
Abstract
Objective: 16-Dehydropregnenolone (16-DHP) is a potential antitumor compound with poor solubility. A liposome entrapped 16-DHP (16-DHP-LM) formulation was developed to surmount its solubility obstacle. The aim of this study is to investigate the pharmacokinetics of 16-DHP-LM and 16-DHP solution in female mice and tissue distribution of 16-DHP-LM in female tumor-bearing nude mice.
Methods: Rotary-evaporated film method was used to prepare 16-DHP-LM. The comparison of pharmacokinetics between 16-DHP-LM and 16-DHP solution in female mice was investigated after intravenous administration at a single dose of 15 mg/kg. The dose proportionality of 16-DHP-LM was also evaluated after intravenous administration of 16-DHP-LM at the doses of 7.5, 15.0 and 30.0 mg/kg. The tissue distribution of 16-DHP-LM in female tumor-bearing nude mice was evaluated after intravenous administration of 16-DHP-LM at a single dose of 30.0 mg/kg.
Results: The pharmacokinetic study indicated that the 16-DHP-LM group had higher area under the plasma concentration-time curve (AUC), lower apparent volume of distribution (Vz) and smaller systemic clearance (CL) than the 16-DHP solution group. For dose proportionality, good linearity of the pharmacokinetics of 16-DHP after intravenous administration of 16-DHP-LM was observed in the regression analysis of the AUC-dose plot (r = 0.99) and the Cmax-dose plot (r = 0.98). The tissue distribution study showed that the main tissue depots for 16-DHP in tumor-bearing nude mice were plasma, liver, spleen and tumor, which was benefit to anti-tumor effect. All these results provided a significant basis for the design of clinical trial of 16-DHP-LM.
Introduction
16-Dehydropregnenolone (16-DHP, ), a sterols compound, was first identified in the circulation of pre-term neonates (Kobayashi et al., Citation1994), and for the first time isolated from the ethanol extract of the whole plant from Solanum lyratum (Sun et al., Citation2006). It has been reported that 16-DHP was a 17α-hydroxylase and 5α-reductase inhibitor (Ling et al., Citation1998). The research by the Central Drug Research Institute of Lucknow (CDRI) showed 16-DHP had significant hypolipidemic effect in normal as well as in hyperlipidemic subjects, and CDRI had developed 16-DHP as an oral antihyperlipidemic agent (Pratap et al., Citation2005). In our previous study, 16-DHP was proved to be a cytotoxic substance against many kinds of human tumor cells (Sun et al., Citation2006), and the in vitro molecular mechanism study showed that 16-DHP inhibited the growth of human cervical carcinoma cells (HeLa cells) in a time- and dose-dependent via inducing ATM–Chk2–p53 activation-mediated G1 arrest and mitochondrial cell apoptosis (Ma et al., Citation2012). Further study indicated that 16-DHP can inhibit the growth of tumor on human cervical cancer xenograft mouse tumor models (Qin et al., Citation2015) with less adverse effect, which had been declared in a patent (Pratap et al., Citation2005). The above findings suggested that 16-DHP maybe a potential antitumor compound for the treatment of human cervical cancer.
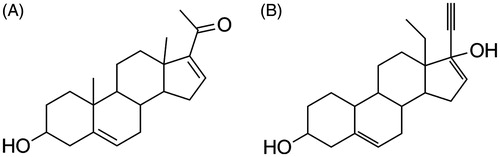
Figure 1. Chemical structure of 16-DHP (A) and levonorgestrel (B).
Pharmacokinetics was necessary and vital in the course of development of new drugs. In recent years, pharmacokinetic behavior of 16-DHP after oral and intramuscular administration have been studied. Nevertheless, the results showed 16-DHP had low bioavailability, quick absorption and rapid elimination (Singh et al., Citation2003; Suryawanshi et al., Citation2006, Citation2011; Yang et al., Citation2011; Kumar et al., Citation2012). For example, its elimination half-lives (t1/2) were 2.7 h and 2.5 h for oral and intramuscular administrations, respectively. Thus, intravenous administration is an alternative, which provides greater bioavailability, faster therapeutic effect and lower individual difference. However, due to its hydrophobic nature, the application of 16-DHP was limited by the poorly aqueous solubility. During the previous study of pharmacokinetics after intravenous administration, researchers tried to dissolve 16-DHP with mixed organic solvents which can cause venous irritation and occasionally thrombophlebitis (Suryawanshi et al., Citation2006; Kumar et al., Citation2012). For this type of drugs, researchers have been trying to improve their water solubility and pharmacokinetic behaviors by means of pharmaceutical method, such as hydroxypropyl-β-cyclodextrin (HP-β-CD). However, the published HP-β-CD formation need to be prepared just prior to administration into the tail vein (Kumar et al., Citation2012). These formulations were not completely satisfying. To surmount this solubility obstacle, liposome was considered to be a potential drug delivery system for many advantages, such as biocompatibility and biodegradation (Robert et al., Citation1983), targeting and specificity (Akhter et al., Citation2013; Arpicco et al., Citation2013), increasing antitumor activity (Sadzuka et al., Citation1998; Guchelaar, Citation2012; Tian et al., Citation2014), improving pharmacokinetics and distribution (Minko et al., Citation2006; Huang et al., Citation2014; Ma et al., Citation2014).
In a previous study, we have developed a freeze-dried liposome formulation for 16-DHP (Deng et al., Citation2015). Since the drug is incorporated into one or more lipid bilayers, its pharmacokinetic profile may be completely altered (Jia et al., Citation2002; Immordino et al., Citation2003). Considering its anti-tumor efficacy on cervical cancer, there is an increasing demand for researching pharmacokinetics of 16-DHP-LM on female. In this paper, the studies of pharmacokinetics and tissue distribution of 16-DHP-LM after intravenous administration on female mice were carried out. The high performance liquid chromatography (HPLC) method with UV detection has been previously developed and validated for the determination of 16-DHP in male rat plasma (Yang et al., Citation2011). In the present study, the assay was partially validated to evaluate the pharmacokinetic difference between 16-DHP and 16-DHP-LM in female mice and the tissue distribution of 16-DHP-LM in the tumor-bearing female nude mice.
Experimental
Chemicals and reagents
16-Dehydropregnenolone (16-DHP, purity ≥99.5%) was synthesized in School of Pharmacy, Shenyang Pharmaceutical University (Shenyang, China). Levonorgestrel (IS, purity >98.0%) was purchased from National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Human cervical carcinoma cells (HeLa cells) were cultured and passaged in our own laboratory. Acetonitrile (HPLC grade), n-hexane (HPLC grade) and ammonium acetate (AR grade) were obtained from Shandong Yuwang Chemical Factory (Shandong, China). Purified water was purchased from Hangzhou Wahaha Group Co., Ltd. (Hangzhou, China).
Animals
Female Kunming mice (18–22 g) and female BALB/c-nu mice (16–18 g) were obtained from the Laboratory Animal Center of Shenyang Pharmaceutical University, and kept in an environmentally controlled room (temperature: 25 ± 2 °C, humidity: 50 ± 5%, 12-h dark-light cycle) for 1 week in sterile conditions. For pharmacokinetics study, the female Kunming mice were fasted overnight but had free access to water before the day of the experiment. For tissue distribution study, female nude mice were inoculated subcutaneously (s.c.) in right flank with 0.2 mL of human cervical carcinoma cell (Hela) suspension with the cell density of 1 × 107. The tissue distribution study was carried out when the weight of tumor-bearing mice reached 18–22 g. All animal-use procedures were in accordance with principles in the Declaration of Helsinki.
Chromatographic conditions
The analysis of 16-DHP was performed by a Shimadzu Prominence HPLC system (Shimadzu, Japan) equipped with LC-Solution workstation, binary pumps LC-20A and detector SPD-20A. Chromatography separation was achieved using a reversed phase Diamonsil C18 column (200 × 4.6 mm i.d., 5 μm, Dikma Technologies, Beijing, China) at a temperature of 30 °C. Isocratic elution mode was applied at the flow rate of 1.0 mL/min with the mobile phase of acetonitrile-10 mM ammonium acetate (49:51, v/v) (for pharmacokinetic study) or acetonitrile-0.1% formic acid aqueous solution (47:53, v/v) (for tissue distribution study). The detection was performed at 238 nm. The injection volume was 20 μL of each sample.
Preparation of 16-DHP solution and 16-DHP liposome
For 16-DHP solution
16-Dehydropregnenolone was weighed and dissolved by an ultrasonic method in the mixture of dimethylacetamide:propylene glycol:polyethylene glycol 400–5% glucose aqueous solutions (10:20:30:40, v/v/v/v) to obtain 16-DHP solution with a concentration of 3 mg/mL. The clear solution was filtered through 0.22 μm sterile membrane filter before use.
For 16-DHP-LM (containing 3 mg/mL of 16-DHP)
The dichloromethane solution composed of lecithin, sodium cholesteryl sulfate and 16-DHP was placed in a round-bottomed flask. The solvent was removed by vacuum rotary evaporation at 60 °C until a dry thin film was formed in the sidewall. The film was then hydrated in a defined volume of phosphate buffered saline (PBS, 10 mM, pH 7.4) until the film was completely dissolved at 60 °C. The suspension was dealt with ultrasonication in ice bath for 2 min. Glucose (7.5%, w/v) was then added to the fresh prepared 16-DHP-LM as cryoprotectant. To obtain a homogenous liposome suspension, the mixture was then filtered through 0.8, 0.45 and 0.22 μm millipore membrane filters successively. The 16-DHP-LM was transferred to the vials and freeze-dried on a laboratory freeze drier for 48 h. The average particle size and zeta potential of the prepared liposome were determined by dynamic light scattering with a Nicomp™ 380 (Particle Sizing System, Santa Barbara, CA), which showed that the liposome had the average sizes of 121.3 ± 48.7 nm with the zeta potential of −41.9 mV. The entrapment efficiency (EE%) determined by previous method was 98.7 ± 0.1% (Deng et al., Citation2015).
The lyophilized preparation was stored at 4 °C and rehydrated with the corresponding amount of water in subsequent experiments. Aseptic procedure was followed during preparation.
Preparation of standard and quality control solutions
Stock solutions of 16-DHP were prepared in acetonitrile at concentrations of 1000 μg/mL (for pharmacokinetic study) and 100 μg/mL (for tissue distribution study). The working standard solutions of 16-DHP for calibration curves (0.1–100.0 μg/mL for plasma, 1.0–100.0 μg/g for tissue) were prepared by serial dilution of stock solution with acetonitrile, respectively. The internal standard working solution at the concentrations of 5.0 (for pharmacokinetic study) and 2.0 μg/mL (or tissue distribution study) were prepared by dissolving the accurately weighed standard compound levonorgestrel (IS) with acetonitrile. QC solutions of three concentrations (0.2, 4.0, 80 μg/mL for pharmacokinetic study; 1.5, 15, 80 μg/mL for tissue distribution study) were prepared for the validation of the developed method. Another set of QC solutions, the same concentrations as the QC solutions in plasma above, were prepared with 16-DHP-LM by rehydrating and diluting the freeze-dried 16-DHP-LM with water for the study of extraction recovery. All the working solutions were kept under 4 °C and brought to room temperature before use.
Sample preparation
Sample extraction was performed using the liquid-liquid extraction (LLE). Plasma (100 μL) or tissue homogenate (200 μL) was added with 10 μL (20 μL for tissue homogenate) of IS solution and 10 μL (20 μL for tissue homogenate) of acetonitrile in a 5 mL Eppendorf tube, then vortexed for 30 s. The mixture sample was then extracted twice with 2 mL of n-hexane each time, and centrifuged at 10 000 rpm for 5 min, the combined supernatant was transferred into another Eppendorf tube and dried under a gentle nitrogen gas stream at 40 °C. The residue was reconstituted in 50 μL (100 μL for tissue homogenate) acetonitrile. After centrifugation at 12 000 rpm for 5 min, 20 μL of the supernatant was injected into HPLC-UV system for analysis.
Method validation
A laboratory scheme based on international guidelines was used for the validation procedure (US DHHS, FDA and CDER, Citation2001).
For pharmacokinetics study
Specificity
Specificity was assessed by comparing chromatograms of six different batches of mouse blank plasma, plasma sample spiked with 16-DHP and IS, and plasma samples after intravenous administration of 16-DHP-LM.
Calibration curve and LLOD
The calibration curves were prepared and assayed in duplicate in three consecutive days to evaluate linearity. The linearity of each calibration curve was determined by plotting the peak area ratio (y) of the analyte to IS versus the nominal concentration (x) of the analyte with weighted (1/x2) least-square linear regression.
The lower limit of quantification (LLOQ) was defined as the lowest concentration on the calibration curve and evaluated by analyzing the samples prepared in six replicates. Acceptable criteria for precision and accuracy of LLOQ were less than 20%.
Precision and accuracy
The intra- and inter-day precision and accuracy of the method were assessed by determining QC samples using six replicates at three concentrations levels on three different validation days. The precision, defined as RSD (%), were obtained using a one-way analysis of variance (ANOVA) by SPSS 16.0 (SPSS Inc., Chicago, IL) and should not exceed 15%. The accuracy, defined as RE (%), required to be within ±15%.
Extraction recovery
The method developed for the pharmacokinetic study of 16-DHP-LM after intravenous administration was aimed to quantify the total 16-DHP, which included free and liposome-entrapped 16-DHP. So the extraction recovery of 16-DHP-LM was necessary to confirm that the 16-DHP can be extracted from the liposome formation.
The extraction recoveries of 16-DHP and 16-DHP-LM were determined by comparing the peak areas of 16-DHP from extracted samples spiked with 16-DHP standard solutions and 16-DHP-LM at three QC levels with those from post-extracted blank plasma samples spiked with 16-DHP standard solutions at the same concentration. Each concentration level was prepared in sextuplicate. Extraction recovery of the IS was determined in the same way at the concentration of 500 ng/mL.
Stability
Prepared sample stability was assessed by triple measurements of low, medium and high QC samples in bench-top (25 °C for 12 h), three complete freeze-thaw cycles (−20 °C to 25 °C) and long-term sample storage (−20 °C for 7 days).
Dilution integrity
The dilution integrity experiment was required if some real subject sample concentrations are expected to be higher than the upper limit of quantification. Dilution integrity experiments were carried out by a 10-fold dilution of the plasma samples to 3.0 μg/mL with blank plasma for six replicates. The acceptable precision and accuracy were required to be within ±15%.
For tissue distribution study
Tissues (heart, liver, spleen, lungs, kidney, brain, tumor, uterus and ovary, intestine, stomach and pancreas) from tumor xenograft nude mice were weighed and homogenized in 5-fold solvent (acetonitrile:0.9% NaCl, 20:80, v/v). Blank tissue homogenates was spiked with 16-DHP stock solutions (100 μg/mL) to prepare quality control samples. Predetermined volumes of the 16-DHP stock solution were added separately to 200 μL homogenates to obtain 16-DHP concentrations of 0.1, 1.5, 0.2, 0.5, 1.0, 1.5, 2.0, 5.0, 8.0 and 10 μg/g. The actual concentration ranges may differ slightly in various tissues. Plasma from female tumor-bearing nude mice was prepared by the same method as pharmacokinetic study.
16-Dehydropregnenolone in the homogenates was extracted using the liquid phase extraction method described above. The specificity, linearity, lower limit of quantification, precision and accuracy, extraction efficiency and stability (25 °C for 24 h, three complete freeze-thaw cycles and long-term sample storage at −20 °C for 30 days) of HPLC method were evaluated. Some tissues including uterus and ovary, intestine, stomach and pancreas were of little quantity, so only specificity and linearity were investigated.
Pharmacokinetic study in female mice and statistical analysis
The female mice were randomly divided into four groups and each time point was based on samples from six different mice. Three groups of mice received doses of 30, 15 and 7.5 mg/kg by injecting 16-DHP-LM via the tail vein, respectively. The fourth group of mice received 16-DHP solution intravenously in the same way at a single dose of 15 mg/kg. Blood samples were collected into heparinized Eppendorf tubes at 0.083, 0.17, 0.25, 0.5, 1, 1.5, 2, 4, 8, 12, 24, and 48 h after administration. After centrifugation at 3000 rpm for 10 min, the supernatant was transferred and stored at −20 °C until analysis.
The plasma concentration versus time curves were plotted and all the pharmacokinetic parameters of 16-DHP, including the peak plasma concentration (Cmax), the AUC from 0 to time (AUC0–t), the AUC from 0 to infinity (AUC0–∞), total plasma clearance (CL), apparent volume of distribution (Vz) and terminal elimination half-life (t1/2) were calculated by the non-compartmental analysis using the DAS 2.1.1 software supplied by the Pharmacological Society of China (Beijing, China). Results were reported as means ± SD (SD: standard deviation). The comparison of pharmacokinetic parameters between administration of the 16-DHP-LM and 16-DHP solution was possessed by SPSS 16.0 (Statistical Package for the Social Science) using independent samples t-test after their natural logarithmic transformation or the Mann–Whitney test, p < 0.05 was considered statistically significant for all the tests.
Tissue distribution of 16-DHP-LM in tumor xenograft nude mice
Sixteen female tumor xenograft nude mice (20 ± 2 g) were used in the tissue distribution studies. After tail vein injection of 16-DHP-LM at a dose of 30 mg/kg, blood sample was collected at 0.17, 2, 8 and 12 h, then the mice were killed and organs (heart, liver, spleen, lung, kidney, brain, tumor, uterus and ovary, intestine, stomach and pancreas) were collected at the same time. The blood samples were prepared as described above. The tissue samples were rinsed in ice-cold normal saline, blotted dry with filter paper, and then stored at −20 °C until analysis. About 0.2 g of the tissue samples (taking 0.1 g tissue sample if the total weight of the tissue is 0.1–0.2 g, and taking all the tissue sample and recording the weight if the total weight of the tissue is less than 0.1 g) were accurately weighed and homogenized with 5-fold solvent (acetonitrile-0.9% NaCl, 20:80, v/v) and then extracted with n-hexane and used for analysis by HPLC system.
The concentration of 16-DHP in plasma and tissue at each point was determined and the AUC of 16-DHP in each tissue was calculated by DAS 2.1.1 software. Drug targeting efficiency (Te = AUCtissue/AUC) was used to evaluate the targeting property of 16-DHP-LM in tumor xenograft nude mice model.
Results and discussions
Method validation
Specificity
The typical chromatograms of blank matrix (plasma or tissue homogenate), blank matrix with 16-DHP and real matrix samples are shown in and , respectively. No endogenous components were found to interfere with 16-DHP and IS peaks, which indicated good selectivity for the method.
Figure 2. Representative HPLC chromatograms of 16-DHP and IS in mice plasma for pharmacokinetic study: (A) blank plasma of mice, (B) plasma spiked with 16-DHP (4 μg/mL) and IS, (C) plasma sample 1.0 h after i.v. of 16-DHP-LM (1, IS; 2, 16-DHP).
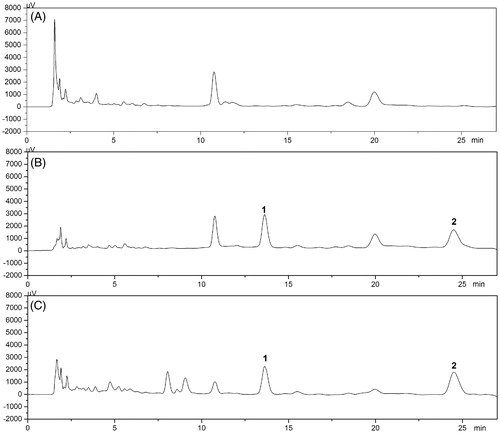
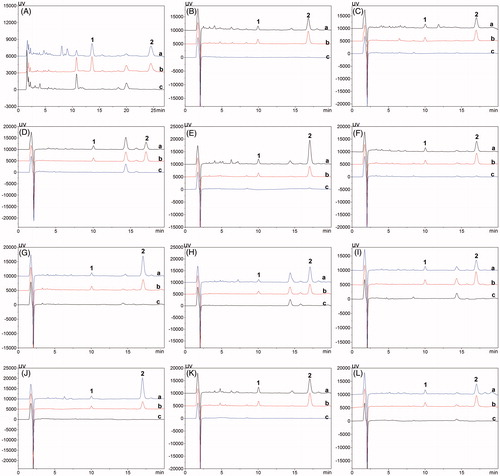
Figure 3. Typical HPLC chromatograms of IS (1, Plasma 5 μg mL−1, tissue 2 μg mL−1) and 16-DHP (2) in Plasma (A), Heart (B), Liver (C), Spleen (D), Lung (E), Kidney (F), Pancreas (G), Intestine (H), Stomach (I), Brain (J), Tumor (K), Uterus and Ovary (L) for tissue distribution study. (a) Biological sample collected after administration at 0.17 h; (b) Blank biological sample spiked with IS and 16-DHP; (c) Blank biological sample.
Calibration curve and LLOD
The linear regressions of the peak area ratios versus concentration were fitted over the concentration range of 0.01–10.0 μg/mL for 16-DHP in plasma, and 0.1–10 μg/g for 16-DHP in tissue. Typical calibration curves for plasma and different tissues are listed in . The correlation coefficient (r) exceeded 0.99, showing a good linearity among the concentration range.
Table 1. Calibration curves of 16-DHP in mouse plasma (μg/mL) and tissue homogenate (μg/g).
An LLOQ of 0.01 μg/mL was obtained for 16-DHP in plasma, which was a significant improvement over the existing HPLC-UV method (Singh et al., Citation2003; Yang et al., Citation2011). The RSD was 11.2%, and the RE was within ±17.3% at LLOQ level, which was within the accepted limits.
Precision and accuracy
The results of the intra- and inter-day precision and accuracy of the analytes in QC samples are summarized in . The intra- and inter-day RSD were below 14.3%, and the relative errors were less than ±6.6%. All the values were within the accepted range.
Table 2. Inter- and intra-precision and accuracy of the method for 16-DHP in mouse plasma and tissue homogenate (n = 6).
Extraction recovery
In the present method, extraction recovery of 16-DHP-LM was assessed to confirm that 16-DHP entrapped in liposome can be extracted by the above plasma sample preparation method. The average extraction recoveries in plasma showed no significant difference between 16-DHP and 16-DHP-LM at three concentration levels. The results were stable and not less than 92.9% for plasma (). Therefore, the QC samples prepared by 16-DHP standard solutions were acceptable during the method validation and the analysis of real samples. The average extraction recoveries of IS was 93.4%. The recoveries of 16-DHP and IS for all the tissue homogenate were over 86.4% and 86.6%, respectively.
Table 3. Recovery of 16-DHP, 16-DHP-LM and IS in mice plasma, 16-DHP and IS in tissue homogenate (n = 6).
Stability
The stability of 16-DHP in mouse bio-sample was investigated under a variety of storage and process conditions. The results showed that 16-DHP was stable in mouse plasma through three freeze-thaw cycles (RE in range −9.8–7.6%), at a temperature of −20 °C for 7 days (RE in range −4.8–4.2%) and at room temperature for 12 h after sample preparation (RE in range −10.2–5.2%). The analyte was also found to be stable in tissue (heart, liver, spleen, lung, kidney, brain and tumor) homogenate through three freeze–thaw cycles (RE in range −11.1–13.7%), at a temperature of −20 °C for 30 days (RE in range −11.0–13.7%) and at room temperature for 24 h after sample preparation (RE in range −13.9 – 13.8%). Therefore, the method can be used for routine analysis of 16-DHP in plasma and tissue homogenate samples.
Dilution integrity
The determination of dilution integrity established the sample dilution factor which conserves linearity in response. Therefore, QC samples of plasma were prepared at a concentration of 30.0 μg/mL in triplicate and then diluted 10-fold with blank plasma to give a concentration of 3.0 μg/mL. The assay precision and accuracy were tested using the same sample pretreatment method. For diluted samples, the precision was 2.8%, and the accuracy was within ±4.5%.
Pharmacokinetic study
The validated method was successfully applied to determine the plasma concentration of 16-DHP in mice after intravenous administration of 16-DHP-LM (7.5, 15 and 30 mg/kg) and 16-DHP solution (15 mg/kg). The mean plasma concentration of 16-DHP versus time profiles are shown in , and the pharmacokinetic parameters are listed in .
Figure 4. The mean plasma concentration-time curves of 16-DHP following intravenous administration of 7.5 (LM-7.5 mg/kg), 15 (LM-15 mg/kg), 30 (LM-30 mg/kg) mg/kg of 16-DHP-LM and 15 mg/kg of 16-DHP solution (S-15 mg/kg) in female mice, the data were presented as mean ± SD (n = 6).
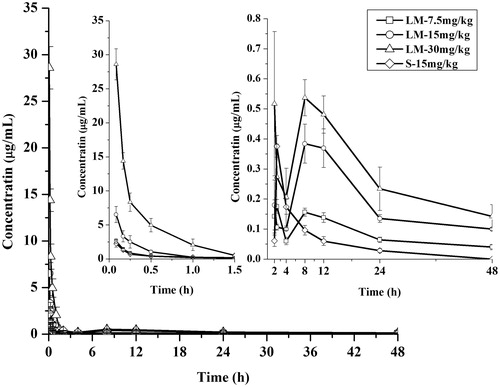
Table 4. Pharmacokinetic parameters for 16-DHP in female mice after intravenous administration of various doses of 16-DHP-LM and 15 mg/kg of 16-DHP solution (mean ± SD; n = 6).
As shown, there were no significant differences in t1/2, Vz and CL showed among the three doses of treatments after intravenous administration of 16-DHP-LM. Cmax values were estimated to be 2.62, 6.52 and 28.61 μg/mL after intravenous dose of 7.5, 15 and 30 mg/kg of 16-DHP-LM, respectively. The AUC increased with increasing doses, and the AUC(0-t) values were 4.75, 10.83 and 23.20 μg h/mL, respectively. These data indicated that the AUC(0-t) and Cmax were dose-dependent. Good correlation was observed in the regression analysis of the AUC-dose plot (r = 0.99) and the Cmax-dose plot (r = 0.98), suggesting that the 16-DHP-LM may have linear pharmacokinetic characteristics in female mice within the dose ranges tested.
Comparison of the mean plasma concentration-time curves of 16-DHP following intravenous administration of 15 mg/kg of 16-DHP-LM and 16-DHP solution showed that the 16-DHP concentration in mice plasma rapidly decreased in the 16-DHP-LM-treated group, but it was higher than that of the 16-DHP-solution-treated group during the first four sampling points. (LM-15 and S-15) showed that the pharmacokinetic parameters of 16-DHP-LM and 16-DHP solution treatment group were significantly different. The plasma Cmax, AUC(0-t) and t1/2 of 16-DHP-LM increased significantly compared with those of 16-DHP solution (p < 0.05). For example, the AUC(0-t) of 16-DHP-LM was 10.83 μg h/mL, which was 3.6 times higher than that of the 16-DHP solution. The t1/2 of 16-DHP-LM was 20.49 h, which was 3.4 times higher than that of the 16-DHP solution. The CL values of 16-DHP-LM were lower than that of the 16-DHP solution. These results showed that the 16-DHP-LM had a long circulation time in plasma, which could be explained by the drug release from liposomes (Shi et al., Citation2010) and the size of the liposomes, which was 120 nm in the current study. It was reported that large liposomes have had short circulation half-life as they can be eliminated very rapidly from circulation by a complement-mediated phenomenon (Bradley et al., Citation1988; Szebeni et al., Citation2000), and end up in the organs of reticuloendothelial system (RES). On the other hand, small liposomes (<200 nm) can circumvent RES uptake because they can reduce the recognition by circulating opsonins (Awasthi et al., Citation2003), and are likely to accumulate between the different cell types within the liver (hepatocytes or Kupffer cells) due to their ability to cross the vascular endothelium and largely to avoid the first phagocytes encountered (Phan et al., Citation2005). The 16-DHP-LM accumulated in the liver might release slowly into blood circulation, which might increase the plasma concentration reflected from the two peaks in the mean plasma concentration-time curves of the 16-DHP-LM group. At the same time, this release characteristic could also help 16-DHP-LM remain a longer blood level compared with 16-DHP solution ().
Tissue distribution of 16-DHP-LM in tumor xenograft nude mouse model
The 16-DHP concentration and distribution in different bio-samples are listed in . As can be seen, 16-DHP was widely and rapidly distributed into most tissues following i.v. administration of 16-DHP-LM. After administration, 16-DHP was first taken up a considerable extent by the lung, liver and tumor, followed by the pancreas, spleen, heart, kidney, and other organs. 16-DHP in the brain could be detected by the HPLC after intravenous injection of 16-DHP-LM, suggesting that 16-DHP could penetrate through the blood brain barrier. Eight hours after the administration of 16-DHP-LM, 16-DHP got the highest distribution in tumor, and the percentage distribution in uterus and ovary was 10.2%, which was beneficial to anti-tumor effect. The percentage distribution in liver was higher in 8 h than 2 h, which may be due to the larger accumulated speed than the elimination in the liver. As shown in , tissue distribution of 16-DHP in all tissues had the highest concentration at 0.17 h. 16-DHP may distribute rapidly to all tissues after intravenous administration, the concentration may reach to the highest level before 0.17 h in some tissue with high blood flow. But in this study, 0.17 h was the first time point, which may not reflect the Tmax in each tissue. This phenomenon also appeared in other literature (Li et al., Citation2015).
Figure 5. Tissue distribution of 16-DHP following intravenous administration of 16-DHP-LM at a dose of 30 mg/kg (n = 4).
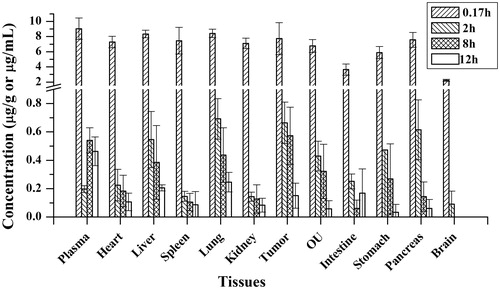
Drug-targeting efficiency (Te = AUCtissue/AUC) was used to evaluate the targeting property of 16-DHP-LM in tumor xenograft nude mouse model, listed in . As can be seen, 16-DHP tends to distribute in plasma, liver, spleen and tumor.
Table 5. Te of 16-DHP in mice plasma and tissues after intravenous administration of 16-DHP-LM.
Conclusions
This study is the first to evaluate the pharmacokinetics and tissue distribution of 16-DHP-LM in mice after intravenous administration. The liposome formation prepared in the present study can improve the pharmacokinetic property of 16-DHP by increasing the t1/2 and reducing the CL of 16-DHP, which makes 16-DHP circulate for a prolonged time compared with solution formation. The dose proportionality study had shown the linear pharmacokinetics of 16-DHP-LM in female mice after intravenous administration at a single dose from 7.5 to 30 mg/kg. For tissue distribution, 16-DHP tends to distribute in plasma, liver, spleen and tumor in tumor-bearing nude mice after intravenous administration of 16-DHP-LM, which was a benefit for its anti-tumor effect. The pharmacokinetic and tissue distribution results suggested that liposome was expected to become clinical formulation for 16-DHP, and provided significant basis for its clinical application.
Declaration of interest
We declare that we have no conflict of interest.
This work was supported by the Project of Liaoning Distinguished Professor (2014), the Program for Liaoning Innovative Research Team in University (No. LT 20121018) and Shenyang Office of Science and Technology (No. F11-148-9-00).
References
- Akhter A, Hayashi Y, Sakurai Y, et al. (2013). A liposomal delivery system that targets liver endothelial cells based on a new peptide motif present in the ApoB-100 sequence. Int J Pharm 456:195–201
- Arpicco S, Lerda C, Pozza ED, et al. (2013). Hyaluronic acid-coated liposomes for active targeting of gemcitabine. Eur J Pharm Biopharm 85:373–80
- Awasthi VD, Garcia D, Goins BA, Phillips WT. (2003). Circulation and biodistribution profiles of long-circulating PEG-liposomes of various sizes in rabbits. Int J Pharm 253:121–32
- Bradley AJ, Devine DV, Ansell SM, et al. (1988). Inhibition of liposome-induced complement activation by incorporated poly(ethylene glycol)-lipids. Arch Biochem Biophys 357:185–94
- Deng ZX, Zhang S, Xie YT, et al. (2015). Preparation and quality evaluation of lyophilized 16-dehydropregnenolone liposome. Herald Med 34:365–70
- Guchelaar GH. (2012). Liposomal drug formulations in cancer therapy: 15 years along the road. Drug Discov Today 17:160–6
- Huang AW, Su ZG, Li S, et al. (2014). Oral absorption enhancement of salmon calcitonin by using both N-trimethyl chitosan chloride and oligoarginines-modified liposomes as the carriers. Drug Deliv 21:388–96
- Immordino ML, Brusa P, Arpicco S, et al. (2003). Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing docetaxel. J Control Release 91:417–29
- Jia L, Garza M, Wong H, et al. (2002). Pharmacokinetic comparison of intravenous carbendazim and remote loaded carbendazim liposomes in nude mice. J Pharm Biomed Anal 28:65–72
- Kobayashi Y, Tagawa N, Saiki K, Watanabe F. (1994). Studies on steroids in fetuses and neonates: identification of 16-dehydropregnenolone in the circulation of pre-term neonates. Biol Pharm Bull 17:1501–4
- Kumar D, Khanna AK, Pratap R, et al. (2012). Dose escalation pharmacokinetics and lipid lowering activity of a novel farnesoid X receptor modulator: 16-dehydropregnenolone. Indian J Pharmacol 44:57–62
- Li Y, Zhang Y, Yang T, et al. (2015). Pharmacokinetics and tissue distribution study of Isovitexin in rats by HPLC-MS/MS. J Chromatogr B 991:13–20
- Ling YZ, Li JS, Kateuya K, et al. (1998). Synthesis and in vitro activity of some epimeric 20α-hydroxy, 20-oxime and aziridine pregnene derivatives as inhibitors of human 17α-hydroxylase/C17,20-lyase and 5α-reductase. Bioorg Med Chem Lett 6:1683–93
- Ma B, Li XT, Li J, et al. (2014). Quantitative analysis of tenuifolin concentrations in rat plasma and tissue using LC–MS/MS: application to pharmacokinetic and tissue distribution study. J Pharm Biomed Anal 88:191–200
- Ma EL, Zhao DM, Li YC, et al. (2012). Activation of ATM–Chk2 by 16-dehydropregnenolone induces G1 phase arrest and apoptosis in HeLa cells. J Asian Nat Prod Res 14:1–9
- Minko T, Pakunlu RI, Wang Y, et al. (2006). New generation of liposomal drugs for cancer. Anticancer Agents Med Chem 6:537–52
- Phan G, Herbet A, Cholet S, et al. (2005). Pharmacokinetics of DTPA entrapped in conventional and long-circulating liposomes of different size for plutonium decorporation. J Control Release 110:177–88
- Pratap R, Gupta RC, Chander R, et al. (2005). U.S. Patent No. 6,875,758. Washington, DC: U.S. Patent and Trademark Office
- Qin Z, Deng ZX, Liu X, et al. (2015). Study on antitumor effect of 16-dehydropregnenolone liposomes. Chin J Pharm, in press
- Robert ES, Denise CY, Mark AR, Vincent HLL. (1983). Effects of topically applied liposomes on disposition of epinephrine and insulin in the albino rabbit eye. Int J Pharm 13:263–72
- Sadzuka Y, Hirotsu S, Hirota S. (1998). Effect of liposomaliazation on the antitumaor activity, side-effects and tissue distribution of CPT-11. Cancer Lett 127:99–106
- Shi S, Chen H, Lin X, Tang X. (2010). Pharmacokinetics, tissue distribution and safety of cinnarizine delivered in lipid emulsion. Int J Pharm 383:264–70
- Singh SK, Mehrotra N, Sabarinath S, Gupta RC. (2003). HPLC-UV method development and validation for 16-dehydropregnenolone, a novel oral hypolipidaemic agent, in rat biological matrices for application to pharmacokinetic studies. J Pharm Biomed Anal 33:755–64
- Sun LX, Fu WW, Ren J, et al. (2006). Cytotoxic constituents from Solanum lyratum. Arch Pharm Res 29:135–9
- Suryawanshi S, Singh SK, Gupta RC. (2006). A sensitive and selective HPLC/ESI-MS/MS assay for the simultaneous quantification of 16-dehydropregnenolone and its major metabolites in rabbit plasma. J Chromatogr B 830:54–63
- Suryawanshi S, Wahajuddin, Gupta RC, Singh SK. (2011). Preclinical pharmacokinetics, dose proportionality, gender difference and protein binding study of 16-dehydropregnenolone, an antihyperlipidemic agent, in rats. J Pharm Pharmacol 63:41–8
- Szebeni J, Baranyi L, Savay S, et al. (2000). Liposome-induced pulmonary hypertension: properties and mechanism of a complement-mediated pseudoallergic reaction. Am J Physiol 279:1319–28
- Tian JL, Wang L, Wang L, Ke X. (2014). A wogonin-loaded glycyrrhetinic acid-modified liposome for hepatic targeting with anti-tumor effects. Drug Deliv 21:553–9
- US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research and Center for Veterinary Medicine. (2001). Guidance for industry: bioanalytical method validation. Available from: http://www/fda.gov/cder/guidance/index.htm [last accessed 18 Apr 2015]
- Yang HY, Zhang WW, Yang WW, et al. (2011). Pharmacokinetic behavior of 16-dehydropregnenolone after intramuscular administration in rats. J Pharm Anal 1:135–8