Abstract
Content: Vascular smooth muscle cells (VSMCs) play a major role in the pathogenesis of atherosclerosis and restenosis, and thus the excessive proliferation of VSMCs contributes to neointimal thickening during atherosclerosis and restenosis. PMC (2,2,5,7,8-pentamethyl-6-hydroxychromane) is the most potent hydrophilic derivative of the α-tocopherols; it acts as a potent anti-inflammatory and free-radical scavenger.
Objective: The present study was designed to examine the inhibitory mechanisms of PMC in VSMC proliferation.
Materials and methods: VSMC proliferation and cytotoxicity were measured by MTT and LDH assays, respectively. The cell cycle and translocation of PKC-α in VSMCs were used by flow cytometry and confocal microscope, respectively. To detect PKC-α translocation and activation in VSMCs, immunoblotting was performed in the present study.
Results: In this study, we demonstrate an anti-proliferative effect of PMC in VSMCs. Concentration-dependent inhibition of serum-induced VSMC proliferation was observed in PMC (20 and 50 μM)-treated cells. PMC pretreatment also arrested VSMC cell cycle progression at the G2/M phase. Furthermore, PMC exhibited obvious inhibitory effects on phorbol 12-myristate 13-acetate (PMA)-induced protein kinase C (PKC)-α translocation and phospho-(Ser/Thr) substrate phosphorylation.
Discussion and conclusion: The inhibitory mechanisms of PMC on VSMC proliferation is mediated, at least in part, by inhibition of PKC-α translocation and causes cell cycle arrest in the G2/M phase. PMC treatment may represent a novel approach for lowering the risk of or improving function in abnormal VSMC proliferation-related vascular diseases.
Introduction
Abnormal proliferation of vascular smooth muscle cells (VSMCs) in the intima plays critical roles in atherosclerosis and related vascular diseases such as restenosis after angioplasty, transplant vasculopathy, and the failure of vein graft bypass (CitationHedin et al., 2004). These vascular proliferative diseases can be initiated by mechanical, biochemical, or immunological injury to vessel walls. Vascular injury triggers a cascade of events which includes endothelial dysfunction, inflammation, and VSMC activation and proliferation. Numerous growth factors and cytokines have been detected in human vascular lesions. These mediators may possibly be released by dysfunctional endothelial cells, inflammatory cells, platelets, and VSMCs, and mediate chemoattraction, cell migration, proliferation, apoptosis, and matrix modulation (CitationRoss, 1995). Basic fibroblast growth factor initiates medial proliferation of VSMCs, whereas platelet-derived growth factor induces subsequent migration of VSMCs toward the intima. Intimal proliferation and matrix accumulation were reported to occur under the influence of platelet-derived growth factor, transforming growth factor-β, angiotensin II, epidermal growth factor, and insulin-like growth factor 1 (CitationDzau et al., 2002).
The intracellular enzyme, protein kinase C (PKC), is a major serine-threonine kinase that was shown to play crucial roles in growth factor- and cytokine-initiated signal transduction. PKC regulates multiple intracellular events, including cellular proliferation and the secretion of various proteins (CitationOkazaki et al., 2000). Molecular cloning has identified at least 11 isozymes of PKC that are further divided into subfamilies based on sequence homology and mode of stimulation. Classical PKCs (α, βI, βII, and γ) are diacylglycerol (DAG)- and calcium-dependent enzymes, whereas novel PKCs (δ, ϵ, θ, and η) require DAG, but not calcium, for their activation. Atypical PKCs (ζ and ι/λ) are not responsive to activation by DAG or calcium, but are activated by other lipid-derived second messengers (CitationChurchill et al., 2008). These multiple isotypes of PKC have been shown to have various tissue distributions and biochemical regulatory and physiological functions that are cell-specific. Of these isotypes, the α isotype appears to be most commonly expressed by VSMCs (CitationMorgan & Leinweber, 1998). In addition, it is also known that PKC-α plays important roles in VSMC proliferation and migration (CitationOkazaki et al., 2000).
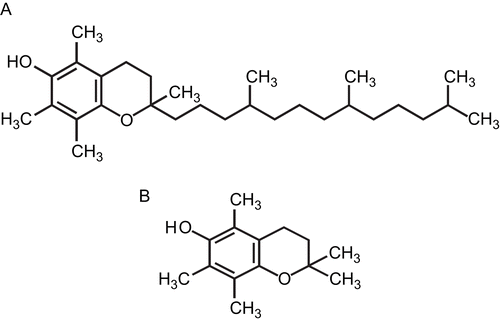
PMC (2,2,5,7,8-pentamethyl-6-hydroxychromane) is the most potent derivative of the α-tocopherols; it can act as a good antioxidant and also inhibits activation of nuclear factor (NF)-κB (CitationSuzuki & Packer, 1993; CitationSheu et al., 1999a, Citation1999b), in which the phytyl chain is replaced by a methyl group (). It is more hydrophilic than other α-tocopherol derivatives, and is reported to have potent free-radical scavenging activity and anti-inflammatory effects (CitationSheu et al., 1999a). An electron spin resonance (ESR) method was conducted on the scavenging activity of PMC on the free radicals formed. PMC (12 μΜ) greatly reduced the ESR signal intensities of superoxide anions and hydroxyl radicals, and methyl radical formation (CitationHsiao et al., 2007).
Figure 1. Chemical structures of (A) α-tocopherol and (B) PMC (2,2,5,7,8-pentamethyl-6-hydroxychromane).
By considering the pivotal roles of VSMC proliferation in the development of restenosis, the present study was designed to examine the inhibitory activity of PMC in VSMC proliferation associated with PKC-α translocation and cell cycle progression in primary cultured rat VSMCs.
Materials and methods
Materials
Wistar rats were purchased from BioLASCO (Taipei, Taiwan). Dulbecco’s modified Eagle’s medium (DMEM), trypsin (0.25%), l-glutamine, and penicillin/streptomycin were all purchased from Gibco (Gaithersburg, MD). Fetal bovine serum (FBS) was purchased from PAA Laboratories (Linz, Austria). PMC, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and phorbol 12-myristate 13-acetate (PMA) were from Sigma (St Louis, MO). The lactate dehydrogenase (LDH)-cytotoxicity assay kit was purchased from BioVision (Mountain View, CA). Propidium iodide (PI) was purchased from Calbiochem (Darmstadt, Germany). RNase A was purchased from BD Bioscience (San Jose, CA). The anti-PKC-α monoclonal antibody (mAb) and anti-phospho-(Ser/Thr) substrate polyclonal antibody (pAb) were from Cell Signaling (Beverly, MA), and the anti-α-actin mAb was purchased from Chemicon International (Temecula, CA). Horseradish peroxidase (HRP)-conjugated secondary Abs were obtained from Amersham (Amersham Buckinghamshire).
VSMC isolation and cultivation
VSMCs were enzymatically dispersed from male Wistar rats (250∼300 g). Thoracic aortas from Wistar rats were removed and stripped of endothelium and adventitia. VSMCs were obtained by modification of the combined collagenase and elastase digestion method (CitationPauly et al., 1997). These cells were grown in DMEM supplemented with 20 mM HEPES, 10% FBS, 1% penicillin/streptomycin, and 2 mM glutamine at 37°C in a humidified atmosphere of 5% CO2. The growth medium was changed every 2∼3 days until cells reached confluency. The growth medium was removed and the monolayer was rinsed with phosphate-buffered saline (PBS). A trypsin-EDTA solution was added and the monolayer was incubated at 37°C for 2 min. The culture dishes were observed under a phase-contrast microscope until the cells had detached. Cells were removed with 10 mL of DMEM and centrifuged at 900 g for 7 min. The pellet was re-suspended in DMEM in a culture dish, and cells from passages 4 to 8 were used in all experiments.
Proliferation assay
VSMCs (2 × 104 cells/well) were seeded on 24-well plates and cultured in DMEM containing 10% FBS for 24 h. The medium was then replaced with serum-free medium for 24 h. Various concentrations of FBS (1%∼2%) and PMC (20 and 50 μM) were added to the culture medium for 48 h. Cell numbers were measured with a colorimetric assay based on the ability of mitochondria in viable cells to reduce the MTT as previously described (CitationMosmann, 1983). The cell number index was calculated as (the absorbance of treated cells/control cells) × 100%.
Cytotoxicity assay
VSMCs (2 × 104 cells/well) were seeded on 24-well plates and cultured in DMEM containing 10% FBS for 24 h. The medium was then replaced with serum-free medium for 24 h. PMC (1∼100 μM) was added to the culture medium for 6 h, and VSMC cytotoxicity was measured with a colorimetric assay based on the activity of LDH released from damaged cells described by the commercial advisory protocol (BioVision, LDH-cytotoxicity assay kit). In order to calculate the cytotoxicity of PMC-treated groups, low- and high-control groups were maintained. The low-control group consisted of VSMCs (2 × 104 cells/well) seeded in 24-well plates without drug treatment. The high-control group consisted of VSMCs (2 × 104 cells/well) seeded in 24-well plates with 1% Triton X-100 incubation. The percentage of cytotoxicity was calculated as the absorbance of (test sample − low control)/(high control − low control).
Immunoblotting assay
Western blot analyses were performed as previously described to determine the expression of PKC-α in VSMCs (CitationHsiao et al., 2004). VSMCs were seeded on 6-cm dishes and cultured in DMEM containing 10% FBS for 24 h. The medium was then replaced with serum-free medium for 24 h. Briefly, cells were treated as per the experimental design for the indicated periods of times. After treatment, cells were washed with ice-cold PBS buffer (pH 7.3) then scraped into lysis buffer. Proteins were extracted with lysis buffer (10 mM Tris-HCl, 140 mM NaCl, 3 mM MgCl2, 0.5% NP-40, 1 mM DTT, 2 mM PMSF, 1 μM aprotinin, and 1 μM leupeptin; pH 7.0) for 30 min. Additionally, phosphatase inhibitors (10 mM sodium fluoride, 1 mM sodium orthovanadate, and 10 mM sodium pyrophosphate) were added to the lysis buffer for the phospho-(Ser/Thr) substrate analysis. Lysates were centrifuged, and the supernatant (50 μg protein) was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and electrophoretically transferred onto polyvinylidene difluoride (PVDF) membranes (0.45 mm; Hybond-P; Amersham). After incubation in blocking buffer (50 mM Tris-HCl, 100 mM NaCl, 0.1% Tween 20, and 5% dry skim milk; pH 7.5) overnight at 4°C and being washed three times with PBS buffer, blots were treated with either an anti-PKC-α or anti-phospho-(Ser/Thr) substrate Ab (1:1000), in PBS buffer for 2 h. They were subsequently washed three times with PBS buffer and incubated with HRP-conjugated goat anti-mouse or anti-rabbit Ab (1:3000) for 1 h. Blots were then washed three times, and the band with peroxidase activity was detected using film exposure with enhanced chemiluminescence detection reagents (ECL+ system, Amersham). Densitometric analysis of specific bands was performed with a Photo-Print Digital Imaging System (IP-008-SD) with analytic software (Bio-1Dlight, V 2000).
Cell fractionation
To determinate PKC-α translocation in VSMCs, cells were separated into cytosolic and membrane fractions as described previously with slight modifications (CitationYano et al., 1999). VSMCs (1 × 106 cells/dish) were seeded on 10 cm dishes and cultured in DMEM containing 10% FBS for 24 h. The medium was then replaced with serum-free medium for 24 h. In brief, cells were treated as per the experimental design for the indicated periods of time. After treatment, cells were washed with ice-cold PBS buffer (pH 7.3) then scraped into buffer A (25 mM Tris-Cl, 0.25 mM sucrose, 10 mM EGTA, 1 mM dithiothreitol, 0.5 mM PMSF, 50 μg/mL leupeptin, and 50 μg/mL peptastatin A; pH 7.5), and centrifuged at 1 × 105 g for 1 h. The supernatant was designated the cytosolic fraction. The pellet was extracted with buffer B (10 mM Tris-Cl, 150 mM KCl, 1% Triton X-100, 0.5 mM PMSF, 50 μg/mL leupeptin, and 50 μg/mL peptastatin A; pH 7.5), and centrifuged at 3 × 104 g for 20 min. The supernatant was saved as the membrane fraction. Protein (15 μg) was run on an SDS-PAGE for immunoblotting as previously described.
Flow cytometric assay
For the cell cycle analysis, VSMCs (2 × 105 cells/dish) were seeded in 6 cm dishes and cultured in DMEM containing 10% FBS for 24 h. The medium was then replaced with serum-free medium for 24 h. VSMCs were pretreated with PMC (20 and 50 μM) for 20 min and then stimulated with 2% FBS for 24 h. After 24 h, cells were detached from the plate using trypsin, washed with PBS, and fixed in 70% ethanol for 30 min on ice. Cells were then washed with PBS and resuspended in a solution containing RNase (50 μg/mL), PI (80 μg/mL) and Triton X-100 (0.2%). Samples were incubated in the dark for 20 min and subjected to a fluorescence-activated cell sorter (FACS) (Beckman Coulter® EPICS® XL™, Fullerton, CA) analysis.
Confocal microscopic analysis
A confocal microscopic analysis was performed to identify and evaluate the translocation of PKC-α in VSMCs. VSMCs were prepared as described above. VSMCs (1 × 105 cells/cover slide) were placed on cover slides and allowed to adhere in a cell culture incubator overnight and then were starved for 24 h. VSMCs were treated as per the design of the experiment, then fixed with 4% paraformaldehyde for 30 min and permeabilized with 80% methanol for 15 min. After incubation with 3% skimmed milk in PBS for 60 min, the preparation was incubated for 1 h with a primary Ab (1:80). Cells were then washed three times with PBS and exposed to the secondary Ab (FITC-conjugated anti-mouse immunoglobin G (IgG) at 1:100, 1% BSA/PBS) for 60 min. Preparations were mounted with mounting buffer (Vector Laboratories, Burlingame, CA) under a glass coverslip on a Leica TCS SP5 Confocal Spectral Microscope Imaging System using an argon/krypton laser (Mannheim, Germany).
Statistical analysis
Experimental results are expressed as the means ± SEM and are accompanied by the number of observations. Data were assessed by the method of analysis of variance (ANOVA). If this analysis indicated significant differences among the group means, then each group was compared using the Newman-Keuls method. A p value of <0.05 was considered statistically significant.
Results
Effects of PMC on VSMC proliferation
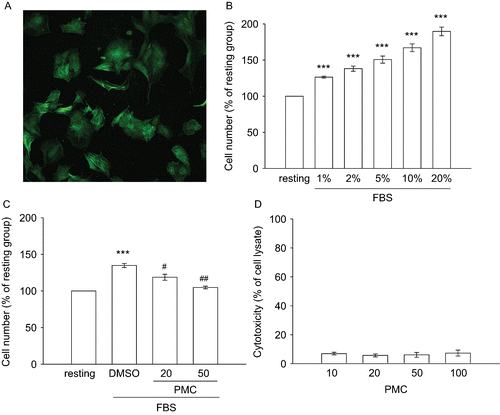
Primary cultured rat aortic VSMCs were identified by staining with smooth muscle α-actin (). In order to evaluate the inhibitory effects of PMC on VSMC proliferation, an MTT assay was utilized to measure the cell number. A concentration-dependent increase (100 ± 0% (resting) to 189.6 ± 6% (20% FBS)) in the cell number of starved VSMCs was observed within 48 h of FBS (1∼20%) stimulation (). In this study, FBS (2%) showed a more significant increase in VSMC proliferation (p < 0.001). In subsequent experiments, we used 2% FBS as a stimulator to explore the inhibitory mechanisms of PMC on VSMC proliferation as shown in . After pretreatment of VSMCs with PMC for 20 min followed by the addition of 2% FBS, we found that PMC (20 and 50 μM) concentration-dependently inhibited VSMC proliferation by approximately 50 and 88%, respectively (). There were no significant differences in the cytotoxicity assay after 6 h of incubation with various concentrations of PMC (1∼100 μM) in VSMCs ().
Figure 2. Effects of PMC on vascular smooth muscle cell proliferation and cytotoxicity. (A) Primary cultured rat VSMCs were identified by immunofluorescence for smooth muscle α-actin (the bar represents 50 μm). VSMCs (2 × 104 cells/well) were starved for 24 h then treated with (B) various concentrations of FBS (1%∼20%), or (C) an isovolumetric solvent control (0.1% DMSO) and PMC (20 and 50 μM) for 20 min, followed by the addition of FBS (2%) for 48 h. The cell number was evaluated by an MTT assay as described in Materials and methods. (D) For the other experiment, the cytotoxicity of PMC toward VSMCs was determined by a lactate dehydrogenase (LDH) assay. VSMCs (2 × 104 cells/well) were treated with various concentrations of PMC (10∼100 μM) for 6 h. Cell-free supernatants were utilized for the LDH assay. Data are presented as the means ± SEM (n = 3). *** P < 0.001, compared to the resting group; #P < 0.05 and ##P < 0.01, compared to the solvent control group.
Effects of PMC on cell cycle progression
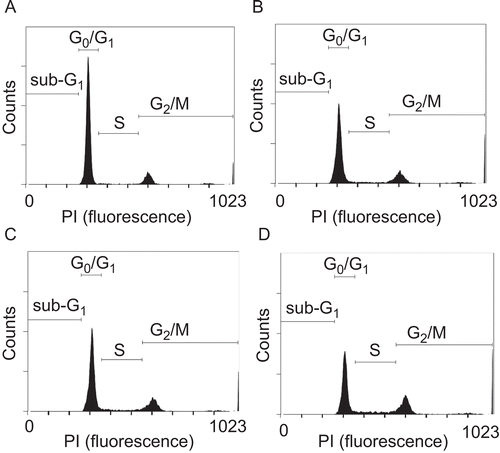
The DNA content was analyzed by PI staining and a FACS analysis to investigate the effects of PMC on cell cycle progression in VSMCs. The FACS analysis showed that PMC (20 and 50 μM) produced a concentration-dependent accumulation of VSMCs in the G2/M phase of the cell cycle as shown in and . After 24 h of stimulation with 2% FBS, we observed an increase in the percentage of cells in the S (from 3.3 ± 0.7% to 9.9 ± 1.2%, p < 0.001, n = 5) and G2/M phases (from 20.4 ± 2.4% to 27 ± 1.8%, p < 0.05, n = 5) but a reduction in the G0/G1 phase (from 75 ± 1.9% to 61.9 ± 2.7%, p < 0.001, n = 5). PMC (50 μM) treatment resulted in an obvious accumulation of cells in the G2/M phase and a reduction of cells in the G0/G1 phase compared to the DMSO-treated group (G2/M phase, 27 ± 1.8% versus 33.6 ± 0.6%; G0/G1 phase, 61.9 ± 2.7% versus 53.5 ± 1.2%, p < 0.05, n = 5) (). These results indicate that PMC arrested cell cycle progression at the G2/M phase in VSMCs.
Table 1. Effects of PMC on FBS (2%)-induced cell cycle progression in VSMCs.
Figure 3. Effects of PMC on FBS-induced cell cycle progression in vascular smooth muscle cells. VSMCs (2 × 105 cells/dish) were (A) untreated, or treated with (B) the isovolumetric solvent control (0.1% DMSO), (C) PMC (20 μM), and (D) PMC (50 μM) for 20 min, followed by the addition of FBS (2%) for 24 h. Representative DNA histograms of propidium iodide fluorescence in cells were assessed by flow cytometry. The profiles are representative examples of five similar experiments.
Effects of PMC on PMA-induced PKC-α translocation
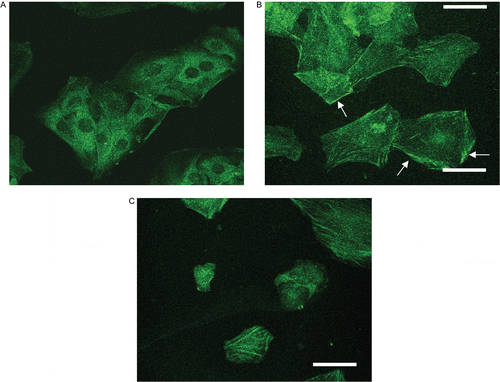
To further investigate the role of PKC-α transduction in PMC-mediated anti-proliferation in VSMCs, levels of PKC-α in the cytosolic and membrane fractions of VSMCs were investigated by an immunoblotting assay. PMA can induce translocation of PKC from the cytosol to membranes, which reflects intracellular PKC activation (CitationKraft & Anderson, 1983). PMA was thus employed as an activator to stimulate PKC-α translocation in this study. As shown in , PMA (1 μM) induced the apparent translocation of PKC-α from the cytosol to membranes on VSMCs after 10 min of stimulation. The membrane-to-cytosol ratio of PKC-α increased from 0.2 ± 0 to 5.3 ± 1.8 after PMA stimulation (). also shows that PKC-α translocation was significantly inhibited by PMC (20 and 50 μM) in a concentration-dependent manner. In addition, the confocal microscopic study provided direct evidence of PKC-α translocation of VSMCs. In resting cells, PKC-α was detected in the cytosol (), and PMA (1 μM) treatment caused more obvious PKC-α translocation from the cytosol to membranes (), which was reduced in the presence of PMC (50 μM) ().
Figure 4. Effects of PMC on PMA-induced PKC-α translocation in vascular smooth muscle cells. VSMCs (1 × 106 cells/dish) were treated with (A) PMA (1 μM) alone for the indicated times (0, 10, and 30 min), or (B) pretreated with an isovolumetric solvent control (0.1% DMSO) and PMC (20 and 50 μM) for 20 min, followed by the addition of PMA (1 μM) to trigger PKC-α translocation. PKC-α distribution in the cytosol (C) and membranes (M) were determined by immunoblotting with a monoclonal antibody which recognizes only PKC-α. Data are presented as the means ± SEM (n = 3). ** P < 0.01, compared to the resting group; #P < 0.05, compared to the solvent control group. The profile (A) is representative example of three similar experiments.
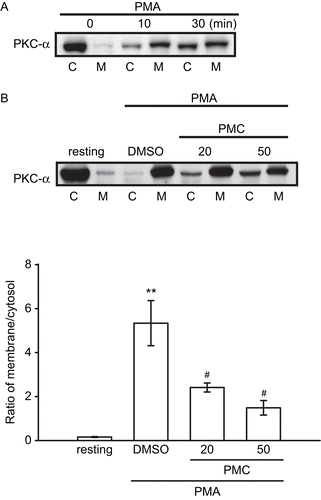
Figure 5. Confocal photographs of PMC on PMA-induced PKC-α translocation in vascular smooth muscle cells. VSMCs (1 × 105 cells/coverslip) were (A) untreated, or treated with (B) isovolumetric solvent control (0.1% DMSO), and (C) PMC (50 μM), followed by the addition of PMA (1 μM). Confocal images are typical of those obtained in three separate experiments demonstrating the distribution of PKC-α (arrows) in VSMCs. The white bar indicates 50 μm.
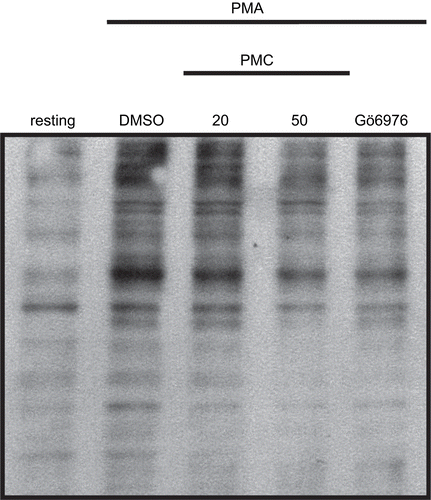
Effects of PMC on PMA-induced phospho-(Ser/Thr) substrate phosphorylation
Since PMC was found to inhibit PKC-α translocation, which may result in lowering PKC-α activation, we thus investigated the influence of PMC on PKC-α activation by measuring the phosphorylation of PKC protein targets. In order to measure the phosphorylation of PKC protein targets, a phospho-(Ser/Thr) substrate Ab was employed in this study. As shown in , an apparent increase in the phosphorylation of phospho-(Ser/Thr) substrates stimulated by PMA (1 μM) was observed compared to the resting group. The presence of PMC (20 and 50 μM) or Gö6976 (3 nM), a PKC-α inhibitor, markedly inhibited these reactions ().
Figure 6. Effects of PMC on PMA-induced phospho-(Ser/Thr) substrate phosphorylation in vascular smooth muscle cells. VSMCs (2 × 105 cells/dish) were treated with an isovolumetric solvent control (0.1% DMSO), PMC (20 and 50 μM), and the PKC-α inhibitor, Gö6976 (3 nM), followed by the addition of PMA (1 μM) as described in Materials and methods. The profile is a representative example of three similar experiments.
Discussion
A series of studies has drawn attention to the regulatory aspects of α-tocopherol on VSMCs and atherosclerosis (CitationMunteanu & Zingg, 2007). However, the mechanism of action of α-tocopherol has proven difficult to define, owing to its highly lipophilic nature. The high lipophilicity of α-tocopherol and its relatively slow cellular uptake (CitationIngold et al., 1987) severely limit its clinical usefulness, especially in emergencies. Therefore, finding a more-hydrophilic analogue is important for exploring the inhibitory mechanisms on VSMC proliferation by α-tocopherol. Of the α-tocopherol analogues studied, PMC is the most potent derivative of α-tocopherol in antioxidation (CitationSuzuki & Packer, 1993). PMC is also more hydrophilic than other α-tocopherol derivatives and has potent free-radical scavenging activity toward iron-induced lipid peroxidation (LPO) in rat brain homogenates than α-tocopherol (CitationSheu et al., 1999b). PMC therefore appears to be a promising therapeutic alternative to α-tocopherol. Furthermore, CitationTafazoli et al. (2005) also showed that pro-oxidant radicals of α-tocopherol analogues can cause intracellular LPO, glutathione (GSH) oxidation, and cytotoxicity. Some α-tocopherol analogues such as α-tocopheryl phosphates and α-tocopheryl esters were found to have cytotoxic effects in VSMCs (CitationKogure et al., 2004; CitationMunteanu et al., 2004), whereas PMC has no cytotoxicity in VSMCs (). PMC is the most effective antioxidant with the lowest pro-oxidant activity and may have therapeutic advantages over the other α-tocopherol analogues (CitationTafazoli et al., 2005).
To examine the inhibitory mechanism responsible for cell proliferation by PMC, the cell cycle distribution was evaluated by flow cytometry. The cell cycle can be divided into distinct phases including a synthesis (S) phase, where DNA is replicated, and a mitosis (M) phase, where cell division occurs. In animal cells, the growth and synthesis of components required for these phases are regulated by extracellular growth factors and occur mainly in two gap phases, G1 (between M and S) and G2 (between S and M) (CitationKastan & Bartek, 2004; CitationMurray, 2004; CitationBjorklund et al., 2006). α-Tocopherol was reported to inhibit cell proliferation by inducing cell cycle arrest at the G1/S phase in VSMCs (CitationAzzi et al., 1993). In the present study we found that loss of the proliferative capacity of VSMCs treated by PMC was associated with G2/M phase arrest, it indicates that PMC may have its unique and different pharmacological activities compared with α-tocopherol.
PKC is a family of Ser/Thr kinases involved in many intracellular signal transduction pathways that regulate gene transcription, differentiation, the cell cycle, cytoskeletal functions, apoptosis, growth factor responses, cell-to-cell interactions, and cell migration (CitationCarter & Kane, 2004). Most PKC proteins are inactivated in the cytosol, but following an extracellular activating signal, they translocate to the cellular membrane compartment (CitationGoodnight et al., 1995). PKC isoforms are typically activated at plasma membranes after complexing with diacylglycerol and specific membrane phospholipids (CitationNishizuka, 1995). Among these PKC isoforms, PKC-α was identified in almost all surveys of VSMCs and appears to be the most prevalent PKC isotype (CitationMorgan & Leinweber, 1998). PKC-α is known to play an important role in VSMC proliferation and migration (CitationOkazaki et al., 2000). PKC isoforms were found to be involved in growth control at the G1/S and G2/M transitions as regulators linking signal transduction pathways to the cell cycle machinery (CitationLivneh & Fishman, 1997). In this study, PMC inhibited PMA-induced PKC-α translocation and phospho-(Ser/Thr) substrate activation in VSMCs (). These results imply that the anti-proliferation and cell cycle regulatory effects of PMC on VSMCs may be mediated, at least in part, by inhibiting PKC-α translocation.
In conclusion, the most important finding of this study demonstrates for the first time that PMC, a potent α-tocopherol derivative, inhibits VSMC proliferation through inhibition of PKC-α activation and causes cell cycle arrest at the G2/M phase. PMC is more hydrophilic than α-tocopherol, and thus it has fewer limitations than α-tocopherol in clinical application. Therefore, PMC may be a potential therapeutic agent for treating VSMC proliferation-related vascular diseases.
Declaration of interest
This work was supported by grants from the National Science Council of Taiwan (NSC97-2320-B-038-016-MY3) and Cathay General Hospital (96-CGH-TMU-01, MR-9614, and MR-9616).
References
- Azzi A, Boscoboinik D, Chatelain E, Ozer NK, Stauble B (1993): d-alpha-Tocopherol control of cell proliferation. Mol Aspects Med 14: 265–271.
- Bjorklund M, Taipale M, Varjosalo M, Saharinen J, Lahdenpera J, Taipale J (2006): Identification of pathways regulating cell size and cell-cycle progression by RNAi. Nature 439: 1009–1013.
- Carter CA, Kane CJ (2004): Therapeutic potential of natural compounds that regulate the activity of protein kinase C. Curr Med Chem 11: 2883–2902.
- Churchill E, Budas G, Vallentin A, Koyanagi T, Mochly-Rosen D (2008): PKC isozymes in chronic cardiac disease: Possible therapeutic targets? Ann Rev Pharmacol Toxicol 48: 569–599.
- Dzau VJ, Braun-Dullaeus RC, Sedding DG (2002): Vascular proliferation and atherosclerosis: New perspectives and therapeutic strategies. Nat Med 8: 1249–1256.
- Goodnight JA, Mischak H, Kolch W, Mushinski JF (1995): Immunocytochemical localization of eight protein kinase C isozymes overexpressed in NIH 3T3 fibroblasts. isoform-specific association with microfilaments, Golgi, endoplasmic reticulum, and nuclear and cell membranes. J Biol Chem 270: 9991–10001.
- Hedin U, Roy J, Tran PK (2004): Control of smooth muscle cell proliferation in vascular disease. Curr Opin Lipidol 15: 559–565.
- Hsiao G, Huang HY, Fong TH, Shen MY, Lin CH, Teng CM, Sheu JR (2004): Inhibitory mechanisms of YC-1 and PMC in the induction of iNOS expression by lipoteichoic acid in RAW 264.7 macrophages. Biochem Pharmacol 67: 1411–1419.
- Hsiao G, Lee JJ, Chen YC, Lin JH, Shen MY, Lin KH, Chou DS, Sheu JR (2007): Neuroprotective effects of PMC, a potent alpha-tocopherol derivative, in brain ischemia-reperfusion: Reduced neutrophil activation and anti-oxidant actions. Biochem Pharmacol 73: 682–693.
- Ingold KU, Burton GW, Foster DO, Hughes L, Lindsay DA, Webb A (1987): Biokinetics of and discrimination between dietary RRR- and SRR-alpha-tocopherols in the male rat. Lipids 22: 163–172.
- Kastan MB, Bartek J (2004): Cell-cycle checkpoints and cancer. Nature 432: 316–323.
- Kogure K, Hama S, Kisaki M, Takemasa H, Tokumura A, Suzuki I, Fukuzawa K (2004): Structural characteristic of terminal dicarboxylic moiety required for apoptogenic activity of alpha-tocopheryl esters. Biochim Biophys Acta 1672: 93–99.
- Kraft AS, Anderson WB (1983): Phorbol esters increase the amount of Ca2+, phospholipid-dependent protein kinase associated with plasma membrane. Nature 301: 621–623.
- Livneh E, Fishman DD (1997): Linking protein kinase C to cell-cycle control. Eur J Biochem 248: 1–9.
- Morgan KG, Leinweber BD (1998): PKC-dependent signalling mechanisms in differentiated smooth muscle. Acta Physiol Scand 164: 495–505.
- Mosmann T (1983): Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immuno Meth 65: 55–63.
- Munteanu A, Zingg JM (2007): Cellular, molecular and clinical aspects of vitamin E on atherosclerosis prevention. Mol Aspects Med 28: 538–590.
- Munteanu A, Zingg JM, Ogru E, Libinaki R, Gianello R, West S, Negis Y, Azzi A (2004): Modulation of cell proliferation and gene expression by alpha-tocopheryl phosphates: Relevance to atherosclerosis and inflammation. Biochem Biophys Res Comm 318: 311–316.
- Murray AW (2004): Recycling the cell cycle: Cyclins revisited. Cell 116: 221–234.
- Nishizuka Y (1995): Protein kinase C and lipid signaling for sustained cellular responses. FASEB J 9: 484–496.
- Okazaki J, Mawatari K, Liu B, Kent KC (2000): The effect of protein kinase C and its alpha subtype on human vascular smooth muscle cell proliferation, migration and fibronectin production. Surgery 128: 192–197.
- Pauly RR, Bilato C, Cheng L, Monticone R, Crow MT (1997): Vascular smooth muscle cell cultures. Meth Cell Biol 52: 133–154.
- Ross R (1995): Cell biology of atherosclerosis. Ann Rev Physiol 57: 791–804.
- Sheu JR, Lee CR, Hsiao G, Hung WC, Lee YM, Chen YC, Yen MH (1999a): Comparison of the relative activities of alpha-tocopherol and PMC on platelet aggregation and antioxidative activity. Life Sci 65: 197–206.
- Sheu JR, Lee CR, Lin CC, Kan YC, Lin CH, Hung WC, Lee YM, Yen MH (1999b): The antiplatelet activity of PMC, a potent alpha-tocopherol analogue, is mediated through inhibition of cyclo-oxygenase. Br J Pharmacol 127: 1206–1212.
- Suzuki YJ, Packer L (1993): Inhibition of NF-kappa B activation by vitamin E derivatives. Biochem Biophys Res Commun 193: 277–283.
- Tafazoli S, Wright JS, O’Brien PJ (2005): Prooxidant and antioxidant activity of vitamin E analogues and troglitazone. Chem Res Toxicol 18: 1567–1574.
- Yano K, Bauchat JR, Liimatta MB, Clemmons DR, Duan C (1999): Down-regulation of protein kinase C inhibits insulin-like growth factor I-induced vascular smooth muscle cell proliferation, migration, and gene expression. Endocrinology 140: 4622–4632.