
Abstract
Nanosuspensions (NS) can enhance the saturation solubility and dissolution velocity of poorly soluble drugs. PEG as a non-ionic surfactant plays an important role in surface modification of nanoparticles for prolonging in vivo circulation. In this study, anti-solvent precipitation method was introduced to prepare gambogenic acid nanosuspensions (GNA-NS) with PVPK30 and PEG2000 as stabilizers to settle the disadvantages of GNA. The obtained nanoparticles were spherical with a mean particle size of 183.7 nm and a zeta potential of −22.8 mV. The entrapment efficiency and drug loading of the resultant formulation were 97.3 and 29.73%. X-ray diffraction analysis confirmed the amorphous phase of GNA in NS. Fourier transform infrared indicated there may be hydrogen bond interaction between the drug and excipients. After lyophilization of GNA-NS, the freeze-dried powder displayed sufficient long-term physical stability at 4 and 25 °C. In comparison to GNA solution, in vitro studies of GNA-NS showed much slower release and higher cytotoxicity in HepG2 cells. What’s more, the pharmacokinetic study in rats revealed that the AUC0–∞ and t1/2 of GNA-NS were increased 2.63- and 1.77-fold than that of the reference formulation. Taken together, in vitro/in vivo evaluations showed NS would be an effectively strategy to change the poor aqueous solubility and prolong the half-life for GNA. The GNA-NS with enhanced bioavailability and drug efficacy provided a promising delivery system for the application of GNA.
Introduction
Gamboge, a well-known herbal medicine derived from the Garcinia hanburyi tree, shows widely pharmacological activities including detoxification, anti-inflammatory and parasiticide effects (Asano et al., Citation1996; Li et al., Citation2010). Its main active components includes gambogenic acid (GNA, ) and gamboge acid (GA, ), and GNA exhibits stronger and broader pharmacological activities (Zhou et al., Citation2008). Previous studies have confirmed that GNA has a broad spectrum anti-tumor effect in breast cancer cell line, lung cancer cells and other cancer cells (Fan et al., Citation2013; Zhou et al., 2013; Wang et al., Citation2014). However, the poor solubility, short biological half-life and excessive vascular irritation of GNA hindered its clinical application (Hua et al., Citation2015). In order to enhance delivery efficiency and bioavailability of GNA, we have used nanotechnology and successfully prepared PEGylated non-ionic surfactant vesicles and solid lipid nanoparticles of GNA in previous researches (Huang et al., Citation2013; Lin et al., Citation2013). Nevertheless, low drug encapsulation efficiency and drug loading, residual organic solvent, especially a large amount of excipients and surfactants used in these nano-carriers will cause many side effects, such as toxicity and hemolysis.
Figure 1. The chemical structure of GNA (a) and GA (b).
Nanosuspensions (NS) as a carrier-free submicron colloidal drug delivery system consist of pure drug with a small amount of stabilizers (Verma et al., Citation2009; Liu et al., Citation2013). By reducing particle size (PS), it confers a series of enhanced characteristics to drugs, such as the safety, stability, dissolution rate and bioavailability (Zhang & Zhang, Citation2014; Zhang et al., Citation2014). NS can be prepared either by top–down approach or by bottom–up approach, and various administration routes such as oral, intravenous, ocular and pulmonary delivery system, were verified well by the corresponding pharmacokinetic studies in recent years (Van et al., Citation2008; Xia et al., Citation2010; Chin et al., Citation2014). As one of the promising formulations to solve the delivery obstacle of poorly soluble drugs, NS have been attracted more and more attention (Liu et al., Citation2012; Hu et al., Citation2014; Li et al., Citation2015). For example, Li et al. (Citation2015) successfully prepared paclitaxel nanosuspensions (PTX-NS) by anti-solvent precipitation method. Compared with Taxol (commercial products of paclitaxel), PTX-NS increased AUC and Cmax by 26- and 16-fold, prolonged t1/2 by 314-fold and had better in vitro and in vivo antitumor activity.
In the process of NS preparation, the small amount of stabilizers were used to stabilize newly formed drug nanoparticles with high surface Gibbs free energy (δG) and prevent nanoparticles aggregation by reducing the interfacial tension (Tian et al., Citation2013; Aditya et al., Citation2015). The non-toxic stabilizers, such as polyvinyl pyrrolidone (PVP), pluronics, tweens, polyethylene glycols (PEGs), polyvinyl alcohols (PVAs) and lecithin, are commonly used. Specially, PEG endowed the prolongation of pharmaceutical circulation time in blood by avoiding the uptake of the mononuclear phagocyte system (MPS) (Wen et al., Citation2012).
The main objective of this study was to: (i) prepare gambogenic acid nanosuspensions (GNA-NS) with PVPK30 and PEG2000 as stabilizers, (ii) characterize the formulation by PS, zeta potential, drug encapsulation efficiency, drug loading, scanning electron microscope (SEM), Fourier transform infrared (FT-IR) and X-ray diffraction (XRD), (iii) investigate in vitro dissolution and the in vitro cytotoxicity using HepG2 cell lines, and (iv) assess the in vivo bioavailability determination in SD rats. This study is the first time to prepare GNA nanoparticles with simple, fast, and easily scaled-up NS method compared to other nano-techniques.
Materials and methods
Materials
GNA [high-performance liquid chromatography (HPLC) purity>99%] was isolated from gamboge in Prof. Wang Xiaoshan’s Laboratory. PVPK30 and PEG2000 were obtained from (Nanjing, China). Cell culture medium (RPMI-1640, Waymouth), 0.25% trypsin–EDTA and heat-inactivated fetal bovine serum (FBS) were obtained from Gibco (Grand Island, NY). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma (Sigma-Aldrich, Seelze, Germany and USA). The human HepG2 cell line was obtained from Institute of Biochemistry and Cell Biology, Institute for Biological Sciences, Chinese Academy of Science (Shanghai, China). Methanol and acetonitrile were of HPLC grade. All other chemicals and solvents were analytical purity grade, and water used in the study was bidistilled and deionized.
Preparation of GNA-NS
GNA-NS was prepared by anti-solvent precipitation method. The procedure was optimized by single factor and the preparation process was described as follows. In brief, 10 mg GNA was dissolved into 4 mL ethanol to obtain uniform organic solution. Meanwhile, 15 mg PVPK30 together with 10 mg PEG2000 were dissolved into 25 mL water to form aqueous stabilizers solution. At room temperature, the prepared organic solution was injected rapidly into aqueous phase under a stirrer (ETS-D4.IKA, Germany) at 1000 rpm. The formed dispersion was then stirred magnetically at room temperature for 6 h in a ventilated cupboard to evaporate ethanol. Finally, GNA-NS was harvested. The different stabilizers and the ratio of drug to stabilizers were tested with other parameters fixed.
HPLC analysis
The concentration of GNA samples were assayed by HPLC method. HPLC was equipped with Shimadzu LC-15C (Shimadzu, Kyoto, Japan), SPD-15C UV-Spectrophotometric detector system. The analytical column was a COSMOSIL C18 column (4.6 × 250 mm, 5 μm; Nacalai Inc, Kyoto, Japan) with temperature at 30 °C. The mobile phase was comprised of methanol and 0.1% phosphoric acid solution (90:10, v/v) and was delivered at a flow rate of 1.0 mL/min. The detection of GNA was carried out at 360 nm and the injection volume was 20 μL.
PS, polydispersity index and zeta potential measurement
The PS, polydispersity index (PDI) and zeta potential (ZP) of GNA-NS were evaluated by dynamic light scattering (DLS) using a Zetasizer (Malvern Instruments, Malvern, UK). Before measurement, all samples were diluted with deionized water to yield a suitable scattering intensity and analyzed at 25 °C in triplicate.
Morphology observation
The morphology of GNA-NS was investigated by a field emission scanning electron microscope (FESEM, JEOL-6700F, Tokyo, Japan). In general, a drop of NS was added to a clean copper and air dried. The copper was fixed on aluminum stubs using double-sided adhesive tape sputter coated with gold before observation.
Entrapment efficiency and drug loading analysis
The encapsulation efficiency (EE%) and drug loading (DL%) of GNA-NS was investigated by ultrafiltration method with ultrafiltration tube. The ultrafiltration tube is consisted of an inner tube with filter membrane (molecular weight cutoff: 100 000) and an outer tube. Briefly, 2 mL NS was added into the inner tube and centrifuged at 3000 rpm for 15 min. The drug molecules passed through the inner tube whereas the drug nanoparticles were cut off. EE% and DL% GNA-NS were calculated using the following equations:
Here, WF is the mass of GNA hasn’t entrapped in the GNA-NS. WA, WPEG2000 and WPVPK30 were the total weight of GNA, PEG2000 and PVPK30 in the system, respectively.
Fourier transform infrared spectroscopic studies
The Fourier transform infrared (FT-IR) spectra of GNA-NS, pure GNA powder, PEG2000 and PVPK30 were obtained using a Bruker Vector-22 FT-IR spectrometer from 4000 to 500 cm−1 at room temperature. The samples were ground with KBr and pressed into a disk shape for measurement.
XRD diffraction
The powder XRD patterns were measured on silica substrate by Philips X’Pert Pro Super Diffractometer Cu Kα radiation (λ = 1.541874 Å), and the operating voltage and current were maintained at 40 kV and 40 mA, respectively. The scanning speed was 10°/min over an angular range of 10–80° of 2θ, with a step size of 0.02°.
Lyophilization and stability studies
Lyophilization was performed to convert NS into dry powders. Prior to the drying process, 5% trehalose (w/w) as cryoprotectant was added to GNA-NS to inhibit nanoparticles agglomeration. Then, 2 mL NS were put into 10 mL vials and freezing in refrigerator (−80 °C) for 24 h. Subsequently, the frozen NS were put into the vacuum adapter of lyophilizer (FD-1-50; Beijing, China) under a pressure of 0.01 mbar for 48 h. Finally, the lyophilized powder was obtained. Since the solid form GNA-NS should be reconstituted for administration, the properties of the redispersed dried powder in terms of PS and ZP were also performed. Storage stability of the lyophilized powder was investigated by storing at 4 and 25 °C for up to 3 months.
In vitro release of GNA-NS
GNA solution was prepared as control to investigate the dissolution profile of GNA-NS. The in vitro drug release profiles of the two formulations were carried out using USP dissolution apparatus II method under sink conditions. Phosphate-buffered saline (PBS, PH7.4) containing 0.5 wt% Tween-80 was used as the release medium. The paddle speed and bath temperature were set at 100 rpm and 37 ± 0.5 °C. Briefly, equivalent amount of GNA for the two formulations were put into two swelled dialysis bag (21 mm, molecular weight cutoff: 8000–14 000 Da). Then, the dialysis bags were tied and immersed into 500 mL dissolution medium. At specific time intervals, 0.5 mL of the medium was withdrawn from receiver compartment and the same amount of pre-warmed fresh media was added. The GNA samples were filtered using 0.22 µm microporous membrane Polyvinylidene Fluoride filters and analyzed by HPLC method under the analytic conditions described above. Each experiment was performed in triplicate and values were reported as the means ± standard deviation (SD).
In vitro cytotoxicity studies
Cytotoxicity of GNA nanoparticles against HepG2 cell line was assessed by MTT assay. Prior to the assay, the cell line was adapted and maintained in RPMI1640 medium supplemented with 10% FBS, 100 units/mL penicillin and 100 units/mL streptomycin under a humidified atmosphere containing 5% CO2 at 37 °C. The cells harvested from the confluent cultures by trypsinization were seeded in 96-well plates with a density of 3 × 104 cells/mL and incubated overnight. Then the cells exposed to different concentrations (0.5, 1, 2, 4, 8 and 16 µM) of GNA solution and GNA-NS for 48 h with PEG2000 and PVPK30 as negative controls. After incubation, 20 μL of MTT solution (5 g/L) was added to each well and incubated for 4 h. The medium was withdrawn and 150 μL of DMSO was added to each well and agitated thoroughly to dissolve the formazan crystals at room temperature. The solution was transferred to 96-well plates and immediately read the absorbance on a microplate reader (PerkinElmer’s 1430 Multilabel Counter) at 490 nm. The cell inhibition rate was calculated as follows: inhibition rate (IR%) = [(mean control absorbance) − (mean experimental absorbance)/mean control absorbance] × 100 (%). All procedures were performed on sterile conditions. The study was carried out in triplicates and the viability assay was presented as the mean ± SD.
In vivo evaluation of GNA-NS
In vivo study was approved by the animal-related ethics regulations of Anhui university of Chinese Medicine. Twelve healthy SD rats weighing 200–220 g were supplied by Experimental animal center of Anhui province (2011-002, Hefei, Anhui, China). Animals were housed in cages for 1 week before experimentation and simultaneously fed with food and water ad libitum. Prior to dosing, the rats were randomly divided into two groups with half male and half female and were fasted overnight but allowed free access to water. Equivalent amount of GNA in GNA-NS and GNA solution were administrated to two groups of rats at a single dose of 8 mg/kg by intraperitoneal injection. At specified time intervals, retro-orbital blood samples were collected into 0.5 mL heparinized centrifuge tubes. The tubes were immediately centrifuged for 10 min at 3000 r/min to separate the plasma. Afterwards, the plasma was processed using acetonitrile protein precipitation method and analyzed by HPLC. The software program 3P97 (Chinese Pharmacological Society, China) was employed to estimate the pharmacokinetic parameters. Data from different experimental groups were compared by using one-way analysis of variance (ANOVA) statistical analysis. All data were expressed as the mean ± SD. The statistical analysis was made using the t-test. A p value < 0.05 was considered statistically significant.
Results and Discussion
Preparation of GNA-NS
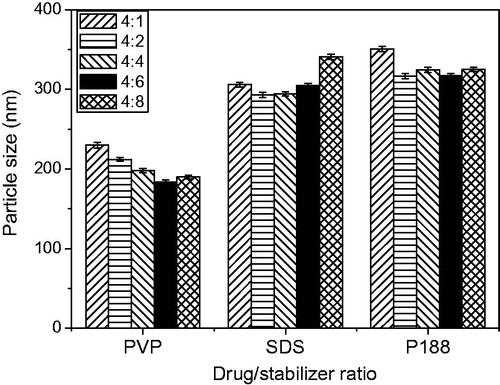
The major challenge for formulating NS is to select suitable stabilizers to efficiently reduce PS and adequately stabilize drug nanoparticles in colloid system (Cerdeira et al., Citation2012). In addition, the amount of stabilizers should be sufficient to cover the particle surface and inhibit nanoparticles aggregation (Gao et al., Citation2008). During the preparation of GNA-NS, GNA (10 mg) dissolved in 4 mL ethanol was injected into three kinds of aqueous stabilizers solution (25 mL) containing the same amount of PVPK30, P188 and SDS with 10 mg PEG2000 which was determined by our preliminary experiments that 1:1 ratio of PEG2000 to GNA could preferable stabilize the system. The influence of different drug/stabilizers ratio on the PS is demonstrated in . Three kinds of stabilizers combined with PEG2000 all can facilitate the size reduction and stabilize the formed nanoparticles, which may relate to fast precipitation of GNA and rapid absorption of stabilizers under high-speed stirring (Bajaj et al., Citation2012). It can be seen from , biocompatible and non-cytotoxic PVPK30 significantly decrease PS of nanoparticles with drug/stabilizer ratio as 4:6. Therefore, the ratio of GNA, PEG2000 and PVPK30 in our resultant formulation is set as 4:4:6.
Figure 2. Influence of different stabilizers and drug/stabilizer ratio on the PS of nanoparticles.
Characterization of GNA-NS
As shown in , PS, PDI and ZP of GNA-NS prepared by the optimized formulation are 183.7 ± 3.1 nm, 0.052 ± 0.01 and −22.8 Mv, respectively. It was well known that ZP was one of the significant characteristics for the evaluation of the stability of colloidal systems. ZP was about −22.8 mV in this study, guaranteeing that nanoparticles could be stably dispersed in GNA-NS (Jacobs et al., Citation2000; Kuo et al., Citation2010). The resultant GNA-NS was found to be a uniform colloidal system with opalescent yellow (), whereas crude GNA is visibly insoluble in water suspension (). The loose lyophilized powder with mannitol as cryoprotectant () can be readily re-dispersed in water to reconstruct homogeneous dispersion (). The PS and ZP of nanoparticles in reconstitution GNA-NS were slightly changed to 198.8 nm and −20.6 mV. In the following experiment, re-dispersed lyophilized GNA-NS was used on as the formulation for characterization and in vitro–in vivo evaluation. SEM image shown in clearly demonstrated that nanoparticles in GNA-NS are spherical in shape with an average diameter about 200 nm. The above results demonstrated that GNA-NS improved the water-solubility by nanosizing GNA particles associated with high surface area and high potential energy (Kumar et al., Citation2014). The EE% and DL% of GNA-NS examined by ultrafiltration centrifugation method are 97 and 29.73%, respectively. The high EE% here would be attributed to the poor water solubility of GNA, leading to almost little amount of dissolved molecules in aqueous solution (Wang et al., Citation2013). And the high DL% of GNA-NS is mainly due to the absence of organic matrix in this colloid system.
Figure 3. PS distribution (a) and zeta potential (b) of GNA-NS.
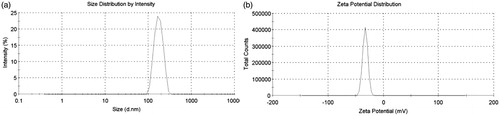
Figure 4. Photographs of GNA-NS (a), crude GNA suspension (b), lyophilized GNA-NS powder (c) and re-dispersed aqueous suspension (d).

Figure 5. SEM images of GNA-NS (a) and crude GNA powder (b).

FT-IR spectra studies
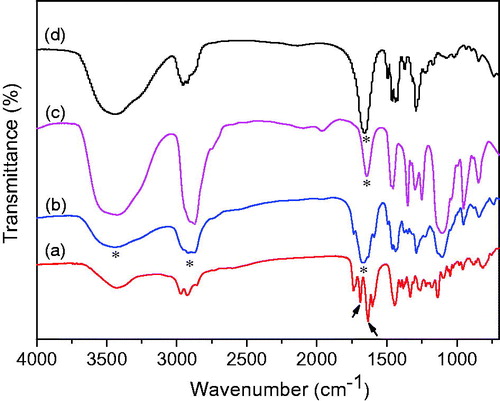
FT-IR spectra shown in were characterized to investigate the interactions between GNA and the excipients, which may affect the dissolution rate of drug (Tran et al., Citation2014). The FT-IR spectrum of GNA () exhibits characteristic bands around ca. 1636.2 and 1688.3 cm-1, which are attributed to the C=O groups on the nuclear parent and carboxylic acid with stretching vibration. Compared to GNA crude drug, these two characteristic bands of the C=O groups were disappeared after transformed to GNA-NS (). Meanwhile, the absorption bands at 1659 () and 1645 cm−1 () assigned to different vibration modes of PVPK30 and PEG2000 have slightly shifted to 1675 cm−1 in GNA-NS. And the C–H vibration bands at 2975, 2930 and 2860 cm−1 accompanied by the O–H absorbance at 3430 cm−1 in the GNA spectrum () have changed to broad peaks in the GNA-NS spectrum (). It can be assumed that there interaction between drug and the excipients. These changes indicated that there are some molecular interactions, such as hydrogen bond, between GNA and excipients in the formation of GNA-NS.
Figure 6. FT-IR spectra of pure GNA (a), GNA-NS (b), PEG2000 (c) and PVPK30 (d).
X-ray powder diffraction
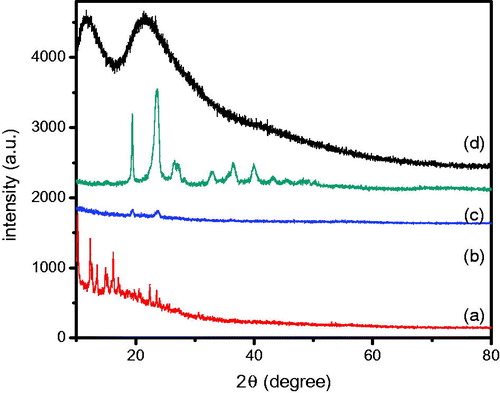
Crystalline state is one of the most important parameters affecting the stability, solubility, dissolution and in vivo efficacy of drugs (He et al., Citation2013). The XRD patters in identified the physical state of GNA in crude GNA and GNA-NS system. showed sharp diffraction peaks near 10–25° for pure GNA, implied good crystallinity in crude powder. However, GNA-NS has no characteristic diffraction peaks for GNA, indicating the dramatically decreased crystallinity of GNA would be likely to transform to amorphous state during the quick precipitation process.
Figure 7. XRD patterns of pure GNA (a), GNA-NS (b), PEG2000 (c) and PVPK30 (d).
Stability of lyophilized GNA-NS
shows the PS of lyophilized GNA-NS powder increased from 198.8 to 203.7 nm at 4 °C and to 209.1 nm at 25 °C during 3 months of storage, accompanied with no obvious changes of zeta potential. Here, the growth inhibition and agglomeration prevention of particles thank to the presence of stabilizers which adsorption at the surface of particles through steric stabilization (Matteucci et al., Citation2006; Zimmermann et al., Citation2009). The powder is more stable than liquid form formulation (Supplementary Table S1) which indicated that the GNA-NS could be converted into solid dosage form.
Table 1. The physical stability of GNA-NS after lyophilization (n = 3).
In vitro release of GNA-NS
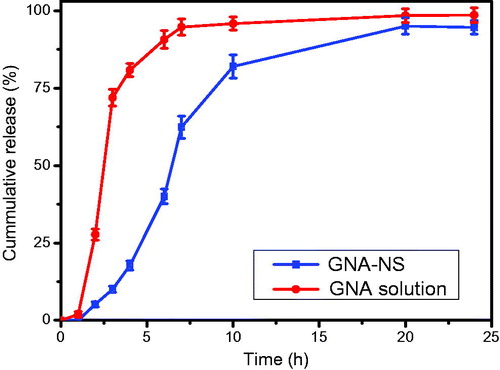
The in vitro dissolution profile of GNA-NS was conducted by the conventional dialysis diffusion technique with GNA solution as control. clearly indicated the faster release of GNA solution than GNA-NS. Approximately 94.7% of the drug in solution has been released within 7 h. On the contrary, 62.4% of the GNA released from GNA-NS in the initial 7 h and achieved to 94% over 24 h. It was found that drug release from GNA-NS close to first-order release equation ln(1 − Mt/97.8) = −0.1633 × t + 0.2047 (Mt means cumulative released quantity, r = 0.9751) which was the prediction of the in vivo sustained release. The difference in vitro drug release rate between GNA-NS and GNA solution inferred that the sustained release properties of GNA-NS by NS technology.
Figure 8. In vitro release profiles of GNA solution and GNA-NS in PBS (0.5% of T-80 in PBS, pH 7.4) at 37 ± 0.5 °C (n = 3).
In vitro anti-tumor studies
The cytotoxicity of GNA solution and GNA-NS on HepG2 cells were assessed using the MTT assay. As illustrated in , the cytotoxicity of GNA solution and GNA-NS toward HepG2 cells was both dose and concentration dependent. The equivalent dose of GNA-NS nanoparticles exhibited superior cytotoxicity than GNA solution. As illustrated in , the IC50 of GNA-NS was 2.22 ± 0.05 μM and the GNA solution was 4.4 ± 0.12 μM. It meant that GNA-NS has significant cytotoxicity (p < 0.05) to HepG2 cells and this enhanced cytotoxicity may be due to nanoparticle uptake.
Figure 9. Cytotoxicity of GNA-NS, GNA solution and blank excipients with various concentrations on HepG2 cells for 48 h. Values are presented as mean ± SD (n = 6).
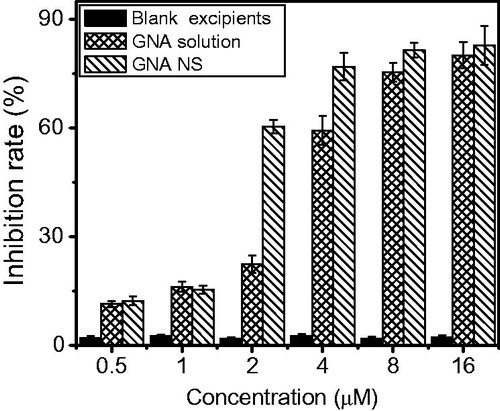
Table 2. IR (%) and IC50 (μM) of HepG2 incubated with solution and GNA-NS for 48 h (n = 6).
In vivo evaluation of GNA-NS
In this study, the in vivo evaluation of GNA-NS was conducted by intraperitoneal administration of GNA-NS to rats with GNA solution as control. The software 3p97 was used to calculate the pharmacokinetic parameters. The plasma drug concentration–time curves (mean ± SD) of the two GNA formulations are presented in and the corresponding pharmacokinetic parameters are listed in . Compared with the GNA solution group, the important pharmacokinetic parameters were remarkably enhanced in GNA-NS. The AUC0–∞ of GNA-NS was increased by 2.63-fold as compared to GNA solution and AUC0–t increased by 1.6-fold, which means GNA-NS delivery system resulted in increased absorption. Meanwhile, the t1/2 and MRT of GNA-NS was longer than that of GNA solution. This enhancement may be attributed to PEG2000, as compared to GNA-NS without PEG modification (Supplementary Figure S1 and Table S2). By characterization of FT-IR, we have confirmed the interaction between GNA and PEG2000. When entered into blood, GNA nanoparticles avoided the recognition of opsonin by the surface adsorption of PEG2000 and resulted a prolonged in vivo circulation time.
Figure 10. Mean concentration–time profiles of GNA-NS and GNA solution in plasma following intraperitoneal administration to rats (n = 6).
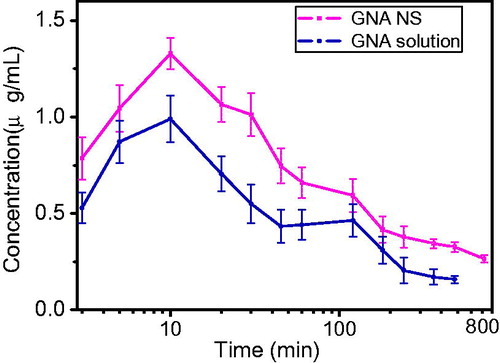
Table 3. Pharmacokinetic parameters of GNA-NS and GNA solution following intraperitoneal administration in rats (mean ± SD, n = 6).
illustrated that the double peaks of maximum concentrations were observed for GNA solution, which may be attributed to the enterohepatic circulation. This was in accordance with the previous study of Huang et al (Huang et al., Citation2013). However, there was no double peaks phenomenon in rats by administration of GNA-NS. As the literature reported, nanoparticulate drug delivery system can overcome the enterohepatic recirculation phenomena (Singh et al., Citation2014). For example, Hong et al. (Citation2014) prepared myricetin NS and the enterohepatic recirculation of myricetin crude drug in rats was subdued by administration of NS formulation.
Conclusions
The present study has successfully developed a novel GNA-NS by simple precipitation method. The SEM morphology and PS analysis manifested that spherical nanoparticles were formed with a mean PS of 183.7 nm. XRD analysis demonstrated that GNA in NS may be in amorphous phase. The lyophilized powder showed good physical stability for 3 months at 25 °C, hinted a transformed forms as solid dosage. As compared to GNA solution, GNA-NS exerted a much slower in vitro dissolution rate and an enhanced cytotoxicity in HepG2 cells. The in vivo pharmacokinetics study in rats revealed that the relative bioavailability of GNA-NS formulation was 263% with longer t1/2 and MRT compared to GNA solution. In conclusion, this research demonstrated that GNA-NS formulation was obviously superior to GNA solution. Hence, GNA-NS with enhanced bioavailability and drug efficacy would be a promising drug delivery system and be worthy of further exploration.
Supplementary material available online
Supplementary Figure S1 and Tables s1 and S2
Supplementary Figure S1 and Tables S1 and S2
Download PDF (39 KB)Acknowledgement
The authors gratefully acknowledge Wang Xiaoshan’s laboratory for the gift samples.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
The authors also acknowledge the funding support from the National Natural Science Foundation of China (Grants 51303006) and the Provincial Natural Science Foundation (1408085MH196, KJ2012ZD09, 10040606Q36) of Anhui Province.
Related Research Data
References
- Aditya NP, Yang H, Kim S, et al. (2015). Fabrication of amorphous curcumin nanosuspensions using-lactoglobulin to enhance solubility, stability, and bioavailability. Colloids Surf B Biointerfaces 127:114–21
- Asano J, Chiba K, Tada M, et al. (1996). Cytotoxic xanthones from Garcinia hanburyi. Phytochemistry 41:815–20
- Bajaj A, Rao MR, Pardeshi A, et al. (2012). Nanocrystallization by evaporative antisolvent technique for solubility and bioavailability enhancement of telmisartan. AAPS PharmSciTech 13:1331–40
- Cerdeira AM, Werner IA, Mazzotti M, et al. (2012). Simultaneous quantification of polymeric and surface active stabilizers of nanosuspensions by using near-infrared spectroscopy. Drug Dev Ind Pharm 38:1360–70
- Chin WWL, Parmentier J, Widzinski M, et al. (2014). A brief literature and patent review of nanosuspensions to a final drug product. J Pharm Sci 103:2980–99
- Fan Y, Peng A, He S, et al. (2013). Isogambogenic acid inhibits tumour angiogenesis by suppressing Rho GTPases and vascular endothelial growth factor receptor 2 signalling pathway. J Chemother 25:298–308
- Gao L, Zhang D, Chen M. (2008). Drug nanocrystals for the formulation of poorly soluble drugs and its application as a potential drug delivery system. J Nanopart Res 10:845–62
- He W, Lu Y, Qi J, et al. (2013). Food proteins as novel nanosuspension stabilizers for poorly water-soluble drugs. Int J Pharm 441:269–78
- Hua X, Liang C, Dong L, et al. (2015). Simultaneous determination and pharmacokinetic study of gambogic acid and gambogenic acid in rat plasma after oral administration of Garcinia hanburyi extracts by LC-MS/MS. Biomed Chromatogr 29:545–51
- Huang X, Chen YJ, Peng DY, et al. (2013). Solid lipid nanoparticles as delivery systems for gambogenic acid. Colloids Surf B Biointerfaces 102:391–7
- Hong C, Dang Y, Lin G, et al. (2014). Effects of stabilizing agents on the development of myricetin nanosuspension and its characterization: an in vitro and in vivo evaluation. Int J Pharm 477:251–60
- Hu X, Chen X, Zhang L, et al. (2014). A combined bottom–up/top–down approach to prepare a sterile injectable nanosuspension. Int J Pharm 472:130–9
- Jacobs C, Kayser O, Müller RH. (2000). Nanosuspensions as a new approach for the formulation for the poorly soluble drug tarazepide. Int J Pharm 196:161–4
- Kumar S, Xu X, Gokhale R, et al. (2014). Formulation parameters of crystalline nanosuspensions on spray drying processing: a DOE approach. Int J Pharm 464:34–45
- Kuo YC, Shih KH, Yang JT. (2010). Capillary electrophoresis of bone marrow stromal cells with uptake of heparin-functionalized poly(lactide-co-glycolide) nanoparticles during differentiation towards neurons. Electrophoresis 31:315–23
- Li Q, Cheng H, Zhu G, et al. (2010). Gambogenic acid inhibits proliferation of A549 cells through apoptosis inducing and cell cycle arresting. Biol Pharm Bull 33:415–20
- Li Y, Wu Z, He W, et al. (2015). Globular protein-coated paclitaxel nanosuspensions: interaction mechanism, direct cytosolic delivery and significant improvement in pharmacokinetics. Mol Pharm 12:1485–500
- Lin T, Fang Q, Peng D, et al. (2013). PEGylated non-ionic surfactant vesicles as drug delivery systems for gambogenic acid. Drug Deliv 20:277–84
- Liu G, Zhang D, Jiao Y, et al. (2013). In vitro and in vivo evaluation of riccardin D nanosuspensions with different particle size. Colloids Surf B Biointerfaces 102:620–6
- Liu Y, Zhang D, Duan C, et al. (2012). Studies on pharmacokinetics and tissue distribution of bifendate nanosuspensions for intravenous delivery. J Microencapsul 29:194–203
- Matteucci M, Hotze M, Johnston K, et al. (2006). Drug nanoparticles by antisolvent precipitation: mixing energy versus surfactant stabilization. Langmuir 22:8951–9
- Singh G, Pai RS. (2014). Optimized PLGA nanoparticle platform for orally dosed trans-resveratrol with enhanced bioavailability potential. Expert Opin Drug Deliv 11:647–59
- Tian X, Li H, Zhang D, et al. (2013). Nanosuspension for parenteral delivery of a p-terphenyl derivative: preparation, characteristics and pharmacokinetic studies. Colloids Surf B Biointerfaces 108:29–33
- Tran TT, Tran PH, Nguyen MN, et al. (2014). Amorphous isradipine nanosuspension by the sonoprecipitation method. Int J Pharm 474:146–50
- Van Eerdenbrugh B, Van den Mooter G, Augustijns P. (2008). Top-down production of drug nanocrystals: nanosuspension stabilization, miniaturization and transformation into solid products. Int J Pharm 364:64–75
- Verma S, Gokhale R, Burgess DJ. (2009). A comparative study of top-down and bottom-up approaches for the preparation of micro/nanosuspensions. Int J Pharm 380:216–22
- Wang Y, Zhang L, Wang Q, et al. (2013). Stability issue of nanosuspensions in drug delivery. J Control Release 172:1126–41
- Wang M, Chen D, Cheng H, et al. (2014). Gambogenic acid kills lung cancer cells through aberrant autophagy. PLoS One 9:e83604
- Wen H, Dong C, Dong H, et al. (2012). Engineered redox-responsive PEG detachment mechanism in PEGylated nano-graphene oxide for intracellular drug delivery. Small 8:760–9
- Xia D, Quan P, Piao H, et al. (2010). Preparation of stable nitrendipine nanosuspensions using the precipitation–ultrasonication method for enhancement of dissolution and oral bioavailability. Eur J Pharm Sci 40:325–34
- Zhang X, Guan J, Ni R, et al. (2014). Preparation and solidification of redispersible nanosuspensions. J Pharm Sci 103:2166–76
- Zhang Y, Zhang J. (2014). Preparation of budesonide nanosuspensions for pulmonary delivery: characterization, in vitro release and in vivo lung distribution studies. Artif Cells Nanomed Biotechnol. [Epub ahead of print]. DOI: 10.3109/21691401.2014.944645
- Zimmermann A, Millqvist-Fureby A, Elema, MR, et al. (2009). Adsorption of pharmaceutical excipients onto microcrystals of siramesine hydrochloride: effects on physicochemical properties. Eur J Pharm Biopharm 71:109–16
- Zhou A, Li QL, Peng DY, et al. (2008). Determination of the content of gambogic acid and gambogenic acid in gamboge by HPLC. J Sep Sci 15:53–4
- Zhou J, Luo Y, Wang J, et al. (2013). Gambogenic acid induction of apoptosis in a breast cancer cell line. Asian Pac J Cancer Prev 14:7601–5