Recessive mutations in zinc finger protein 469 (ZNF469) cause the brittle cornea syndrome (Mendelian Inheritance in Man #229200), characterized by fragile thin ectatic corneas that are easily ruptured, blue sclera, and extraocular skeletal and/or connective tissue manifestations (e.g., frail skin, scoliosis, joint laxity, hearing loss).Citation1–3 In all children and particularly in adults confirmed to be homozygous for one of four reported ZNF469 mutations,Citation1–3 there were clinically-evident extraocular skeletal and/or connective tissue manifestations. We document a novel homozygous ZNF469 mutation in an adult with corneal fragility but lacking clinical evidence for extraocular manifestations.
The patient, a 37-year-old Saudi Arabian male with decreasing vision and a history for congenital glaucoma, had undergone unspecified ocular surgery in his left eye during early childhood. At 12 years of age he sustained a blunt trauma to his left eye and underwent multiple anterior segment and posterior segment ocular surgeries but soon thereafter had complete loss of vision in that eye (). Visual acuity was 20/200 in the right eye (no improvement with pinhole) and no light perception in the left eye, which was phthisical. Slit-lamp examination of the right eye was significant for a thin ectatic cornea with an old inferotemporal Descemet break that resembled Haab striaeCitation4 (). Following cycloplegia (tropicamide 1%), retinoscopy was attempted but scissoring of the retinoscopic reflex precluded accurate refraction. Indirect ophthalmoscopy revealed an unremarkable fundus. The optic nerve had a central round 0.4 cup that did not appear glaucomatous. Orbscan (Bausch & Lomb, Rochester, New York, USA) data from the right eye were as follows: 13.5 mm white to white, 233 microns central corneal thickness, central irregular astigmatism with ectasia. IOLMaster (Zeiss, Oberkochen, Germany) data from the right eye were as follows: white-to-white 12.9 mm, keratometry 42.51 D/53.91 D @143, anterior chamber depth 5.5 mm, axial length 27.37 mm. This ophthalmic examination suggested a diagnosis of brittle cornea syndrome rather than congenital glaucoma.
FIGURE 1 Clinical image showing that, at presentation, the patient had phthisis in the left eye. The cornea in the right eye appears large and the pupil is pharmacologically dilated. A Descemet membrane break can be seen inferotemporally. The sclera does not appear blue.
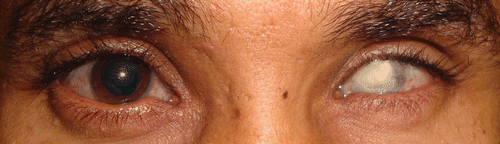
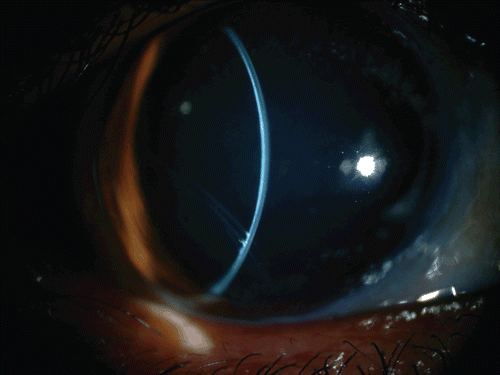
FIGURE 2 A slit-lamp image of the right eye showing the large and ectatic cornea, and the deep anterior chamber. The old inferotemporal Descemet membrane break can be seen.
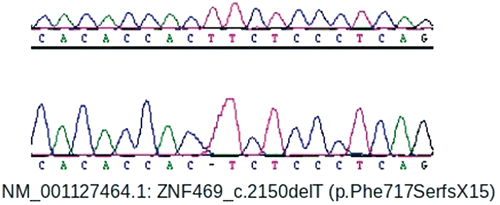
Directed history revealed no history of frail skin, excessive skin scarring, joint laxity, or hearing loss. By history as a child the patient was considered to have blue sclera. Directed general physical examination revealed a well-nourished adult male with normal vital signs and black hair. The sclera did not appear blue (). There was no dysmorphism of the face, hands, feet, or back. Oral inspection revealed poor dental hygiene and a slightly arched palate. His skin had unremarkable texture, thickness, elasticity and no evidence for abnormal striations or excessive scarring. There was no joint hypermobility (Beighton score 2/9Citation5) or scoliosis. After informed consent (KFSHRC RAC #2070023), 5cc of venous blood was collected for ZNF469 sequencing using previously-described methods.Citation3 This revealed a novel homozygous base-pair deletion predicted to cause a loss of 3195 amino acids (out of 3925 amino acids in the normal protein), including conserved ZNF domains (NM_001127464.1: c.2149delT, p.Phe717SerfsX15) (). This premature protein truncation is further upstream than that caused by either of the two previously-reported homozygous ZNF469 (c.5943delA, c.9531delG).Citation1
FIGURE 3 A DNA chromatogram showing that the patient harbored a novel homozygous base-pair deletion which caused a frameshift and premature protein truncation.
The four previously-reported ZNF469 mutations are two base-pair deletionsCitation1 and two missense variants.Citation2,Citation3 All previously-reported individuals homozygous for one of these four mutations had clinically-evident non-ocular findings, particularly by adulthood.Citation1–3 The c.5943delA mutation was identified in five unrelated patients of Tunisian Jewish ancestry with chestnut hair.Citation1 Their phenotypes were described as including both typical ocular and typical extraocular findings of the brittle cornea syndrome. The c.9531delG mutation was identified in affected members of a Palestinian family.Citation1 Their phenotypes included a minimum of both corneal fragility and joint hyperlaxity. The c.10016G>A mutation was reported in two affected brothers from a Norwegian family who were carefully phenotyped as adults.Citation2 At 42 and 48 years of age, both were blind from retinal detachments and secondary glaucoma, had chestnut hair, velvety skin, scoliosis, dental anomalies, hearing loss, and minor cardiac defects. Ancillary testing revealed reduced bone mineral density. The c.4174G>T mutation was reported in siblings from a Syrian family with corneal fragility and extraocular findings.Citation3 At 13 years old the brother had bilateral phthisis, thin velvety skin that was prone to scarring, talipes valgus, and hallux valgus. At 8 years old the sister had significant corneal ectasia, thin velvety skin, prominent lumbar lordosis, and joint hyperflexibility.
Although the function of ZNF469 is unknown, the prominent corneal findings associated with homozygous mutations in the gene suggest that ZNF469 has a role in synthesis and/or organization of corneal collagens. This is also supported by the 30% homology of ZNF469 to the helical parts of collagens highly expressed in the cornea (COL1A2, COL4A1, and COL1A1).Citation1 Recently, the brittle cornea syndrome has been associated with homozygous mutations in a second gene, PR-domain-containing protein 5 (PRDM5), a transcription regulator that modulates extracellular tissue development and maintenance.Citation6,Citation7 Because homozygous mutations in both ZNF469 and PRMD5 cause strikingly similar phenotypes, the two genes are likely involved in the same regulatory pathway. There is experimental evidence that supports this. Fibroblasts with PRMD5 mutations disrupt the expression of several key extracellular matrix components, and similar extracellular matrix down-regulation is observed in skin-derived fibroblasts from brittle cornea syndrome patients with ZNF469 mutations.Citation6
It is unclear why our patient as an adult had no clinically-evident extraocular manifestations while all other children and adults with documented homozygous ZNF469 (or PRMD5) mutations do. This is not expected to be specific to the particular mutation because this particular mutation severely truncates the ZNF469 protein, more so than the previously-reported ZNF469 mutations that result in premature protein truncation. It is likely that other genetic and/or epigenetic factors involved in the ZNF469 (and PRMD5) pathway were responsible for our patient’s lack of clinically-evident extraocular findings. Ancillary systemic testing would have been interesting to perform in this patient but was not feasible because of a lack of funds for such testing in a patient without extraocular signs or symptoms.
ACKNOWLEDGMENTS
Financial support: This study was funded in part by a KACST grant (08-MED497-20) to FSA.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Abu A, Frydman M, Marek D, et al. Deleterious mutations in the zinc-finger 469 gene cause brittle cornea syndrome. Am J Hum Genet 2008;82(5):1217–1222.
- Christensen AE, Knappskog PM, Midtbo M, et al. Brittle cornea syndrome associated with a missense mutation in the zinc-finger 469 gene. Invest Ophthalmol Vis Sci 2010;51(1):47–52.
- Khan AO, Aldahmesh MA, Mohamed JN, et al. Blue sclera with and without corneal fragility (brittle cornea syndrome) in a consanguineous family harboring ZNF469 mutation (p.E1392X). Arch Ophthalmol 2010;128(10):1376–1379.
- Khan AO. Conditions that can be mistaken as early childhood glaucoma. Ophthalmic Genet 2011;32(3):129–137.
- Beighton P, Solomon L, Soskolne CL. Articular mobility in an African population. Ann Rheum Dis 1973;32(5):413–418.
- Burkitt Wright EM, Spencer HL, Daly SB, et al. Mutations in PRDM5 in brittle cornea syndrome identify a pathway regulating extracellular matrix development and maintenance. Am J Hum Genet 2011;88(6):767–777.
- Aldahmesh MA, Mohamed JY, Alkuraya FS. A novel mutation in PRDM5 in brittle cornea syndrome. Clin Genet 2012;81(2):198–199.