Abstract
Context: Earlier findings demonstrated that pretreatment of Herba Cistanches [the dried whole plant of Cistanche deserticola Y.C. Ma (Orobanchaceae)], a “Yang-invigorating” Chinese tonic herb, stimulated the ATP-generation capacity (ATP-GC) in mitochondria isolated from rat heart ex vivo. The enhancement of mitochondrial ATP-GC by Herba Cistanches was associated with induction of glutathione antioxidant status and protection against ischemia/reperfusion (I/R) injury in rat hearts.
Objectives: This study investigated the relationship between enhancements in mitochondrial ATP-GC and glutathione antioxidant status in H9c2 cardiomyocytes using a semipurified fraction of Herba Cistanches (HCF1).
Materials and methods: HCF1 (10–300 ng/mL) was tested for its effects on mitochondrial ATP generation, glutathione antioxidant status and protection against oxidant injury in H9c2 cardiomyocytes and rat hearts.
Results and discussion: HCF1 at 30 ng/mL increased mitochondrial ATP-GC and ADP-stimulated state 3 respiration (by 50 and 100%, respectively) in H9c2 cardiomyocytes. The stimulation of mitochondrial respiration was associated with the induction of mitochondrial uncoupling (27%) and enhancement of cellular glutathione redox cycling as well as protection against hypoxia/reoxygenation (hypox/reoxy)-induced apoptosis (by 60%). While HCF1 treatment increased reactive oxygen species generation from mitochondrial respiration in H9c2 cardiomyocytes, pretreatment with antioxidants (DMTU) abrogated the HCF1-induced cellular responses and the associated cytoprotective effect. HCF1 pretreatment (1.14 and 3.41 mg/kg × 14) also protected against myocardial I/R injury in rats (by 13 and 32%), presumably mediated by the induction of glutathione antioxidant response.
Conclusion: The long-term intake of HCF1 may offer a prospect for the prevention of ischemic heart disease.
Introduction
Ischemic heart disease (IHD) remains the leading killer worldwide, particularly in industrialized countries. Myocardial infarction, a consequence arising from reperfusion of the previously ischemic myocardium following surgical/therapeutic intervention, is the main cause of death in patients suffering from IHD (CitationHofstra et al., 2000; CitationSaraste et al., 1997). The search for therapeutic agents aimed at reducing the extent of myocardial ischemia/reperfusion (I/R) injury has become an area of intense research. Recent studies in our laboratory have shown that a “Yang-invigorating” Chinese tonic herb, Herba Cistanches the dried whole plant of Cistanche deserticola Y.C. Ma (Orobanchaceae) (Moriya et al., Citation1995a,Citationb), enhanced mitochondrial ATP generation and antioxidant capacity in rat hearts (CitationLeung & Ko, 2008; CitationSiu & Ko, 2010). The beneficial effect of Herba Cistanches was associated with protection against myocardial I/R in rats (CitationSiu & Ko, 2010). However, the active ingredient(s) and mechanism underlying the cardioprotective action of Herba Cistanches remains to be investigated.
Herba Cistanches (Cistanche or Rou Cong Rong in Chinese) is one of the most commonly used “Yang-invigorating” tonic herbs in traditional Chinese medicine (CitationJiang et al., 2009). The herb is clinically prescribed for the reinforcement of vital functions of the “kidney” in the context of Chinese medicine. Recently, our laboratory has demonstrated that the pharmacological action of “Yang-invigoration” in Chinese medicine involves the enhancement of mitochondrial ATP-generation capacity (ATP-GC) (CitationKo et al., 2004, 2006). In the present study, as an approach to investigate the mechanism underlying the cardioprotective action of Herba Cistanches, we examined the relationship between enhancements in mitochondrial ATP-GC (CitationWong et al., 2011) or mitochondrial respiration, and glutathione antioxidant status in H9c2 cardiomyocytes, using a semipurified and biologically active fraction (HCF1) obtained from silica gel column chromatography of the ethanol extract of Herba Cistanches. In addition, cyto-/cardio-protective effects of HCF1 on hypoxia/reoxygenation (hypox/reoxy)-induced apoptosis in H9c2 cardiomyocytes and I/R injury in rats were investigated.
Materials and methods
Herbal material
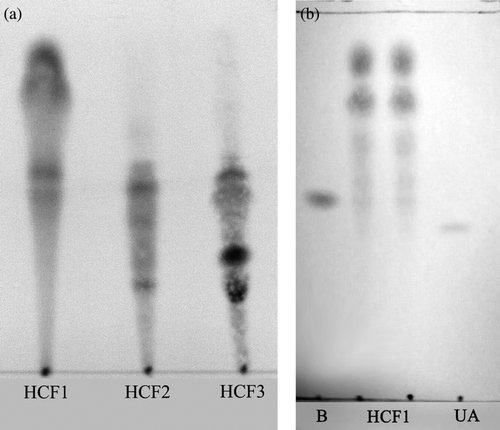
Herba Cistanches (mainly the rhizome of the dried plant) was purchased from a local (Hong Kong-based) herbal dealer, Lee Hoong Kee. The herb was authenticated by Lee Koong Kee and a voucher specimen (HKUST00301) was deposited in the Division of Life Science, Hong Kong University of Science and Technology (HKUST). Herba Cistanches ethanol extract (HCE) was shown to enhance mitochondrial ATP-GC and confer cardioprotection against I/R injury. In brief, HCE was obtained by ethanol extraction of ground herbal material by heating under reflux at 78°C for 2 h, as previously described (CitationLeung & Ko, 2008), with the yield being 14% (w/w). HCE was further fractionated by silica gel column chromatography with 30% acetone in petroleum ether as eluting solvent. The fractionation was monitored by thin layer chromatography (TLC) () and three fractions were obtained [HCF1 (at a yield of 1.14%, with reference to HCE), HCF2 (0.89%) and HCF3 (0.63%)], with the respective TLC chromatogram shown in . All the extracts were dried by evaporating the solvent under reduced pressure at 50°C, and the dried extracts were stored at 4°C until use.
Figure 1. Silica gel thin layer chromatography (TLC) analysis of Herba Cistanches fractions. (a) HCF1, HCF2 and HCF3 fractions were analyzed using TLC plates coated with silica gel 60 UV254 (0.2 mm) with acetone: petroleum ether (30:70, v/v) as developing solvent. (b) HCF1 was analyzed using β-sitosterol (B) and ursolic acid (UA) for comparison.
Cell culture
H9c2 cells, a subclone of the original clonal cell line derived from embryonic BD1X rat heart tissue (CitationHescheler et al., 1991), were purchased from the American Tissue Culture Centre (ATCC). The cells were cultured as monolayers in Dulbecco’s Modified Eagle’s medium (DMEM) (Gibco BRL Life Technologies, Grand Island, NY, US), supplemented with 10% (v/v) fetal bovine serum (FBS), 100 IU/mL of penicillin (Sigma, St. Louis, MO), 100 μg/mL of streptomycin and 17 mM NaHCO3. All cells were grown under an atmosphere of 5% (v/v) CO2 in air at 37°C.
Measurement of ATP-GC in situ
H9c2 cardiomyocytes were seeded at a density of 2.5 × 104 cells/well into 24-well microtiter plates. After cell attachment, cells were incubated with herbal extracts for 4 h at 37°C. The control group was given vehicle [DMSO (0.2%, v/v, final concentration)] only. After the incubation, the ATP-GC assay was performed as previously described (CitationLeung & Ko, 2008). In brief, the removal of herbal extract-containing medium was followed by cell membrane permeabilization with digitonin (50 μg/mL) in an incubation buffer (120 mM KCl, 5 mM KH2PO4, 2 mM EGTA, 10 mM HEPES, 0.1 mM MgCl2, 0.5% BSA, pH 7.4). Pyruvate (15 mM), malate (5 mM) and ADP (10 μM) were added for mitochondrial ATP generation at 37°C, which was monitored at increasing time intervals ranging from 0 to 15 min. The reaction was terminated by the addition of 60 μL of perchloric acid (PCA) (30%, w/v), and the reaction mixtures were then centrifuged at 540×g for 10 min at 4°C, and the supernatants were measured for ATP content by a bioluminescence assay (ATPlite, Perkin Elmer, CA, USA). Data of herbal extract preincubated groups were expressed as percent control.
Measurement of mitochondrial respiration
After 4 h of incubation with HCF1 at a final concentration of 30 ng/mL, cells were trypsinized and suspended in assay buffer (120 mM KCl, 5 mM KH2PO4, 2 mM EGTA, 10 mM HEPES, 0.1 mM MgCl2, 0.5% (w/v) BSA, pH 7.4) and stored at 37°C. Mitochondrial respiration was measured with a Clarke-type electrode (Hansatech Instruments Ltd., Norfolk, UK) (CitationSgobbo et al., 2007). An aliquot (1 mL) of suspended cells (1.5 × 106 cells/mL) (CitationComelli et al., 2007) were placed in the air tight liquid-phase oxygen electrode chamber. The system was maintained at 30°C through a constant temperature water-jacketing system. After the equilibration, cells were permeabilized by digitonin (50 μg/mL). Pyruvate (1.67 mM), malate (5 mM) and ADP (60 μM) were added to allow mitochondrial State 3 respiration. Mitochondrial State 4 respiration was then induced by the addition of complex V inhibitor, oligomycin (1 mg/mL). Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) (1 μM), a chemical uncoupler, was used as the positive control in the measurement. The rate of mitochondrial respiration was estimated by the rate of oxygen consumption by the cell suspension in the oxygen electrode chamber (nmol/O2/min). For HCF1-treated cells, the rate of mitochondrial respiration was normalized to a respective mean control value from drug-untreated samples and expressed as percent control.
Measurement of mitochondrial reactive oxygen species production
After 4 h incubation with HCF1 at 30 ng/mL, cells were trypsinized and permeabilized by digitonin (50 μg/mL). The cells were then centrifuged at 1000×g for 2 min at 4°C to remove cell debris, and the supernatant was centrifuged at 12 000×g for 10 min at 4°C. The pellet was suspended in the incubation buffer (0.1 mM EGTA, 5 mM KH2PO4, 3 mM MgCl2, 145 mM KCl, 30 mM HEPES, pH 7.4) to give a mitochondria-rich fraction for the measurement of mitochondrial reactive oxygen species (ROS) production, using 2′,7′-dichlorofluorescein diacetate (DCFDA) as a probe (CitationLeung et al., 2005). An aliquot (50 μL) of mitochondria-rich fraction (adjusted to 1 mg protein/mL) was added to the wells of a black microtiter plate and then incubated with 60 μL of DCFDA (17.5 μM in incubation buffer) for 10 min at 37°C. An aliquot (40 μL) of incubation buffer and 50 μL of substrates (20 mM pyruvate and 10 mM malate) were then added in each well. Fluorescence emission (Ex 485 nm and Em 535 nm) was monitored every 5 min for 30 min at 37°C. ROS generation was calculated from fluorescence intensities after subtracting the fluorescence value of a blank sample containing only incubation buffer, substrate solution, and DCFDA. ROS generation over the 30-min period was estimated by computing the area under the curve (AUC) of the graph plotting fluorescence intensity against time (0–30 min), and the AUC of HCF1-treated samples were normalized with respective drug-untreated control samples and expressed as percent control.
Measurement of cellular reduced glutathione level
Cellular glutathione (GSH) levels were determined enzymatically using 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) and glutathione reductase (GR), in a protocol modified from CitationGriffith (1980). After HCF1 incubation, an aliquot (100 µL) of 3% (w/v) 5-sulfosalicyclic acid (SSA) was added for the preservation of cellular thiols and deproteinization. The samples were then centrifuged at 540 × g for 10 min at 4°C. The assay solution containing 0.63 mM DTNB and 0.053 mM NADPH in phosphate buffer (100 mM, 5 mM Na2EDTA, pH 7.5) was preincubated at 37°C for 3 min. For the measurement of cellular total glutathione, an aliquot (30 µL) of supernatant was added to the wells of a 96-well microtiter plate, then 180 µL of prewarmed assay solution with 0.525 U/mL GR was added and the changes in absorbance at 412 nm were monitored spectrophotometrically for 5 min. The concentration of total glutathione was estimated from a calibration curve using GSH (dissolved in 3% SSA) as standard and expressed in nmol/mg protein. For the measurement of cellular oxidized glutathione (GSSG) levels, an aliquot (50 µL) of supernatant was mixed with 2-vinylpyridine and triethanolamine, the reaction mixtures were incubated at room temperature for 1 h. After the incubation, an aliquot (30 µL) of reaction mixture was added to the wells of a 96-well microtiter plate, then 180 µL of prewarmed assay solution with 0.525 U/mL GR was added and the changes in absorbance at 412 nm were monitored spectrophotometrically for 5 min. The concentration of GSSG was estimated from a calibration curve using GSSG (dissolved in 3% SSA) as standard and expressed in nmol/mg protein. The GSH level was calculated by subtracting twice the amount of GSSG from total glutathione and data were expressed as percent control.
Hypox/reoxy-induced apoptosis
After 4 h incubation with HCF1 at 30 ng/mL, the cells were washed twice with Krebs-Ringer Bicarbonate buffer (KRB; 115 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 24 mM NaHCO3, 10 mM HEPES, pH 7.4). Aliquots (1.5 mL) of KRB supplemented with 0.01% (w/v) BSA were added to the cells immediately prior to hypoxia. A Billups-Rothenberg modular incubator chamber (Billups-Rothenberg, CA, USA) was used to produce an in vitro hypox/reoxy challenge. In essence, cells were placed in the sealed chamber and the chamber was flushed with nitrogen for 15 min at a flow rate of 20 mL/min. After closing all sealable connectors, the chamber was transferred to an incubator and the cells in the chamber were subjected to a 2 h period of hypoxia at 37°C.
Reoxygenation was initiated by opening the chamber and then replacing the KRB with fresh DMEM medium. The cells were then cultured in the incubator under an atmosphere of 5% (v/v) CO2 in air at 37°C for 16 h.
Measurement of caspase 3 activity
The extent of apoptotic cell death was determined by TruPoint™ Caspase-3 Assay Kit (Perkin Elmer, CA, USA). After reoxygenation, cells were lysed with 300 µL of lysis buffer (20 mM Tris–HCl, 2 mM EDTA, 3 mM EGTA, 1% Triton X-100, 2 mM dithiothreitol (DTT), pH 7.5) for 15 min at 4°C. An aliquot (5 µL) of cell lysate was added into the well of 384-black microtiter plate, and the reaction was initiated by adding 10 µL of caspase DTT reaction buffer (10 mmol DTT) and 5 µL of TruPoint caspase 3 substrate (800 nmol). The blank sample contained only 15 µL of caspase DTT reaction buffer and 5 µL of TruPoint caspase 3 substrate. Fluorescence emission (Ex 340 nm and Em 615 nm) was measured (initial reading) at 37°C with a Victor2 multilabel counter. The reaction mixtures were then incubated in the dark at 37°C for 1 h and the fluorescence emission was measured again (final reading).
The signal of caspase 3 activity (S) was calculated from final fluorescence intensity after subtracting the initial fluorescence value. The caspase 3 activity was then estimated by the ratio of mean signal of sample to mean signal of blank. The caspase 3 activity of hypox/reoxy cell samples was normalized with non-hypox/reoxy samples and expressed as percent control.
Animal care
Female adult Sprague-Dawley rats (10-week-old; 250–300 g) were maintained under a 12 h dark/light cycle at about 22°C and allowed water and food ad libitum. Experimental protocols were approved by the Research Practice Committee at the Hong Kong University of Science & Technology, Hong Kong. Animals were randomly divided into groups, with five animals in each. In the experiment, rats were treated intragastrically with HCF1 at a daily dose of 1.14 and 3.42 mg/kg (equivalent to 0.1 and 0.3 g/kg HCE) for 14 consecutive days and were sacrificed 24 h after the last dosing for the isolated heart experiment. Control animals were treated with the vehicle (olive oil) only. Hearts were isolated from control or HCF1-pretreated rats and then subjected to 40 min of ischemia followed by 20-min reperfusion, as previously described (CitationChiu & Ko, 2004). The degree of myocardial I/R injury was assessed by measuring the extent of LDH leakage in coronary effluent (CitationChiu & Ko, 2004).
Measurement of I/R-induced LDH leakage
The LDH activity of coronary effluent was assayed by adding 60 µL of the sample into the wells of a 96-well microtiter plate, and the reaction was initiated by 140 µL substrate mixture (1.4 mM pyruvate and 0.2 mM NADH in 0.1 M phosphate buffer, pH 8.0). Absorbance changes of the reaction mixture in a final volume of 200 µL were monitored spectrophotometrically at 340 nm for 4 min at 37°C. The enzyme activity was estimated by using an extinction coefficient for reduced form of NADH at 340 nm of 6.22 × 103 M−1·cm−1 and expressed as U/L of coronary effluent. One unit (U) represents the activity of enzyme that can catalyze the oxidation of 1 μmol NADH per minute. The extent of myocardial damage was estimated by computing the AUC of the graph plotting the percent LDH leakage with respect to the mean preischemia value measured during the equilibration period against the reperfusion time (1–10 min).
The extent of I/R-induced myocardial damage could be quantified by noting the differences in the extent of LDH leakage between I/R and non-I/R hearts. The extent of LDH leakage (AUC) of the non-I/R hearts would serve as basal value for the 40-min ischemia and 20-min reperfusion experiment.
The degree of protection against I/R injury was estimated by the equation:
Tissue ATP level
Ventricular tissue samples (~70 mg) of perfused and I/R hearts were homogenized with 5% (v/v) PCA (4 μL/mg tissue) at 4°C. After centrifugation at 2150×g for 10 min at 4°C, the supernatant was diluted five-fold with 5% PCA. An aliquot (120 μL) of the supernatant was neutralized with 90 μL of 1.4 M KHCO3, followed by mixing and centrifugation. The resulting supernatant was analyzed for the ATP content as described before.
Preparation of mitochondrial fraction
Heart ventricular tissues were excised and rinsed with ice-cold isotonic buffer (320 mM sucrose, 1 mM EDTA, 50 mM Tris–HCl, pH 7.4). A 10% (w/v) cardiac homogenate was prepared by homogenizing the minced ventricular tissue with a Teflon glass homogenizer at 4000 rpm for 10 complete strokes. Heart homogenate was centrifuged at 600×g for 10 min at 4°C to remove cell nuclei and debris. The supernatant was then centrifuged at 9200×g for 30 min at 4°C to sediment the mitochondria. The pellet was re-suspended in 0.5 mL of isotonic buffer to reconstitute the mitochondrial fraction.
Measurement of ATP-GC ex vivo
An aliquot (50 μL) of mitochondrial fractions (adjusted to 1 mg protein/mL) was mixed with 50 μL of substrate solution (3 mM pyruvate and 3 mM malate) and 25 μL of pretreated ADP (30 mM) solution to allow mitochondrial ATP generation. The ATP content was then measured as described before (CitationLeung et al., 2005).
Measurement of mitochondrial GSH level
Mitochondrial GSH level was determined enzymatically using DTNB and GR as described before (CitationLeung et al., 2005). An aliquot (210 µL) of the mitochondrial fraction was mixed with 90 µL of 10% SSA, and the supernatant was used for the measurement of GSH.
Statistical analysis
All data were expressed as mean ± standard error of the mean (SEM), unless otherwise specified. Data were analyzed by one-way analysis of variance (one-way ANOVA) and intergroup difference was detected by least significant difference (LSD) when p < 0.05.
Results
Effect of HCF1 on mitochondrial respiration
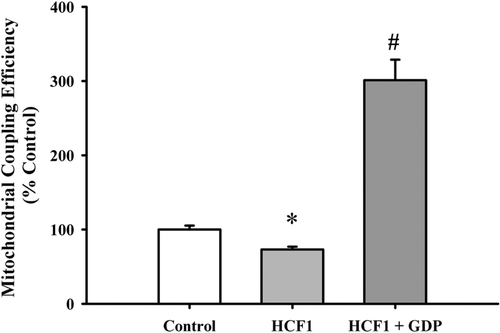
As shown in , incubation with HCE (50–150 µg/mL, 4 h) significantly increased the ATP-GC at all tested concentrations in H9c2 cardiomyocytes, with the extent of stimulation being 70% at 150 µg/mL. Fractions isolated from HCE were tested for their effects on ATP-GC in H9c2 cardiomyocytes. HCF1 increased the ATP-GC in a concentration-dependent manner (5–30 ng/mL), with the maximum stimulation being 50% (). HCF2 () and HCF3 () also maximally stimulated the ATP-GC at 3 and 20 µg/mL, respectively. HCF1, which had a higher yield than those of HCF2 and HCF3, was used for further studies. To confirm the HCF1-induced increase in ATP-GC, the rate of mitochondrial respiration was determined (). HCF1 enhanced mitochondrial respiration in H9c2 cardiomyocytes, as evidenced by a significantly higher rate of State 3 respiration (100%), when compared with the untreated control. State 4 respiratory rate was observed immediately after State 3 respiration when the added ADP was completely phosphorylated to ATP. HCF1 pretreatment (30 ng/mL) significantly enhanced mitochondrial State 4 respiration in H9c2 cardiomyocytes, with the stimulation being 1.7-fold higher than the untreated control (). HCF1 pretreatment (30 ng/mL) significantly decreased the respiratory control ratio (RCR) in H9c2 cardiomyocytes by 27%, with the uncoupling effect being completely abrogated by GDP (500 μM), a specific inhibitor of uncoupling proteins ().
Table 1. Effect of HCF1 on mitochondrial respiration in H9c2 cells.
Figure 2. The effect of Herba Cistanches ethanol extract (HCE) on ATP generation capacity (ATP-GC) in H9c2 cells. H9c2 cells were incubated with HCE at the indicated concentrations for 4 h. ATP-GC was measured as described in Materials and Methods. Data were expressed in percent control with respect to the herbal extract-untreated control (control AUC2 (arbitrary unit) value = 851.03 ± 6.97). Values given are mean ± SEM, with n = 6. *Significantly different from the Control group.
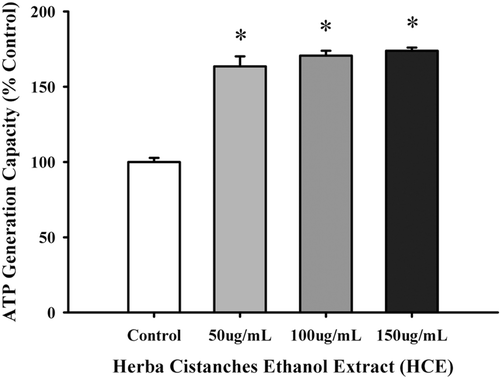
Figure 3. The effect of Herba Cistanches fractions on ATP-GC in H9c2 cells. Data were expressed in percent control with respect to the untreated control [Control AUC2 (arbitrary unit) values = 850.02 ± 4.66 (HCF1), 861.07 ± 7.47 (HCF2), 882.97 ± 5.44 (HCF3)]. Values given are mean ± SEM, with n = 6. *Significantly different from the respective control group.
![Figure 3. The effect of Herba Cistanches fractions on ATP-GC in H9c2 cells. Data were expressed in percent control with respect to the untreated control [Control AUC2 (arbitrary unit) values = 850.02 ± 4.66 (HCF1), 861.07 ± 7.47 (HCF2), 882.97 ± 5.44 (HCF3)]. Values given are mean ± SEM, with n = 6. *Significantly different from the respective control group.](/cms/asset/2e3f057f-5a14-4795-98c2-90f85451534d/iphb_a_710242_f0003_b.gif)
Figure 4. The effect of HCF1 pretreatment on mitochondrial uncoupling in H9c2 cells. H9c2 cells were incubated with HCF1 for 4 h. Mitochondrial respiration rates were measured using Clarke-type electrode as described in Materials and Methods. The respiratory control ratio was estimated by calculating the ratio of State 3 to State 4 respiration. Data were expressed in percent control with respect to the untreated control. Values given are mean ± SEM, with n = 3. *Significantly different from the control group; # significantly different from the HCF1-pretreated group.
Effect of HCF1 on mitochondrial ROS generation
HCF1 preincubation significantly increased the mitochondrial ROS production by 16%, when compared with the control group. The extent of increase was completely suppressed by KCN, a complex IV inhibitor ().
Table 2. Effect of HCF1 on mitochondrial reactive oxygen species (ROS) production in H9c2 cells.
Effect of HCF1 on cellular antioxidant status
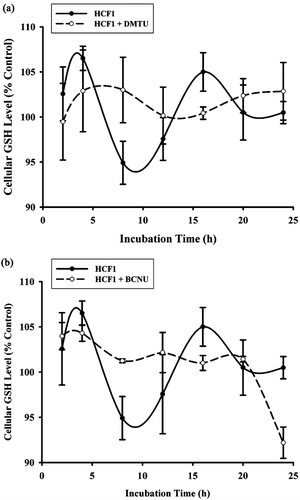
The effect of HCF1 on cellular antioxidant status was also examined. The measurement of cellular GSH level is a reliable indicator of cellular antioxidant status. The incubation with HCF1 at increasing concentrations (30–300 ng/mL) induced a time-driven cyclic variation of cellular GSH level in H9c2 cardiomyocytes, with a concentration-dependent increase in the amplitude of oscillation (94–138%) (). The time-driven cyclic variation of cellular GSH was abrogated by the coincubation of HCF1 with antioxidant, DMTU () and BCNU, a specific inhibitor of GR ().
Figure 5. The effect of HCF1 pretreatment on cellular reduced glutathione (GSH) level in H9c2 cells. H9c2 cells were incubated with HCF1 (30–300 ng/mL) for increasing period of time and the cellular GSH level was measured. Data were expressed in percent control with respect to the time-matched untreated control (initial control GSH level = 20.74 ± 0.53 nmol/mg protein). Values given are mean ± SEM, with n = 4.
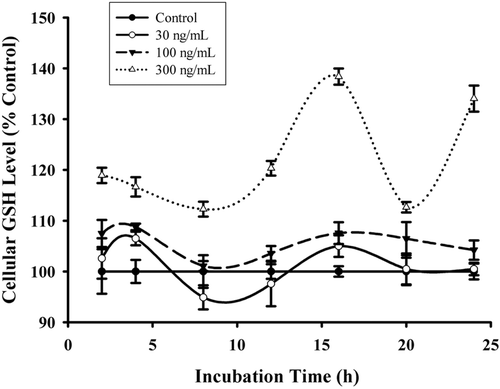
Figure 6. Effects of BCNU and DMTU on HCF1-induced changes in cellular GSH levels in H9c2 cells. DMTU, an antioxidant, and BCNU, a specific inhibitor of glutathione reductase (GR) were coincubated with HCF1 (30 ng/mL). Data were expressed in percent control with respect to the time-matched untreated control (initial control GSH level = 20.73 ± 0.52, 20.35 ± 0.56 nmol/mg protein, respectively). Values given are mean ± SEM, with n = 4.
Effect of HCF1 on hypox/reoxy-induced apoptosis
Caspase 3 activity can serve as an indirect measure of cell apoptosis. Hypox/reoxy challenge significantly increased the caspase 3 activity by 40%, when compared with the non-challenged control (). HCF1 significantly suppressed the stimulation of caspase 3 activity, with the degree of protection being 62%, when compared with the untreated and challenged control (). The cytoprotection afforded by HCF1 against apoptosis was abrogated by both DMTU and BCNU coincubations ().
Table 3. Effect of DMTU and BCNU on HCF1-induced protection against hypox/reoxy-induced apoptosis in H9c2 cells.
Figure 7. The effect of HCF1 on hypoxia/reoxygenation (hypox/reoxy)-induced caspase 3 activity in H9c2 cells. H9c2 cells were incubated with HCF1 for 4 h. Cells were then subjected to hypox/reoxy challenge, and the caspase 3 was measured thereafter. Data were expressed in percent control with respect to the non-hypox/reoxy control [control caspase 3 activity (arbitrary unit) = 12.44 ± 0.11]. Values given are mean ± SEM, with n = 3. *Significantly different from the non-hypox/reoxy control group; #Significantly different from HCF1-pretreated and non-hypox/reoxy group.
![Figure 7. The effect of HCF1 on hypoxia/reoxygenation (hypox/reoxy)-induced caspase 3 activity in H9c2 cells. H9c2 cells were incubated with HCF1 for 4 h. Cells were then subjected to hypox/reoxy challenge, and the caspase 3 was measured thereafter. Data were expressed in percent control with respect to the non-hypox/reoxy control [control caspase 3 activity (arbitrary unit) = 12.44 ± 0.11]. Values given are mean ± SEM, with n = 3. *Significantly different from the non-hypox/reoxy control group; #Significantly different from HCF1-pretreated and non-hypox/reoxy group.](/cms/asset/6f081d13-f1fd-4306-bf6f-7776a794e7f5/iphb_a_710242_f0007_b.gif)
Effect of HCF1 on myocardial I/R injury
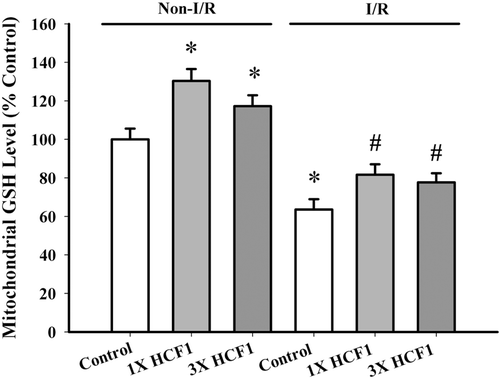
Long-term, low-dose HCF1 treatment (1.14 and 3.42 mg/kg) protected against I/R injury in rat hearts, with the degree of protection being 13 and 32%, respectively (). HCF1 pretreatment significantly increased mitochondrial GSH level by 30 and 18%, respectively, when compared with the untreated control, in non-I/R hearts (). A significant depletion of mitochondrial GSH was observed in I/R rat hearts, with the extent of depletion being 36%. HCF1 pretreatment partially reversed the I/R-induced depletion in mitochondrial GSH (back to 81 and 79% of the non-I/R control value).
Table 4. Effect of long-term HCF1 treatment on myocardial ischemia/reperfusion (I/R) injury in rats.
Figure 8. The effect of long-term HCF1 pretreatment on mitochondrial GSH level. Rats were orally treated with HCF1 at daily doses of 1.14 and 3.42 mg/kg for 14 consecutive days. Mitochondrial GSH level was measured as described. Data were expressed in percent control with respect to the non-I/R control (Non-I/R control GSH level = 2.281 ± 0.15 nmol/mg protein). Values given are mean ± SEM, with n = 5. *Significantly different from the non-I/R control group; #Significantly different from the I/R control group.
HCF1 pretreatment significantly decreased the tissue ATP level in non-I/R rat hearts (by 35 and 23%, respectively). I/R challenge decreased the tissue ATP content in both control (52%) and HCF1-pretreated (26 and 34%, respectively) rat hearts, when compared with the respective untreated and HCF1-treated non-I/R hearts (). HCF1 pretreatment increased the ATP-GC in mitochondria isolated from non-I/R rat hearts, with the extent of stimulation being 12 and 25%, respectively, when compared with the untreated control (). An increase in ATP-GC was observed after I/R challenge in both control (25%) and HCF1-pretreated (44 and 20%) rat heart mitochondria.
Figure 9. The effect of long-term HCF1 pretreatment on tissue ATP content in rat hearts. Animals were orally treated with HCF1 at daily doses of 1.14 (1×) and 3.42 (3×) mg/mL for 14 consecutive days. The basal tissue ATP content was determined as described in Materials and methods. Data were expressed in percent control with respect to the non-ischemia/reperfused (I/R) control (control tissue ATP content = 1273 ± 239 nmol/mg protein tissue). Values given are mean ± SEM, with n = 5. *Significantly different from the non-I/R control group.
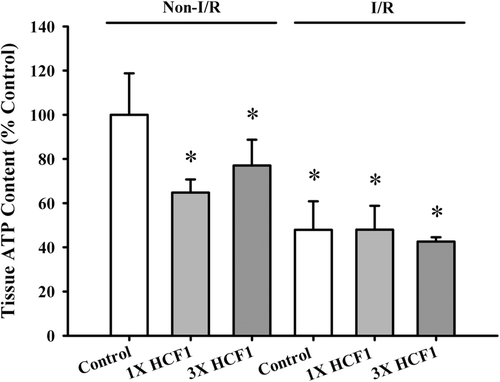
Figure 10. The effect of long-term HCF1 pretreatment on ATP-GC in mitochondria isolated from control and I/R rat hearts. Rats were orally treated with HCF1 at daily doses of 1.14 and 3.42 mg/kg for 14 consecutive days. Heart mitochondria were isolated and subjected to the measurement of ATP-GC in situ. Data were expressed in percent control with respect to the non-I/R control [Non-I/R control AUC2 (arbitrary unit) value = 1000.02 ± 53.74]. Values given are mean ± SEM, with n = 5. *Significantly different from the non-I/R control group; #Significantly different from the I/R control group.
![Figure 10. The effect of long-term HCF1 pretreatment on ATP-GC in mitochondria isolated from control and I/R rat hearts. Rats were orally treated with HCF1 at daily doses of 1.14 and 3.42 mg/kg for 14 consecutive days. Heart mitochondria were isolated and subjected to the measurement of ATP-GC in situ. Data were expressed in percent control with respect to the non-I/R control [Non-I/R control AUC2 (arbitrary unit) value = 1000.02 ± 53.74]. Values given are mean ± SEM, with n = 5. *Significantly different from the non-I/R control group; #Significantly different from the I/R control group.](/cms/asset/affd39f8-988b-4059-b2ba-95140125c9b7/iphb_a_710242_f0010_b.gif)
Discussion
Previous findings in our laboratory have demonstrated that “Yang-invigorating” Chinese tonic herbs invariably increased the ATP-GC in both isolated rat hearts ex vivo and cultured H9c2 cardiomyocytes in vitro (CitationLeung & Ko, 2008; CitationWong et al., 2011). In the present study, the ethanol extract (HCE) but not the aqueous extract of Herba Cistanches was used for investigation because the former contains a wide range of non-polar and polar ingredients but not only water-soluble ingredients. The finding that HCE increased the ATP-GC in H9c2 cardiomyocytes confirmed the presence of active “Yang-invigorating” compounds in the fraction. Using an ATP-GC assay as an activity monitor, a semipurified fraction (HCF1) was isolated from HCE by silica gel column chromatography. HCF1, which can stimulate ATP-GC in H9c2 cardiomyocytes, is a relatively non-polar fraction, with the degree of purification being 1500-fold with reference to HCE. The fraction was initially analyzed by HPLC-MS and major markers of Herba Cistanches (echinacoside and acteocide) were not detected in the fraction (data not shown) (CitationHui et al., 2003; CitationJiang & Tu, 2009). However, silica gel thin layer chromatography revealed the presence of β-sitosterol in HCF1 (). β-Sitosterol is a hydrophobic compound that shares a similar chemical structure with cholesterol, an important component of membrane lipids (CitationDempsey et al., 1956; Citationvan Rensburg et al., 2000). The finding that HCF2 and HCF3 fractionated from HCE could also stimulate ATP-GC in H9c2 cardiomyocytes suggests the presence of active ingredients with varied molecular structures in HCE. Future studies will investigate whether or not β-sitosterol can stimulate ATP-GC in H9c2 cardiomyocytes.
HCF1 pretreatment was found to be capable of increasing mitochondrial respiration, inducing mitochondrial uncoupling and eliciting a cellular antioxidant response. Based on these experimental observations, we hypothesize that the HCF1-induced increase in mitochondrial respiration can cause a parallel increase in electron leakage from the electron transport chain. The resultant increase in mitochondrial ROS generation can in turn trigger cellular responses such as mitochondrial uncoupling and glutathione antioxidant response, a process known as mitohormesis (CitationTurrens, 2003; CitationBrookes et al., 2004; CitationRistow & Zarse, 2010; CitationRistow & Schmeisser, 2011). The findings that HCF1 pretreatment significantly increased the ROS generation in a mitochondria-rich fraction isolated from H9c2 cardiomyocytes and that the HCF1-induced ROS production was abrogated by mitochondrial complex IV inhibitor support the hypothesis. In addition, the HCF1-induced decrease in coupling efficiency was reversed by the coincubation with Trolox (data not shown), indicating the involvement of ROS in the process. In this regard, the induction of mitochondrial uncoupling has been shown to serve as a feedback mechanism for limiting the generation of ROS during mitochondrial respiration (CitationRousset et al., 2004). Superoxide generated during mitochondrial respiration is capable of activating the uncoupling protein-mediated (or GDP-sensitive) mitochondrial uncoupling, thereby decreasing the membrane potential and ROS production (CitationEchtay et al., 2002).
HCF1 pretreatment also induced a time-driven cyclic variation of cellular GSH, indicative of glutathione redox cycling. The enhanced cyclic variation of GSH induced by HCF1 was also abolished by the coincubation with DMTU, a thiol-containing antioxidant, suggesting the involvement of mitochondrial ROS production in the HCF1-induced cellular antioxidant response. The finding that the HCF1-induced cyclic change in cellular GSH was suppressed by BCNU, a specific GR inhibitor, supported the involvement of GR-mediated redox cycling of GSH in the process. Conceivably, the HCF1-induced increase in mitochondrial ROS production can deplete cellular GSH, thereby activating the glutathione redox cycling. Hence, the cyclic change in cellular GSH levels is likely a result of the interplay between ROS generation and GR-mediated regeneration of GSH in HCF1-treated cells. The enhancement of glutathione redox cycling by HCF1 was associated with the significant protection against hypox/reoxy-induced apoptosis in H9c2 cardiomyocytes, as indirectly assessed by caspase 3 activation. Consistently, the cytoprotection against apoptosis afforded by HCF1 was abrogated by DMTU or BCNU co-incubation in H9c2 cardiomyocytes. In this connection, the cellular glutathione redox cycling has been shown to play a determinant role in cytoprotection against oxidant injury (CitationCircu et al., 2009; CitationCircu & Aw, 2010).
In order to confirm the protective effect of HCF1 on oxidant injury, the cardioprotective effect of HCF1 was examined in a rat model of myocardial I/R injury (CitationArmstrong, 2004; CitationVenardos et al., 2007). Long-term, low-dose HCF1 treatment protected against myocardial I/R injury in rats. The cardioprotection against I/R injury afforded by HCF1 pretreatment was associated with an enhancement in mitochondrial glutathione status in both non-I/R and I/R rat hearts. The ability of HCF1 pretreatment to partially reverse the mitochondrial GSH depletion induced by I/R challenge likely involved the upregulation of cellular glutathione redox cycling through the intermediacy of mitochondrial ROS production, as was the case in H9c2 cardiomyocytes (CitationYim & Ko, 1999; CitationRamires & Ji, 2001). While the reduction of tissue ATP level by long-term, low-dose HCF1 pretreatment in non-I/R rat hearts might be due to the induction of a sustained mitochondrial uncoupling, the significant reduction of tissue ATP level in ischemic/reperfused rat hearts was likely due to the impaired mitochondrial electron transport and oxidative phosphorylation (CitationTalbot et al., 2004; CitationBrennan et al., 2006). The reduced extent of I/R-induced myocardial ATP depletion in HCF1-pretreated rat hearts, as observed in the present study, was likely related to the increased ATP-GC that compensated the enhanced ATP consumption for cellular homeostasis during reperfusion (CitationPlaschke et al., 1998; CitationYoung, 2008).
Conclusion
In conclusion, HCF1 pretreatment enhanced mitochondrial ATP-GC in both H9c2 cardiomyocytes and mitochondria isolated from rat hearts. The associated increase in mitochondrial ROS production in HCF1-treated cells would in turn induce mitochondrial uncoupling and cellular antioxidant response. The resultant upregulation of cellular antioxidant mechanism preserves the integrity of cellular components, particularly under oxidative stress condition. The sustained increases in electron transport and ROS generation induced by HCF1 may therefore result in a higher resistance of cardiomyocytes to oxidant injury. The long-term intake of HCF1 can therefore offer a promising prospect for the prevention of IHD. Future studies will investigate whether HCF1 can stimulate the glutathione redox cycling and protect against oxidant injury in organs such as the brain, liver and kidney in rats.
Declaration of interest
The authors report no declarations of interest.
References
- Armstrong SC. (2004). Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc Res, 61, 427–436.
- Brennan JP, Southworth R, Medina RA, Davidson SM, Duchen MR, Shattock MJ. (2006). Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation. Cardiovasc Res, 72, 313–321.
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. (2004). Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am J Physiol, Cell Physiol, 287, C817–C833.
- Chiu PY, Ko KM. (2004). Schisandrin B protects myocardial ischemia-reperfusion injury partly by inducing Hsp25 and Hsp70 expression in rats. Mol Cell Biochem, 266, 139–144.
- Circu ML, Aw TY. (2010). Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med, 48, 749–762.
- Circu ML, Moyer MP, Harrison L, Aw TY. (2009). Contribution of glutathione status to oxidant-induced mitochondrial DNA damage in colonic epithelial cells. Free Radic Biol Med, 47, 1190–1198.
- Comelli M, Metelli G, Mavelli I. (2007). Downmodulation of mitochondrial F0F1 ATP synthase by diazoxide in cardiac myoblasts: A dual effect of the drug. Am J Physiol Heart Circ Physiol, 292, H820–H829.
- Dempsey ME, Farquhar JW, Smith RE. (1956). The effect of beta sitosterol on the serum lipids of young men with arteriosclerotic heart disease. Circulation, 14, 77–82.
- Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham JC, Brand MD. (2002). Superoxide activates mitochondrial uncoupling proteins. Nature, 415, 96–99.
- Griffith OW. (1980). Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal Biochem, 106, 207–212.
- Hescheler J, Meyer R, Plant S, Krautwurst D, Rosenthal W, Schultz G. (1991). Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ Res, 69, 1476–1486.
- Hofstra L, Liem IH, Dumont EA, Boersma HH, van Heerde WL, Doevendans PA, De Muinck E, Wellens HJ, Kemerink GJ, Reutelingsperger CP, Heidendal GA. (2000). Visualisation of cell death in vivo in patients with acute myocardial infarction. Lancet, 356, 209–212.
- Hui RH, Hou DY, Li TC, Cuan CX. (2003). Analysis of volatile components in Herba Cistanches. Chinese J Anal Chem, 5, 601–603.
- Jiang Y, Li SP, Wang YT, Chen XJ, Tu PF. (2009). Differentiation of Herba Cistanches by fingerprint with high-performance liquid chromatography-diode array detection-mass spectrometry. J Chromatogr A, 1216, 2156–2162.
- Jiang Y, Tu PF. (2009). Analysis of chemical constituents in Cistanche species. J Chromatogr A, 1216, 1970–1979.
- Ko KM, Mak DH, Chiu PY, Poon MK. (2004). Pharmacological basis of ‘Yang-invigoration’ in Chinese medicine. Trends Pharmacol Sci, 25, 3–6.
- Ko KM, Leon TY, Mak DH, Chiu PY, Du Y, Poon MK. (2006). A characteristic pharmacological action of ‘Yang-invigorating’ Chinese tonifying herbs: enhancement of myocardial ATP-generation capacity. Phytomedicine, 13, 636–642.
- Leung HY, Chiu PY, Poon MK, Ko KM. (2005). A yang-invigorating Chinese herbal formula enhances mitochondrial functional ability and antioxidant capacity in various tissues of male and female rats. Rejuvenation Res, 8, 238–247.
- Leung HY, Ko KM. (2008). Herba Cistanche extract enhances mitochondrial ATP generation in rat hearts and H9c2 cells. Pharm Biol, 46, 418–424.
- Moriya A, Tu P, Karasawa D. (1995a). Pharmacognostical studies of Cistanchis Herba (II): Comparison of the components of Cistanche plants. Nat Med, 49, 394–400.
- Moriya A, Tu P, Karasawa D, Arima H, Deyama T, Kegasawa K. (1995b). Pharmacognostical studies of Cistanchis Herba (1). Comparison of the morphology of Cistanche plants. Nat Med, 49, 383–393.
- Plaschke K, Bardenheuer HJ, Weigand MA, Martin E, Hoyer S. (1998). Increased ATP production during long-term brain ischemia in rats in the presence of propentofylline. Eur J Pharmacol, 349, 33–40.
- Ramires PR, Ji LL. (2001). Glutathione supplementation and training increases myocardial resistance to ischemia-reperfusion in vivo. Am J Physiol Heart Circ Physiol, 281, H679–H688.
- Ristow M, Schmeisser S. (2011). Extending life span by increasing oxidative stress. Free Radic Biol Med, 51, 327–336.
- Ristow M, Zarse K. (2010). How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp Gerontol, 45, 410–418.
- Rousset S, Alves-Guerra MC, Mozo J, Miroux B, Cassard-Doulcier AM, Bouillaud F, Ricquier D. (2004). The biology of mitochondrial uncoupling proteins. Diabetes, 53 Suppl 1, S130–S135.
- Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M, Voipio-Pulkki LM. (1997). Apoptosis in human acute myocardial infarction. Circulation, 95, 320–323.
- Siu AH, Ko KM. (2010). Herba Cistanche extract enhances mitochondrial glutathione status and respiration in rat hearts, with possible induction of uncoupling proteins. Pharm Biol, 48, 512–517.
- Sgobbo P, Pacelli C, Grattagliano I, Villani G, Cocco T. (2007). Carvedilol inhibits mitochondrial complex I and induces resistance to H2O2 -mediated oxidative insult in H9C2 myocardial cells. Biochim Biophys Acta, 1767, 222–232.
- Talbot DA, Duchamp C, Rey B, Hanuise N, Rouanet JL, Sibille B, Brand MD. (2004). Uncoupling protein and ATP/ADP carrier increase mitochondrial proton conductance after cold adaptation of king penguins. J Physiol (Lond), 558, 123–135.
- Turrens JF. (2003). Mitochondrial formation of reactive oxygen species. J Physiol (Lond), 552, 335–344.
- van Rensburg SJ, Daniels WM, van Zyl JM, Taljaard JJ. (2000). A comparative study of the effects of cholesterol, beta-sitosterol, beta-sitosterol glucoside, dehydroepiandrosterone sulphate and melatonin on in vitro lipid peroxidation. Metab Brain Dis, 15, 257–265.
- Venardos KM, Perkins A, Headrick J, Kaye DM. (2007). Myocardial ischemia-reperfusion injury, antioxidant enzyme systems, and selenium: A review. Curr Med Chem, 14, 1539–1549.
- Wong HS, Leung HY, Ko KM. (2011). ‘Yang-invigorating’ Chinese tonic herbs enhance mitochondrial ATP generation in H9c2 cardiomyocytes. Chinese Med, 2, 1–5.
- Yim TK, Ko KM. (1999). Schisandrin B protects against myocardial ischemia-reperfusion injury by enhancing myocardial glutathione antioxidant status. Mol Cell Biochem, 196, 151–156.
- Young LH. (2008). AMP-activated protein kinase conducts the ischemic stress response orchestra. Circulation, 117, 832–840.