Abstract
Context: Hypertrophic scarring following surgical procedures, trauma and especially burns can lead to severe functional and cosmetic impairment, causing a decrease in the quality of life. Although a wide choice of treatments is offered, few therapeutic methods are universally accepted because of their side effects.
Objective: The effects of the essential oil (EO) extracted from rhizomes of Ligusticum chuanxiong Hort. (Umbelliferae) in human hypertrophic scar fibroblasts (HSFs) are investigated for the first time.
Materials and methods: Chemical composition of hydrodistilled EO obtained from rhizomes of Ligusticum chuanxiong was analyzed by gas chromatography-mass spectrometry (GC-MS). The effects of EO on cell viability, apoptosis rate, mitochondrial membrane potential (MMP), lactate dehydrogenase (LDH), reactive oxygen species (ROS) and caspase-3 in HSFs were investigated.
Results: The experimental results showed that EO significantly inhibited cell viability, elicited morphological changes and induced apoptosis in HSFs. EO also evidently increased the loss of MMP, the levels of LDH release and cellular ROS production, and the activity of caspase-3.
Discussion and conclusion: EO-induced apoptosis was at least partially carried out via destruction of the intracellular antioxidant system and elicitation of excessive ROS accumulation in HSFs, which impaired mitochondrial membranes and elicited caspase-3 activation. EO could be an effective cure for human hypertrophic scar.
Introduction
The skin wound healing process comprises inflammation, granulation tissue formation and tissue reconstruction (CitationMartin, 1997). In this process dermal fibroblasts, which can migrate to the wound site, proliferate, synthesize extracellular matrix components such as collagen, form granulation tissue (CitationGrinnell, 1994) and generate mechanical forces initiating wound contraction (CitationGabbiani et al., 1972), play a key role. However, excessive proliferation and accumulation of fibroblasts in the wound position will cause abnormal wound contraction or contractures (CitationRedden & Doolin, 2003) and form a hypertrophic scar or keloid, resulting in cosmetic and functional problems (CitationVande & Rudolph, 1985; CitationFang & Alexander, 1990; CitationBrissett & Sherris, 2001). When an exuberant scar occurs there are many treatment choices. The therapeutic methods include both conservative procedures such as corticosteroid injections, pressure therapy, splinting, and serial casting, and non-conservative procedures such as surgical excisions. Unfortunately there is no universal consensus about the therapeutic mode causing complete and permanent improvement of a hypertrophic scar or keloid with few side effects (CitationReish & Eriksson, 2008; CitationBloemen et al., 2009).
It has been generally recognized that traditional Chinese herbal medicines play a unique therapeutic role in the treatment of many diseases (CitationZhang et al., 2008a; CitationDu et al., 2009; CitationWu et al., 2009; CitationXing et al., 2009). In particular, the essential oils from many plant species have become popular in recent years, and the investigations on their bioactivities and mechanisms of action have been carried out for human health (CitationZhang et al., 2007, Citation2008b; CitationSamarth et al., 2008).
Rhizoma chuanxiong (RCX), the dry rhizome of Ligusticum chuanxiong Hort. (Umbelliferae), is one of the well-known traditional Chinese medicines and possesses efficacy in promoting the circulation of blood and qi, expelling wind and alleviating pain. RCX has been used for the clinical treatment of pain, migraine, rheumatic arthralgia, inflammation, menstrual disturbance, and cardiovascular and cerebrovascular diseases for hundreds of years (CitationChina Pharmacopoeia Committee, 2005; CitationChan et al., 2006; CitationPeng et al., 2009). The volatile compounds in this herbal drug are considered an important part of its pharmacological effects mentioned above. In addition, there are several investigations reported suggesting that the EO from RCX could protect against DNA damage and apoptosis induced by ultraviolet B in mammalian cells (CitationJeong et al., 2009). Ethyl ether extract of RCX blocks endothelial cell damage induced by hydrogen peroxide (CitationHou et al., 2004). The polysaccharides from RCX also have antioxidant effects and cytotoxicity in human liver carcinoma cell line (CitationYuan et al., 2008). Until now, to the best of our knowledge, there have been no reports on induction of apoptosis by EO from the rhizomes of Ligusticum chuanxiong in hypertrophic scar fibroblasts (HSFs).
In a previous investigation we screened a large number of plant extracts by MTT assay and observed that EO extracted from RCX markedly inhibited the viability of HSFs and elicited cell death. The present study was designed to substantiate whether the essential oil extracted from RCX could induce apoptosis in HSFs and to explore the possible mechanism of action.
Materials and methods
Preparation of essential oil
Rhizoma chuanxiong (RCX) was collected from Pengzhou, Sichuan, China, and identified as the rhizome of Ligusticum chuanxiong by Lu-Ping Qin, a pharmacognosist from the Department of Pharmacognosy, School of Pharmacy, Second Military Medical University, Shanghai. A voucher specimen of Ligusticum chuanxiong was deposited with the number SY3202 at the Herbarium of the university. The essential oil was extracted by hydrodistillation. The dried rhizomes were broken into grains, 200 g of which were immersed in 2000 mL of distilled water and boiled in a distillation apparatus for 6 h. The yield of EO was 0.25% (v/w). It was dissolved in DMSO and diluted to the desired concentrations before utilization, with the concentration of DMSO kept below 1%.
Reagents
3-[4,5-Dimethyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide (MTT) was purchased from Amresco (Solon, OH). Dulbecco’s modified Eagle medium (DMEM) was obtained from Thermo Fisher Scientific (Waltham, MA), rhodamine 123 from Sigma (St Louis, MO), and fetal bovine serum (FBS) from Hyclone (Thermo Fisher). 0.25% Trypsinase was purchased from Gibco Invitrogen (Carlsbad, CA), dimethyl sulfoxide (DMSO) from Fisher Scientific, and lactate dehydrogenase assay kits from Nanjing Jianchen Bioengineering Institute, China. A reactive oxygen species (ROS) detection kit, 2',7'-dichlorofluorescein diacetate (DCFH-DA) and a caspase-3 assay kit were purchased from Beyotime Institute of Biotechnology, Haimen, China.
GC-MS analysis
GC-MS analysis was performed for determination of EO composition with a Finnigan Voyager gas chromatograph fitted with a fused silica VF-5 ms capillary column (30 m×0.25 mm; coating thickness 0.25 μm) using the following temperature program. The initial temperature was 50°C for 2 min, raised to 300°C at 15°C/min, holding for 10 min. The injector temperature was 250°C and the MS source helium was used as the carrier gas at a flow rate of 1 mL/min with a split ratio of 30:1 (v/v). The gas chromatograph was coupled to a Finnigan Voyager mass selective detector. The ionization source temperature was 200°C and the ionization energy was 70eV.
Cell culture and treatment
The fibroblast was established as a primary cell line from hypertrophic scar tissue, confirmed pathologically, and obtained from patients (aged 16–30 years) recovering from severe burns. The areas of hypertrophic scarring were manifested by raised, erythematous, pruritic, thickened, and non-compliant scars confined to the injury site, suggesting an actively hyperplastic phase. There was no local infection or ulceration and no case was treated with glucocorticosteroids or radiotherapy.
The hypertrophic scar specimens were washed three times under sterile conditions with phosphate-buffered saline (PBS) solution containing a combination of 1% penicillin, streptomycin sulfate. The epidermis and subcutaneous fat were removed. The remaining dermal specimen was minced into approximately 0.5-1 mm3 pieces with sterile eye scissors, and placed evenly in 75 mm3 culture flasks (Corning, NY) precoated with a thin layer of FBS in order to allow tissue pieces to adhere to the flask wall firmly. They were cultured in a saturated humidity incubator at 37°C with 5% CO2 atmosphere for 6 h, and then DMEM containing 20% FBS was added to the flasks and culture continued. Following sufficient outgrowth of fibroblasts, the tissue was removed and cell passage was carried out. The culture medium with 10% FBS was changed every 3–4 days. The growth status and morphological changes of the cells were observed with an inverted phase-contrast microscope (Olympus, Tokyo, Japan). Successive cultures were passed at confluence. Three to six passages were performed in this study. Hypertrophic scar fibroblasts (HSFs) at the same passage were used to examine the effects of EO on cell proliferation and apoptosis.
HSFs in the logarithmic phase were prepared as a single-cell suspension (5 × 104 cells/mL) for inoculation into 96-well plates. The suspension (100 μL) was added to each well. The wells were randomly divided into four groups (n = 10). After 24 h incubation at 37°C in 5% CO2 atmosphere, HSFs were treated with or without different concentrations of EO.
To test any effect of DMSO on the growth of HSFs, DMSO solution was added to DMEM at a final concentration of 1%. No difference in cell growth was observed between the cells treated with or without DMSO, indicating that DMEM containing 1% DMSO did not influence the growth of HSFs.
Cell viability determination
Before analysis, HSFs were cultured with or without EO for 12, 24, and 48 h, respectively. At the end of incubation, 20 μL of 0.5% MTT was added to each well, followed by incubation at 37°C in 5% CO2 atmosphere for 4 h. The medium was then removed carefully, and 150 μL of DMSO was added to each well. The plates were shaken gently for 10 min to dissolve blue formazan crystals. Absorbance was measured at 570 nm using an ELx-800 universal microplate reader (Bio-Tek, Winooski, VT). Cell viability was expressed as a percentage of the value in the control group. The morphological features of HSFs were also photographed using an Olympus C-5060 digital camera.
LDH release assay
Cytotoxicity was quantitatively assessed by measuring the activity of LDH released from damaged cells into culture medium. LDH activity was measured according to the method reported by CitationHarauchi and Hirata (1993). Before analysis, HSFs were cultured with or without EO for 24 and 48 h, respectively. At the end of incubation, the culture supernatant was collected for the measurement of extracellular enzyme activity with an automatic biochemistry analyzer (Hitachi, Tokyo) using a commercially available assay kit (Roche, Tokyo). LDH activity was in proportion to the number of damaged fibroblasts.
Apoptosis detection
Apoptotic rates were analyzed by flow cytometry using an Annexin V-FITC apoptosis detection kit (Nanjing Keygen Biotechnology, China) according to the manufacturer’s instructions. Briefly, 5 × 105 HSFs were harvested after treatment with EO for 12 h, washed with ice-cold PBS, resuspended in 500 μL of binding buffer, followed by the addition of 5 μL Annexin V stock solution and incubation for 10 min at 4°C. Aliquots of 5 μL of propidium iodide (PI) were added to the cells and they were immediately analyzed using a FACSCalibur flow cytometer (BD, San Jose, CA).
Measurement of mitochondrial membrane potential
To measure the mitochondria membrane potential (MMP), HSFs were treated with different concentrations of EO for 12 h. Rhodamine 123 (10 μg/mL) was added to the cells and incubated in a CO2 incubator for 30 min. The cells were rinsed twice with PBS to remove unbound dye. Fibroblasts (5 × 105) per sample were analyzed with flow cytometry and the experiment was independently repeated three times. The mean fluorescence intensity in HSFs reflected the status of MMP depolarization.
Determination of cellular reactive oxygen species
DCFH-DA was used to measure the level of ROS induced by EO. DCFH-DA easily diffuses into cells, produces DCFH, and then changes into the highly fluorescent compound 2,7-dichlorofluorescein (DCF) in the presence of ROS. Therefore, the fluorescence intensity is proportional to the level of intracellular ROS production. After 12 h of the treatment with EO, the cells were harvested, rinsed twice with PBS, and incubated with 10 μmol/L of DCFH-DA for 30 min at 37°C. The level of ROS production was reflected by the fluorescence intensity of HSFs and measured with an automated 96-well plate reader (Thermo, Waltham, MA) using an excitation wavelength of 485 nm and an emission wavelength of 530 nm.
Caspase-3 assay
Caspase-3 activity can be determined by catalysis of chromophore p-nitroaniline (pNA) release from Ac-DEVD-pNA. Assays were performed according to the manufacturer’s instructions. To evaluate the activity of caspase-3 in HSFs, cell lysates were prepared after treatment with different concentrations of EO for 12 h. Assays were carried out on 96-well plates by incubating 10 μL protein of cell lysate per sample in 80 μL reaction buffer containing 10 μL caspase-3 substrate (Ac-DEVD-pNA, 2 mM). Lysates were incubated at 37°C for 2 h. Samples were measured at 405 nm using an ELx-800 universal microplate reader (Bio-Tek).
Statistical analysis
All results were presented as the mean ± SD. Data were analyzed using a SPSS 13.0 statistical package. Data for multiple comparisons were performed by one-way ANOVA followed by Dunnett’s test. A value of P <0.05 was considered statistically significant.
Results
Essential oil components
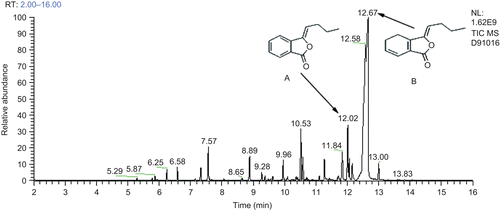
The GC-MS analysis showed that dozens of compounds were detected from the essential oil and the major components were ligustilide and butylidenephthalide with relative contents of 67.027 and 5.831%, respectively ().
Figure 1. Fingerprintof the essential oil analyzed by GC-MS. (A) Butylidenephthalide (3-butylidene-1(3H)-isobenzofuranone), (B) Ligustilide (3-butylidene-4,5-dihydro-1(3H)-isobenzofuranone).
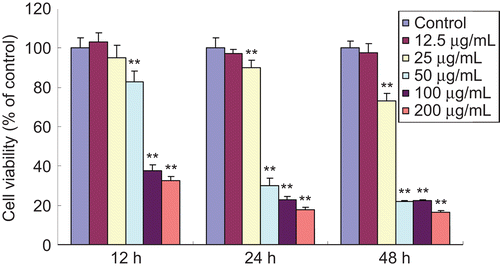
Inhibitory effects of EO on cell viability in hypertrophic scar fibroblasts
Following the treatment with various concentrations of EO for 12, 24, and 48 h, cell viability was determined by MTT assay. As shown in , EO significantly inhibited the viability of HSFs in a time- and dose-dependent manner. The proliferation of HSFs treated with EO (25–200 μg/mL) evidently decreased, compared with the control group, except at the low-dose group, which showed no statistical significance.
Figure 2. Inhibitory effects of EO on cell viability in HSFs at different times. Cell viability was determined by MTT assay and the experiment was repeated three times. When HSFs were treated with EO for 12, 24 and 48 h, the cell viability significantly decreased in a time- and dose-dependent manner. **P <0.01 compared with the control group. Data are expressed as the mean ± SD, n = 10.
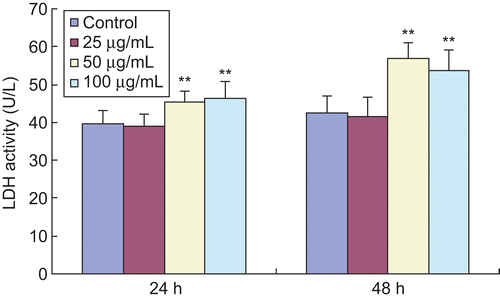
Facilitating effects of EO on LDH release in HSFs
LDH release is another indicator for cell toxicity. When cell membranes are destroyed, intracellular LDH will be released into the medium. Therefore, LDH release can be reflected by determination of LDH activity in the medium. It is shown in that treatment with EO led to a significant increase of LDH activity in the medium compared with the control group. But the low-dose EO (25 μg/mL) did not increase LDH activity at 24 and 48 h.
Figure 3. Effects of EO on LDH activity in HSFs. The LDH activity released by HSFs was measured with an automatic biochemistry analyzer. After treatment with 100 and 200 μg/mL EO for 24 and 48 h, the LDH activity in the medium was markedly enhanced, suggesting an increase in LDH release of HSFs. **P <0.01 compared with the control group. Data are presented as the mean ± SD, n = 10.
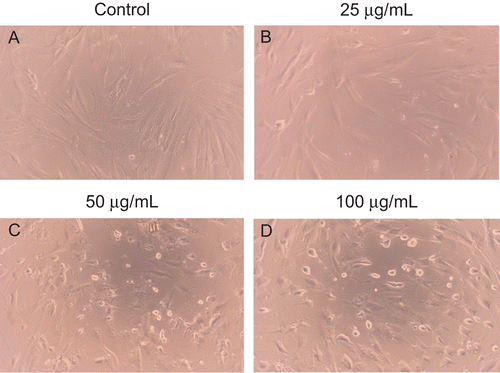
Induction of apoptosis in HSFs
Morphological analysis revealed that cell death evidently increased with the prolongation of exposure time and the increase of dose when HSFs were treated with different concentrations of EO for different times. HSFs treated without EO exhibited a long, fusiform, bipolar shape, and adhered and aligned densely in similar directions. Little cell death was observed in the control group. However, HSFs treated with EO for 12, 24, and 48 h rounded, shrank, floated and aligned loosely, and intercellular space increased. displayed morphological changes of HSFs treated with EO for 12 h.
Figure 4. The morphological alterations to HSFs treated with indicated concentrations of EO for 12 h. (A) Little cell death was observed in the control group. (B–D) The number of cell death significantly increased in a dose-dependent manner when HSFs were treated with 25, 50 and 100 μg/mL EO for 12 h. Cell morphology was assessed using an Olympus inverted phase-contrast microscope with an Olympus C-5060 digital camera. The morphological alterations to HSFs treated with EO for 24 and 48 h were not displayed.
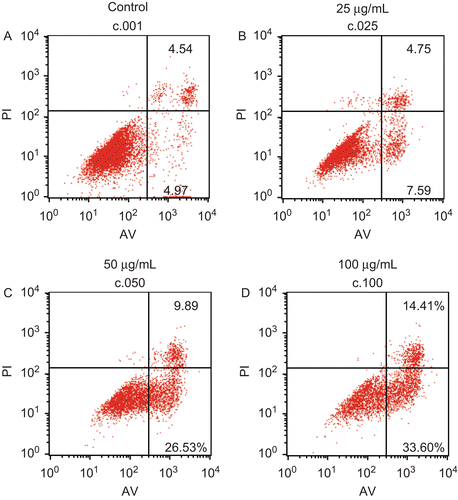
In order to substantiate cell apoptosis, flow cytometry assay was carried out. The percentage of apoptotic cells could be analyzed with a flow cytometer when these cells were stained with both PI and Annexin V (AV). AV-negative and PI-negative cells were used as the control. The AV-positive and PI-negative population represent early apoptotic cells, while cells positive for AV as well as for PI are considered as late apoptotic or necrotic. As shown in , analysis of the cell population showed distinct sets of population. Most HSFs were vital (90.32%) in the control group (). However, when HSFs were incubated with various concentrations of EO (25, 50, 100 μg/mL) for 12 h, there was a significant increase in the percentage of early apoptotic HSFs (AV positive but PI negative) in a dose-dependent manner from 4.97% in the control up to 7.59%, 26.53% and 33.6% in the treated groups. The increase in the percentage of late apoptotic HSFs (AV and PI double-positive cells) is further evidence for induction of apoptosis by EO, apparent at concentrations of 25, 50, and 100 μg/mL ().
Figure 5. Effects of EO on apoptosis in HSFs discriminated by AV and PI double staining. After 12 h of treatment with EO, HSFs were labeled with AV and PI and analyzed by a FACSCalibur flow cytometer. The lower left quadrant shows vital cells (double negative). The lower right quadrant indicates early apoptotic cells (AV positive but PI negative). The upper right quadrant represents late apoptotic cells or necrotic cells (double positive).
Effects of EO on MMP loss, cellular ROS level and caspase-3 activity in HSFs
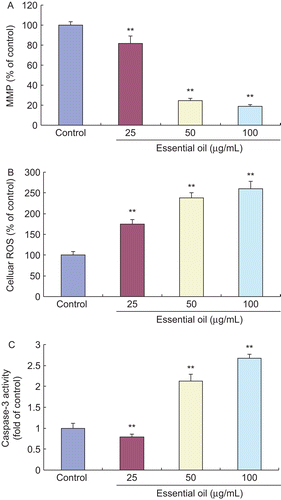
Mitochondria play a pivotal role in cell apoptosis. Loss of mitochondrial membrane potential (MMP) is a crucial step, which causes excessive ROS accumulation, whereas excessive ROS further triggers cell apoptosis. Caspase-3 is well known to act as an important mediator of apoptosis and contributes to morphological changes of apoptosis. Therefore, we determined MMP loss, cellular ROS generation and caspase-3 activity in HSFs treated with EO for 12 h to investigate the mechanism of action. As shown in , the treatment with EO for 12 h significantly increased the loss of MMP, the level of cellular ROS generation and the activity of caspase-3 in HSFs compared with the control group, suggesting that EO elicited apoptosis in HSFs through inducing the loss of MMP, causing the accumulation of excessive ROS, and triggering the activation of caspase-3.
Figure 6. Effects of EO on MMP loss, cellular ROS level and caspase-3 activity in HSFs. After 12 h of treatment with 25, 50 and 100 μg/mL EO, the loss of MMP, the level of cellular ROS and the activity of caspase-3 in HSFs significantly increased when compared with the control group, which showed statistical differences. **P <0.01 versus control. Data are presented as the mean ± SD. (A) 5 × 105 fibroblasts per sample were analyzed with a flow cytometer following the treatment with 10 μg/mL Rhodamine 123 for 30 min. The experiment was independently repeated three times. (B) The level of cellular ROS was reflected by the fluorescence intensity of HSFs and measured with an automated 96-well plate reader using an excitation wavelength of 485 nm and an emission wavelength of 530 nm; n = 10. (C) Caspase-3 activity in HSFs was measured at 405 nm using an ELx-800 universal microplate reader; n = 10.
Discussion
Hypertrophic scar formation following surgical procedures, trauma and especially burns is a significant clinical concern for patients and clinicians (CitationBloemen et al., 2009). It can result in severe functional and cosmetic impairment, and symptoms of pruritus and pain, which are responsible for a decrease in quality of life (CitationAtkinson et al., 2005; CitationBock et al., 2006; CitationVan Loey et al., 2008). The abnormal biological behavior of fibroblasts plays a central role in hypertrophic scar formation and development, which can change into myofibroblasts and facilitate both collagen deposition and wound contraction (CitationTuan & Nichter, 1998). Hypertrophic scar fibroblasts (HSFs) have a higher capacity for proliferation, cytokine production, and collagen synthesis than normal fibroblasts (CitationNiessen et al., 1999). Therefore, inhibitory proliferation with induction of apoptosis in fibroblasts is an important treatment for hypertrophic scarring. In the current study, HSFs from human skin were employed to estimate the effects of the essential oil (EO) from the rhizomes of Ligusticum chuanxiong on cell proliferation and apoptosis.
During the process of cell apoptosis, mitochondria play a pivotal role (CitationPreston et al., 2001), whereas loss of mitochondrial membrane potential (MMP) is a crucial step, which causes glutathione (GSH) exhaustion and excessive ROS accumulation, and subsequently triggers the release of apoptotic factors such as cytochrome c and apoptosis-inducing factor (AIF) from the mitochondrial intermembrane space into cytosol, where these factors elicit the propagation of the apoptotic cascade and execution of cell death (CitationKroemer et al., 2007). On the other hand, excessive ROS accumulation also impairs mitochondrial membranes in reverse, resulting in more loss of MMP.
Living cells constantly produce ROS such as H2O2 and O2- in metabolic processes, and mitochondria are the main source of ROS generation in aerobic cells (CitationOtt et al., 2007). Under physiological conditions, an appropriate level of intracellular ROS is very important for the maintenance of both redox balance and cell proliferation (CitationMurrell et al., 1990; CitationNicotera et al., 1994; CitationPreeta & Nair, 1999; CitationMartin & Barrett, 2002). Nevertheless, excessive ROS will result in cellular injuries, including lipid peroxidation, protein oxidation, enzyme inactivation, and oxidative DNA damage (CitationAmes, 1989; CitationMorel et al., 1990; CitationRanderath et al., 1996; CitationMallis et al., 2001).
In this study, the experimental results showed that EO significantly inhibited cell viability and induced cell apoptosis. The release of LDH evidently increased, as excessive ROS injured the cellular membrane integrity. Investigating the mechanism of action by which HSFs were subjected to apoptosis in response to the treatment with the EO, we observed that the EO evidently induced MMP collapse, and increased ROS and caspase-3 levels, suggesting that the EO destroyed the intracellular antioxidant system. Therefore, we speculate that the induction of apoptosis by the essential oil from Rhizoma Chuanxiong (RCX) in HSFs is at least partially carried out through eliciting loss of MMP, causing accumulation of excessive ROS, and triggering activation of caspase-3. GSH exhaustion and cytochrome c release are likely to participate in this process.
In conclusion, the essential oil extracted from the rhizomes of Ligusticum chuanxiong significantly promoted apoptosis in hypertrophic scar fibroblasts from human skin through inducing the loss of MMP, eliciting the accumulation of excessive ROS, and triggering the activation of caspase-3. Our investigation directly provides evidence for the clinical application of the essential oil, which has favorable potency for the development of an inhibitory agent of hypertrophic scarring, though clinical studies are required.
Declaration of interest
This work was supported by a grant from the Key Programs for Basic Research of Shanghai (No. 08JC1405700).
Related Research Data
References
- Ames BN. (1989). Endogenous DNA damage as related to cancer and aging. Mutat Res, 214, 41–46.
- Atkinson JA, McKenna KT, Barnett AG, McGrath DJ, Rudd M. (2005). A randomized, controlled trial to determine the efficacy of paper tape in preventing hypertrophic scar formation in surgical incisions that traverse Langer’s skin tension lines. Plast Reconstr Surg, 116, 1648–1656.
- Bloemen MC, van der Veer WM, Ulrich MM, van Zuijlen PP, Niessen FB, Middelkoop E. (2009). Prevention and curative management of hypertrophic scar formation. Burns, 35, 463–475.
- Bock O, Schmid-Ott G, Malewski P, Mrowietz U. (2006). Quality of life of patients with keloid and hypertrophic scarring. Arch Dermatol Res, 297, 433–438.
- Brissett AE, Sherris DA. (2001). Scar contractures, hypertrophic scars, and keloids. J Biol Chem, 17, 263–272.
- Chan SSK, Choi AOK, Jones RL, Lin G. (2006). Mechanisms underlying the vasorelaxing effects of butylidenephthalide, an active constituent of Ligusticum chuanxiong, in rat isolated aorta. Euro J Pharmacol, 537, 111–117.
- China Pharmacopoeia Committee. (2005). Chinese Pharmacopoeia, Vol. 1, Beijing: Chemical Industry Press, 28.
- Du J, Sun LN, Xing WW, Huang BK, Jia M, Wu JZ, Zhang H, Qin LP. (2009). Lipid-lowering effects of polydatin from Polygonum cuspidatum in hyperlipidemic hamsters. Phytomedicine, 16, 652–658.
- Fang CH, Alexander JW. (1990). Wound contraction following transplantation of microskin autografts with overlaid skin allograft in experimental animals. J Biol Chem, 16, 190–192.
- Gabbiani G, Hirschel BJ, Ryan GB, Statkov PR, Majno G. (1972). Granulation tissue as a contractile organ. A study of structure and function. J Biol Chem, 135, 719–734.
- Grinnell F. (1994). Fibroblasts, myofibroblasts, and wound contraction. J Biol Chem, 124, 401–404.
- Harauchi T, Hirata M. (1993). Effects of p-phenylenediamines and adriamycin on primary culture of rat skeletal muscle cells. Toxicol Lett, 66,35–46.
- Hou YZ, Zhao GR, Yang J, Yuan JY, Zhu GG, Hiltunen R. (2004). Protective effect of Ligusticum chuanxiong and Angelica sinensis on endothelial cell damage induced by hydrogen peroxide. Life Sci, 75, 1775–1786.
- Jeong JB, Ju SY, Park JH, Lee JR, Yun KW, Kwon ST, Lim JH, Chung GY, Jeong HJ. (2009). Antioxidant activity in essential oils of Cnidium officinale Makino and Ligusticum chuanxiong Hort. and their inhibitory effects on DNA damage and apoptosis induced by ultraviolet B in mammalian cell. Cancer Epidemiol, 33, 41–46.
- Kroemer G, Galluzzi L, Brenner C. (2007). Mitochondrial membrane permeabilization in cell death. Physiol Rev, 87, 99–163.
- Mallis RJ, Buss JE, Thomas JA. (2001). Oxidative modification of H-Ras: S-thiolation and S-nitrosylation of reactive cysteines. Biochem J, 355, 145–153.
- Martin KR, Barrett JC. (2002). Reactive oxygen species as double-edged swords in cellular processes: Low-dose cell signaling versus high-dose toxicity. Hum Exp Toxicol, 21, 71–75.
- Martin P. (1997). Wound healing-aiming for perfect skin regeneration. J Biol Chem, 276, 75–81.
- Morel I, Lescoat G, Cillard J, Pasdeloup N, Brissot P, Cillard P. (1990). Kinetic evaluation of free malondialdehyde and enzyme leakage as indices of iron damage in rat hepatocyte cultures: Involvement of free radicals. Biochem Pharmacol, 39, 1647–1655.
- Murrell GA, Francis M J, Bromley L. (1990). Modulation of fibroblast proliferation by oxygen free radicals. Biochem J, 265, 659–665.
- Nicotera TM, Privalle C, Wang TC, Oshimura M, Barrett JC. (1994). Differential proliferative responses of Syrian hamster embryo fibroblasts to paraquat-generated superoxide radicals depending on tumor suppressor gene function. Cancer Res, 54, 3884–3888.
- Niessen FB, Spauwen PH, Schalkwijk J, Kon M. (1999). On the nature of hypertrophic scars and keloids: A review. Plast Reconstr Surg, 104, 1435–1458.
- Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. (2007). Mitochondria, oxidative stress and cell death. Apoptosis, 12, 913–922.
- Peng C, Xie XF, Wang L, Guo L, Hu TL. (2009). Pharmacodynamic action and mechanism of volatile oil from rhizoma Ligustici Chuanxiong Hort. On treating headache. Phytomedicine, 16, 25–34.
- Preeta R, Nair RR. (1999). Stimulation of cardiac fibroblast proliferation by cerium: A superoxide anion-mediated response. J Mol Cell Cardiol, 31, 1573–1580.
- Preston TJ, Abadi A, Wilson L, Singh G. (2001). Mitochondrial contributions to cancer cell physiology: Potential for drug development. Adv Drug Deliv Rev,49, 45–61.
- Randerath K, Randerath E, Smith CV, Chang J. (1996). Structural origins of bulky oxidative DNA adducts (type II I-compounds) as deduced by oxidation of oligonucleotides of known sequence. Chem Res Toxicol, 9, 247–254.
- Redden RA, Doolin EJ. (2003). Collagen crosslinking and cell density have distinct effects on fibroblast-mediated contraction of collagen gels. J Biol Chem, 9,290–293.
- Reish RG, Eriksson E. (2008). Scar treatments: Preclinical and clinical studies. J Am Coll Surg, 206, 719–730.
- Samarth RM, Panwar M, Kumar M, Soni A, Kumar M, Kumar A. (2008). Evaluation of antioxidant and radical-scavenging activities of certain radioprotective plant extracts. Food Chem, 106,868–873.
- Tuan TL, Nichter LS. (1998). The molecular basis of keloid and hypertrophic scar formation. Mol Med Today, 4,19–24.
- Van Loey NE, Bremer M, Faber AW, Middelkoop E, Nieuwenhuis MK. (2008). Itching following burns: Epidemiology and predictors. Br J Dermatol, 158, 95–100.
- Vande Berg JS, Rudolph R. (1985). Cultured myofibroblasts: A useful model to study wound contraction and pathological contracture. J Biol Chem, 14, 111–120.
- Wu JG, Wu JZ, Sun LN, Han T, Du J, Ye Q, Zhang H, Zhang YG. (2009). Ameliorative effects of arctiin from Arctium lappa on experimental glomerulonephritis in rats. Phytomedicine, 16, 1033–1041.
- Xing WW, Wu JZ, Jia M, Du J, Zhang H, Qin LP. (2009). Effects of polydatin from Polygonum cuspidatum on lipid profile in hyperlipidemic rabbits. Biomed Pharmacother, 63, 457–462.
- Yuan JF, Zhang ZQ, Fan ZC, Yang JX. (2008). Antioxidant effects and cytotoxicity of three purified polysaccharides from Ligusticum chuanxiong Hort. Carbohydr Polym, 74,822–827.
- Zhang H, Han T, Sun LN, Huang BK, Chen YF, Zheng HC, Qin LP. (2008b). Regulative effects of essential oil from Atractylodes lancea on delayed gastric emptying in stress-induced rats. Phytomedicine, 15, 602–611.
- Zhang H, Han T, Yu CH, Rahman K, Qin LP, Peng C. (2007). Ameliorating effects of essential oil from Acori graminei rhizoma on learning and memory in aged rats and mice. J Pharm Pharmacol 59,301–309.
- Zhang H, Xing WW, Li YS, Zhu Z, Wu JZ, Zhang QY, Zhang W, Qin LP. (2008a). Effects of a traditional Chinese herbal preparation on osteoblasts and osteoclasts. Maturitas, 61, 334–339.