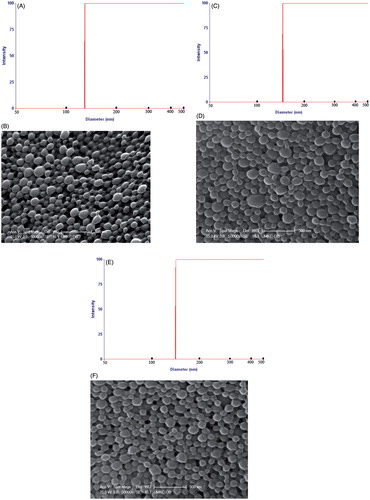
Figure 4. Zeta potential distribution of NP. (A) LLGCV-NP, (B) LDGCV-NP, and (C) DLGCV-NP.
Cellular uptake of NP in HCECs
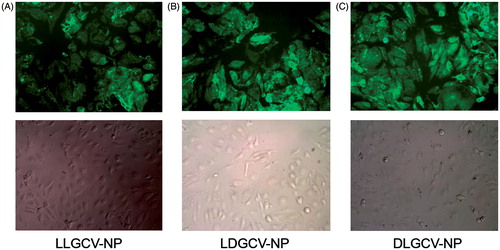
Free drug molecules taken up by cells were collected in the supernatant by centrifugation. The prodrug and it degradants within NP were obtained after dissolving NP and sedimentating cell pellets and polymer. The amount of prodrugs and their degradants from bioconversion were confirmed by HPLC method. About 60% of the prodrug was taken up by HCEC cells within 4 h (). Cellular uptake and intracellular accumulation of prodrugs notably increased during the first 3 h, and a slower cellular uptake ensued. After cell uptake, prodrugs released from NP bioreversed into parent drug GCV. LLGCV showed the highest degradation rate, followed by LDGCV and DLGCV (). d-Val moiety at the terminal position exhibited higher stability with low enzymatic hydrolysis in HCEC cells. DLGCV can be directly bioreversed into parent drug GCV by hydrolysis of the ester bond with esterases at l-Val position, because l-Val is much more susceptible to enzymatic degradation than d-Val. Cellular enzymes can directly hydrolyze LLGCV to generate parent GCV. There are multiple possible means of drug accumulation inside cells with NP. For example, NP may be directly taken up by cells and then retained or released. Under in-vivo conditions, NP could release the prodrug in the cell cytoplasm first at the precorneal site and then the particles can penetrate corneal cells. To delineate the mechanism, we utilized FITC-labeled NP for uptake studies. Intracellular uptake of FITC-labeled NP was further visualized by fluorescence microscope in HCEC cells (). Intracellular accumulation of FITC-labeled NP was observed and the fluorescence intensity observed in cells increased notably within 3 h.
Figure 5. Time-dependent uptake efficiency of (♦) LLGCV-NP, (▪) LDGCV-NP and (▴) DLGCV-NP in HCEC cells. Each data point shown is the average of three samples. Each data point is expressed as mean ± SD (n = 3).
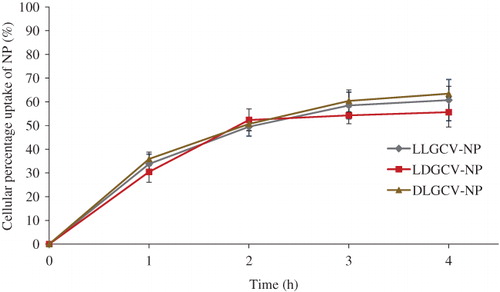
Figure 6. Bioconversion pathway of prodrugs of GCV in HCEC cell homogenate. (A) LLGCV-NP, (B) LDGCV-NP and (C) DLGCV-NP. (♦) LLGCV, (▪) LDGCV, (▴) DLGCV, (×) LGCV, (*) DGCV, and (•) GCV. Each data point is expressed as mean ± SD (n = 3).
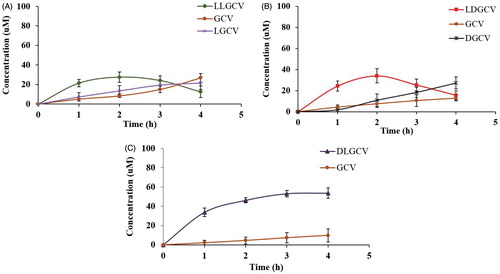
Figure 7. Fluorescence microscope images of HECE cells treated with FITC-labeled (A) LLGCV-NP, (B) LDGCV-NP and (C) DLGCV-NP at 3 h.
Discussion
Delivery of GCV through the cornea to treat herpetic keratitis caused by the HSV-1 type of viruses has limitations due to its poor ocular absorption. The corneal epithelium is rich in membrane lipid and tight intercellular junctions, which significantly hinder hydrophilic GCV transport through the cornea. Gunda et al. (Citation2006) have reported that lipophilic dipeptide monoester prodrugs of GCV including VVGCV can significantly enhance permeability across cornea. Enhanced bioavailability of VVGCV was contributed by higher solubility, better stability and excellent affinity to influx peptide transporters present on the corneal epithelium. The esterases and peptidases present in the corneal tissues can break down VVGCV into its amino acid metabolite and parent GCV molecules to generate higher drug concentration in corneal tissues. Earlier studies from our laboratory indicated that four stereoisomeric dipeptide prodrugs of ACV including L-Val-L-Val-ACV (LLACV), L-Val-D-Val-ACV (LDACV) and D-Val-L-Val-ACV (DLACV) exhibited different affinities to the peptide transporter, permeabilities and bioreversion rates in Caco-2 and rPCEC cell lines, as well as intestine, liver and ocular tissues (Talluri et al., Citation2008; Jwala et al., Citation2011).
It was revealed that incorporation of one d-Val in a dipeptide diminished, but does not negate its affinity towards peptide transporters but significantly improve enzymatic stability of stereoisomeric dipeptide prodrugs to a large extent depending on the position of d-amino acid in a dipeptide conjugate (Tamura et al., Citation1996; Friedrichsen et al., Citation2001). Novel stereoisomeric dipeptide prodrugs of GCV were synthesized in this study. Previous reports from Gunda et al. (Citation2006) clearly explained the bioconversion mechanism of dipeptide prodrugs. The dipeptide monoester prodrug of GCV is initially degraded by the peptidases to amino acid ester of GCV, which is further hydrolyzed by esterases to regenerate the parent drug GCV. Based on uptake studies, it appears that a primary mechanism for bioconversion of stereoisomeric dipeptide prodrugs LLGCV and LDGCV to GCV occurs in a two-step process. The first step is the hydrolysis of terminal peptide bond cleavage by peptidases to generate the amino acid derivative of GCV and, in next step, esterases can cleave the ester bond in Val-GCV monoester to regenerate parent GCV (Gunda et al., Citation2006). However, in the case of DLGCV, the majority of the prodrug is directly cleaved to parent GCV by esterases. It has been reported that the enzymatic degradation of ester depended largely on the structure of the amino substituents (Patel et al., Citation2005).
Therapeutic amounts of prodrugs should be available in the precorneal space. Following transport across corneal epithelium via peptide transporters, the prodrug should readily convert to parent drug GCV within corneal tissue before elimination. Incorporation of VVGCV in PLGA NP will protect VVGCV from precorneal enzymatic degradation and provide prolonged prodrug release that may in turn reduce the dosing frequency. However, hydrophobic nature of PLGA NP may cause particle aggregation when suspended in simple aqueous buffers. NP may be cleared by precorneal drainage associated with rapid tear turnover, blinking, lachrymation and drainage. Moreover, NPs prepared by biodegradable PLGA polymers are known to exhibit burst release. A simple solution to avoid these barriers would be the application of thermogelling PLGA–PEG–PLGA polymer solution as the vehicle for VVGCV-loaded PLGA NP. Such formulations may provide sustained corneal delivery for prodrugs of GCV following topical administration.
It is well known that particle size, surface charge and polymer properties may influence cellular binding, uptake and intracellular NP trafficking. Small particle size such as LLGCV-NP (116 nm) and LDGCV-NP (134 nm) exhibits higher cell uptake relative to DLGCV-NP (143 nm). Pinocytosis is the internalization of extracellular fluid and its content by cells. The uptake of prodrug-loaded PLGA NP can be taken up by this predominant pathway of cellular internalization of NP without direct interaction of NP with the cell membrane, because the bulk extracellular fluid is internalized (Yameen et al., Citation2014; Adjei et al., Citation2014). However, NPs that interact with cell membranes enable higher uptake through this mechanism than NPs that do not (Adjei et al., Citation2014). During the process of cellular internalization of NP, surface energy may facilitate by lowering bending energy to induce curvature of the lipid bilayer. The process induces shell formation on the NP surface in the endocytosis process. NP of diameter < 100 nm may not have sufficient surface energy to provide enough bending energy to form the curvature of the designated NP size (Khin & Feng, Citation2005). NP internalization process is also further confirmed by computer simulation (Decuzzi & Ferrari, Citation2007). The microscopic images verify that NPs are actually internalized by HCEC cells and distributed throughout the cytoplasm instead of being localized on the cell plasma membrane. Incorporation of a d-isomer into a dipeptide results in high affinity towards PEPT-1 and also improve higher enzymatic stability (Tamura et al., Citation1996; Friedrichsen et al., Citation2001).
NP can minimize precorneal enzymatic and cellular degradation of prodrugs after topical administration. Such formulations may form a depot in the cul-de-sac to provide a robust concentration gradient for controlled release and efficient permeation of prodrugs across the cornea.
Prodrug-loaded NP formulations can be selectively applied depending on the location and severity of infection. For acute HSV-1 keratitis, LLGCV-NP may be the therapeutic choice based on its quicker bioreversion. LDGCV-NP demonstrated similar results as LLGCV-NP and may be used for acute and superficial keratitis. Furthermore, DLGCV possesses the highest enzymatic stability in the corneal cells and hence DLGCV loaded NP may provide the most prolonged release in the cellular environment and may be useful for chronic eye diseases. It may even reduce recurrence of HSV-1 keratitis. Since the system has higher stability, it may penetrate deeper into ocular structures such as Bowman's membrane, stroma and retina, and therefore can be useful for cytomegalovirus (CMV) retinitis. The formulation of LLGCV, LDGCV and DLGCV-NP may be promising for patients with the deeper and wide spread ocular infections caused by HSV-1.
Conclusions
In summary, LLGCV, LDGCV and DLGCV released from NP exhibited superior absorption, bioreversion and ocular bioavailability in a corneal model. Prodrugs released in this formulation can be retained on the corneal surface longer following topical administration into the cul-de-sac. In vivo pharmacokinetic studies of aqueous humor concentrations of these formulations following topical administration will be carried out in the future.
Acknowledgements
We sincerely thank Dr. James B Murowchick from the Department of Geosciences at University of Missouri-Kansas City for assistance with X-ray pictures of compounds and NP. Authors would also like to thank Dr. Piyush Jain for his helpful discussions.
Declaration of interest
No competing financial interests exist.
This work was supported by Missouri Life Sciences Research Grant No. MLSRB0017025 and National Institutes of Health grants R01 EY 10659-12.
References
- Adjei IM, Sharma B, Labhasetwar V. (2014). Nanoparticles: cellular uptake and cytotoxicity. Adv Exp Med Biol 811:73–91
- Billaud G, Thouvenot D, Morfin F. (2009). Drug targets in herpes simplex and Epstein Barr Virus infections. Infect Disord Drug Targets 9:117–25
- Biron KK. (2006). Antiviral drugs for cytomegalovirus diseases. Antiviral Res 71:154–63
- Decuzzi P, Ferrari M. (2007). The role of specific and non-specific interaction in receptor-mediated endocytosis of nanoparticles. Biomaterials 28:2915–22
- Duan R, De Vries RD, Van Dun JM, et al. (2009). Acyclovir susceptibility and genetic characteristics of sequential herpes simplex virus type 1 corneal isolates from patients with recurrent herpetic keratitis. J Infect Dis 200:1402–14
- Friedrichsen GM, Jakobsen P, Taub M, et al. (2001). Application of enzymatically stable dipeptides for enhancement of intestinal permeability: synthesis and in vitro evaluation of dipeptide-coupled compounds. Bioorg Med Chem 9:2625–32
- Gunda S, Hariharan S, Mitra AK. (2006). Corneal absorption and anterior chamber pharmacokinetics of dipeptide monoester prodrugs of ganciclovir (GCV): in vivo comparative evaluation of these prodrugs with Val-GCV and GCV in rabbits. J Ocul Pharmacol Ther 22:465–76
- Jwala J, Boddu SH, Shah S, et al. (2011). Ocular sustained release nanoparticles containing stereoisomeric dipeptide prodrugs of acyclovir. J Ocul Pharmacol Ther 27:163–172
- Kansara V, Hao Y, Mitra AK. (2007). Dipeptide monoester ganciclovir prodrugs for transscleral drug delivery: targeting the oligopeptide transporter on rabbit retina. J Ocul Pharmacol Ther 23:321–34
- Kennedy DP, Clement C, Arceneaux RL, et al. (2011). Ocular herpes simplex virus type 1: is the cornea a reservoir for viral latency or a fast pit stop? Cornea 30:251–9
- Khin YW, Feng SS. (2005). Effect of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials 26:2713–22
- Macha S, Duvvuri S, Mitra AK. (2004). Ocular disposition of novel lipophilic diester prodrugs of ganciclovir following intravitreal administration using microdialysis. Curr Eye Res 28:77–84
- Macha S, Mitra AK. (2002). Ocular disposition of ganciclovir and its monoester prodrugs following intravitreal administration using microdialysis. Drug Metab Dispos 30:670–5
- Majumdar S, Nashed YE, Patel K, et al. (2005). Dipeptide monoester ganciclovir prodrugs for treating HSV-1-induced corneal epithelial and stromal keratitis: in vitro and in vivo evaluations. J Ocul Pharmacol Ther 21:463–74
- Patel K, Trivedi S, Luo S, et al. (2005). Synthesis, physicochemical properties and antiviral activities of ester prodrugs of ganciclovir. Int J Pharm 305:75–89
- Perng GC, Jones C. (2010). Towards an understanding of the herpes simplex virus type 1 latency-reactivation cycle. Interdiscip Perspect Infect Dis 2010:262415
- Refai H, Tag R. (2011). Development and characterization of sponge-like acyclovir ocular minitablets. Drug Deliv 18:38–45
- Remeijer L, Maertzdorf J, Buitenwerf J, et al. (2002). Corneal herpes simplex virus type 1 superinfection in patients with recrudescent herpetic keratitis. Invest Ophthalmol Vis Sci 43:358–63
- Shiota H, Naito T, Mimura Y. (1987). Anti-herpes simplex virus (HSV) effect of 9-(1,3-dihydroxy-2-propoxymethyl)guanine (DHPG) in rabbit cornea. Curr Eye Res 6:241–5
- Talluri RS, Gaudana R, Hariharan S, et al. (2009). Pharmacokinetics of stereoisomeric dipeptide prodrugs of acyclovir following intravenous and oral administrations in rats: a study involving in vivo corneal uptake of acyclovir following oral dosing. Ophthalmol Eye Dis 1:21–31
- Talluri RS, Samanta SK, Gaudana R, et al. (2008). Synthesis, metabolism and cellular permeability of enzymatically stable dipeptide prodrugs of acyclovir. Int J Pharm 361:118–24
- Tamura K, Bhatnagar PK, Takata JS, et al. (1996). Metabolism, uptake, and transepithelial transport of the diastereomers of Val-Val in the human intestinal cell line, Caco-2. Pharm Res 13:1213–18
- Taylor JL, Punda-Polic V, O'Brien WJ. (1991). Combined anti-herpes virus activity of nucleoside analogs and interferon. Curr Eye Res 10:205–11
- Tirucherai GS, Dias C, Mitra AK. (2002). Corneal permeation of ganciclovir: mechanism of ganciclovir permeation enhancement by acyl ester prodrug design. J Ocul Pharmacol Ther 18:535–48
- Tirucherai GS, Mitra AK. (2003). Effect of hydroxypropyl beta cyclodextrin complexation on aqueous solubility, stability, and corneal permeation of acyl ester prodrugs of ganciclovir. AAPS PharmSciTech 4:E45
- Tomicic MT, Bey E, Wutzler P, et al. (2002). Comparative analysis of DNA breakage, chromosomal aberrations and apoptosis induced by the anti-herpes purine nucleoside analogues aciclovir, ganciclovir and penciclovir. Mutat Res 505:1–11
- Xu F, Sternberg MR, Kottiri BJ, et al. (2006). Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA 296:964–73
- Yameen B, Choi WI, Vilos C, et al. (2014). Insight into nanoparticle cellular uptake and intracellular targeting. J Control Release 190:485–99
- Yang X, Shah SJ, Wang Z, et al. (2015). Nanoparticle-based topical ophthalmic formulation for sustained release of stereoisomeric dipeptide prodrugs of ganciclovir. Drug Deliv 7:1–11