Abstract
Context: The extraction method and the crude wound healing effects of sacchachitin from Ganoderma tsugae Murr. (Ganodermataceae) has been cited. However, its purity is still largely limited.
Objective: An improvement of the fractionation protocol to purify the sacchachitin from Ganoderma lucidum L. (Ganodermataceae) (SGL) is needed.
Methods: Fruiting bodies were extracted with double distilled water and subsequently the residue treated with 95% ethanol and then 40% ethanol. After being filtered, the pH of the supernatant was adjusted to 4.0 with 1 N HCl and lyophilized. The supernatant was added (3:1 v/v) ethanol, the precipitate was collected, 2% NaOH was added and refluxed. The supernatant was collected with pH adjusted to 4.0, then treated with 10% potassium hydroxide (KOH) with repeating acid precipitation and (3:1) ethanol precipitation twice more to obtain the sacchachitin.
Results: SGL had a hexosamine content 16.3% (w/w), firmly linked to a talomannan. Its Fourier Transform Infrared Spectroscopy (FTIR) spectrum revealed specific absorption (in cm–1) νO–H 3455.5 b,s, amide νC=O 1678.5, and amide I° δN–H 1550.4. The percentage deacetylation degree was 37.6 and 39.4% for SGL and MSC, respectively. As contrast, MSC contained only 6.6% of hexosamine with a low protein/carbohydrate ratio 0.35 comparing to 0.82 for SGL. SGL was only moderately strong antioxidant regarding the anti-DPPH, antihydroxyl free radical, and antisuperoxide anion capabilities, exhibiting an IC33 values of 10 mg/mL (the highest scavenging capability never exceeding 33%), 0.9 mg/mL, and 4.8 mg/mL, respectively.
Conclusion: We have successfully isolated the pure sacchachitin from the fruiting bodies of G. lucidum that exhibits potent antioxidative activity and may be useful in fabrication of the artificial skin composite substitute.
Introduction
Ganoderma lucidum L. (Ganodermataceae) enjoys special veneration in Asia, where it has been used as a medicinal mushroom in Traditional Chinese Medicine for more than 2000 years, making it one of the oldest mushrooms known to have been used in medicine (CitationSanodiya et al., 2009). It belongs to basidiomycete white rot fungus mostly used as an official medicine in many provinces of China (CitationLee, 1578) and other Asian countries including Japan and Korea (Russell & Paterso, 2006). G. lucidum is famous for its folkloric names Língzhī (in Chinese), or Munnertake, Sachitak, or Reishi (in Japanese), or Yeongji (in Korean). In Chinese, the word Língzhī actually means “the herb of spiritual potency” or “the mushroom of immortality” (CitationDavid, 1986). But as often noted, Língzhī commonly denotes one general form for mushroom G. lucidum and its close relative Ganoderma tsugae Murr.
Preclinical studies have established that the polysaccharide fraction of G. lucidum has a diversity of potent medicinal effects (Russell & Paterso, 2006). Currently, Língzhī has been officially listed in the American Herbal Pharmacopoeia and Therapeutic Compendium.
The term “sacchachitin” was first used by CitationSu et al. (1997) for description of special glucosamine derivative isloated from G. tsuga. This Su’s sacchachitin is a copolymer made from β-1,3-glucan (approximately 60%) and N-acetylglucosamine (approximately 40%), having a filamental structure. This pulpy white residue was tested for use as a skin substitute, thereby wound healing was found faster, compared to the commercialized skin substitute, the chitin sheet made of crab shell (BeschitinTM). Furthermore, many researchers indicated that sacchachitin exhibits potent antibacterial (CitationYang et al., 2001; CitationLin, 2001; CitationWang & Ng, 2006; MoradaCitationLi et al., 2006) and antiviral activities (CitationWang & Ng, 2006; MoradaCitationLi et al., 2006).
Accumulating studies have shown that Ganoderma mushrooms possess antitumor, immunomodulatory, immunotherapeutic, antiagglutination (CitationLiu et al. 2006), hypotensive, hypocholesterolemic, and hypoglycemic bioactivities (CitationBensky et al., 2004). Ganoderma mushrooms are useful preventive nutraceutics against cancer metastasis (CitationLee et al., 1984), its potency is comparable to Lentinan prepared from Shiitake mushrooms (CitationSuga et al., 1984). The mechanism by which G. lucidum affected cancer is still unclear. Different cancer stages can be affected differently. In general, it affects the cancer development by inhibiting angiogensis, migration, metastasis of the cancer cells, and inducing apoptosis of tumor cells (CitationPaterson, 2006). Moreover, the Ganderma compounds inhibit 5α-keto-reductase activity that is responsible for the biosynthesis of dihydrotestosterone (CitationLiu et al., 2006), implicating its therapeutic value in prostate cancer.
Its active constituents include polysaccharides, terpenes, and others isolated from the fruiting bodies and mycelia of this fungus (CitationPaterson, 2006; CitationLindequist et al., 2005). Ganoderic acid is a moderate hepatoprotective agent (CitationLi & Wang, 2006). In addition, Ganderma-produced sterols inhibit lanosterol 14α-demethylase activity required for the biosynthesis of cholesterol (CitationHajjaj et al., 1986).
CitationZhu et al. (2007) found G. lucidum polysaccharides enhanced the function of immunological effector cells in immunosuppressed mice. More recently, CitationChen et al. (2009) indicated that the polysaccharides obtained from the fruiting bodies of G. lucidum effectively scavenged free radicals of DPPH, superoxide anions, and hydroxyls in vitro, and activated the antioxidative enzymes like SOD, GPX, and CAT, and enhanced immunity as indicated by levels of IL-1β, IL6, and TNF-α in rats affiliated with uterine cancer.
Materials and methods
Chemicals
Deinoinzed water, ethanol (95%), ethanol 40%, NaOH (2%), KOH (10%), H2O2 (5.6%), anhydrous sodium carbonate, acetylacetone, ethanol, authentic glucosamine HCl, p-dimethylaminobezaldehyde, absolute ethanol, and 95% ethanol were purchased from Wako Pure Chemicals (Osaka, Japan). For determination of hexosamine, the following reagents and solutions were prepared: 1.5 N Na2CO3 solution and 4% acetylacetone solution in 1.5 N Na2CO3. These two solutions must be freshly prepared before use. Ehrlich reagent: 0.267 g of p-dimethylaminobenzaldehyde were dissolved in 10 mL of mixed solvent of 95% ethanol: HCl (=1:1) to make 2.76% Ehrilch reagent. Standard glucosamine HCl solution: 0.2407 g of glucosamine HCl were accurately weighed and dissolved in 10 mL water to obtain a final concentration of 20000 µg/mL (stock solution 1), 50 mL of which was diluted with double distilled water to make a total volume of 1000 mL (stock solution 2, c = 100 μg/mL). These stock solutions were stored in a 4°C refrigerator immediately when prepared.
Source of the fruiting bodies of G. lucidum
The fruiting bodies of G. lucidum were purchased from the local Ganoderma supplier in Hsin-Chu County of Taiwan. The classification of taxonomy was carried out by Dr. C. H. Su with the Fungal Strain Research and Protection Lab, Food Industrial Research Centre, in which the voucher has been filed out.
Proximate composition analysis
The proximate analysis was performed according to the methods described in the Association of Official Analytical Chemists (CitationAOAC, 1995).
Fractionation of polysaccharides and preparation of sacchachitin
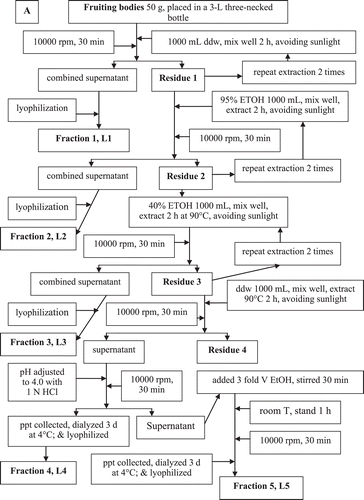
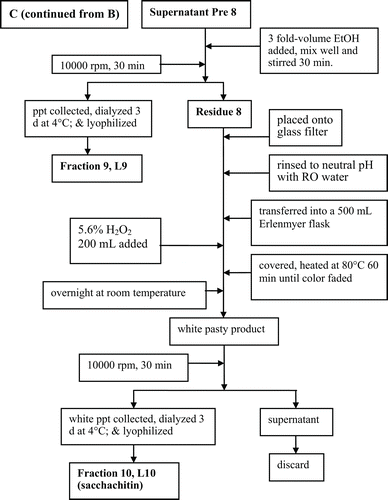
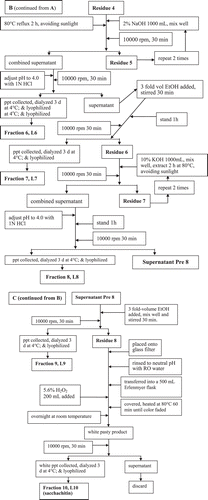
The process shown in was followed to isolate and purify the sacchachitin from the fruiting bodies of G. lucidum L (SGL). In parallel, CitationSu et al. (1997) was followed to prepare the contrast sample “mimic sacchachitin Su et al. (MSC)”.
Figure 1. Flowchart showing the process used for purification of sacchachitin from G. lucidum.
FTIR spectra of the SGL and MSC
The KBr (IR grade) tablet of sample was prepared. Briefly, the two sacchachitin samples (each 50 mg) were separately mixed with KBr powder (IR grade) at a ratio KBr/sacchachitin = 200:1, macerated thoroughly to obtain a completely homogeneous state and then the KBr/sacchachitin tablet was fabricated. The IR spectrum was taken with the Nicolet Type Protege 460 IR dual beam Spectrophotometer ESP against the KBr blank. The running conditions were: accumulation, 5; resolution, 4 cm–1 zero filling, ON; apodiation, cosine; gain, Auto (CitationDavid, 1986); scanning speed, Auto (2 mm/s).
Degree of deacetylation
The degree of deacetylation (DD) of these two isolated products was determined according to CitationLin et al. (1992). Briefly, desiccated chitin samples (each 0.5 g) were accurately weighed and thoroughly dissolved in 0.1 M HCl. The solution was then titrated with 0.1 M NaOH. The degree of deacetylation was calculated according to Eqs 1 and 2:
where C1 is the concentration of HCl (in M). C2 is the concentration of NaOH (in M). V1 is the volume of 0.1 M HCl (in mL). V2 is the volume of 0.1 M NaOH consumed (in mL); G, the weight of chitin sample. W is the water concentration of chitosan sample (in %), and 0.016 is the weight of NH2 equivalent to 1 mL 0.1 M HCl (aq). Substitution of the percentage of functional NH2 obtained from Eq. 1 into Eq. 2 to obtain
where 9.94 is the theoretical NH2 percentage equivalent.
Assay for carbohydrate
The content of carbohydrate was determined by the phenol-H2SO4 method.
Assay for protein
Protein (% w/w) in each extract was determined by the Bradford protein assay (CitationKer et al., 2010).
Assay for hexosamine
Hexosamine content (% w/w) in each extract was determined by the CitationJohnson (1971) hexosamine assay.
The monosaccharide composition in partially purified sacchachitin (PPS)
Hydrolysis, reduction, and derivatization of monosaccharides
The method of CitationKer et al. (2010) was used; in principle, the PPS was subjected to complete acid hydrolysis. The hydrolyzed mixture of monosaccharides was acetylated to form acetylated monosaccharides and analyzed with GC/MS. The quantity in mol % of each individual monosaccharide was calculated from the corresponding percent peak area by comparing with the blank. Practically, the method of CitationKer et al. (2005) was followed. PPS (2 mg) was accurately weighed and transferred into the Cole–Parmer reactor, to which 2 mL of 2 M trifluoroacetic acid was added. The mixture was heated at 120°C with the Cole–Parmer heater for 24 h while shaken vigorously every 30 min during heating until completely hydrolyzed. The hydrolyzed mixture was subjected to a stream of nitrogen until dried. The residue was dissolved twice with NH4OH solution. Each time 100 μL of 1 M NH4OH containing 1 mg/mL of deoxyribose was used as the internal standard. The reduction was conducted in the Cole–Parmer reactor with 0.2 g of NaBH4/10 mL of DMSO. The remaining procedures were conducted following CitationKer et al. (2005). The final deduced product was transferred into a 1 mL reaction vessel, lyophilized and analyzed with GC/MS as described (CitationKer et al., 2005).
GC/MS analysis
GC/MS was conducted in the Restek Rtx 225 column (l × i.d. = 30 m × 0.32 mm). The temperature was programmed, initially at 60°C for 1 min, raised at an elevation rate 8°C/min until 220°C, and held at this temperature for 10 min. The temperatures at the injection port and the detector were set equally at 230°C. Hydrogen was used as the carrier gas and operated at a flow rate of 2.0 mL/min. The reference values of retention time of the standard monosaccharides in CitationKer et al. (2010) are followed.
Preparation of sample solution
Glucosamine HCl powder (10 mg) was accurately weighed and transferred into a 250 mL Erlenmeyer flask. To the sample, 80 mL of hot double distilled water (80°C) was added and stirred vigorously while heating on the heater at 90–100°C until dissolved. The solutions were made to 100 mL with double distilled water to obtain the sample solution (c = 100 µg/mL).
Preparation of standard glucosamine solution
The stock solution 2 of authentic glucosamine (c = 100 μg/mL) was successively diluted to obtain 5, 10, 25, 50, 100, and 250 μg/mL, respectively.
Establishment of calibration curve
The glucosamine HCl standard and sample solutions (0.5 mL each) were respectively transferred into test tubes having screw type capping. To each tube, 0.5 mL of freshly prepared acetylacetone solution was added. After capping, the reaction mixture was incubated at 100°C in a water bath for 30 min when the reaction was completed; the reaction fluids were cooled to room temperature for 10 min. To each tube, 2.5 mL of absolute alcohol was added and mixed well, and then added 0.5 mL of freshly prepared Ehrlich reagent. The mixtures were agitated and incubated at 60°C for 25 min to facilitate the color formation (pink color). On standing for 10 min at ambient temperature, the optical density was read at 528 nm.
Mean molecular weight of different fractions
The method originally reported by CitationYagi et al. (1986) [later slightly modified by CitationKer et al. (2010)] was used for determination of the mean molecular weight (MW) of polysaccharides by gel permeation chromatography (GPC). Briefly, to 20 mg of each fraction (L1–L10) and the “mimic sacchachitin Su et al.”) 1 mL of 10% NaOH and sufficient double distilled water were added to make a final volume of 5 mL. The mixture was thoroughly agitated to facilitate the dissolution and centrifuged at 2500 rpm for 10 min to remove the insoluble precipitate. The supernatant was decanted, each 3 mL of which was eluted with 0.05 N NaOH solution containing 0.02% NaN3 at a flow rate 0.5 mL/min on a Sephadex G-100 column (id × ℓ = 2.5 × 100 cm). The fraction collector (ISCO Retriever 500, Isco., Lincoln, NE, USA) was used to collect the eluents, 6 mL per tube. The collection was stopped at the 100th tube. The calibration curve was established using authentic dextrans having molecular masses of 8.8, 40, 500, 2000, and 5000–40 000 kDa (Sigma, St. Louis, MO, USA), respectively. The molecular mass distribution and the average molecular mass was calculated against this linear correlation between the logarithm of each standard molecular mass and the ratio the eluted to the void-volume (CitationKer et al., 2010).
Calculation
The linear correlation of standard glucosamine solution has two linear ranges which is feasible for the concentration range 0–100 µg/mL (Eq. 3) and 0–250 μg/mL (Eq. 4), respectively.
(R2 = 0.9975, feasible for concentration range 0–100 μg/mL).
(R2 = 0.9996, feasible for concentration range 0–250 μg/mL).
Results and discussion
Proximate composition of the whole fruiting bodies
The proximate composition of the G. lucidum fruiting bodies was very similar to many other common mushrooms, particularly regarding the low fat (3.0%), high crude fiber (29.2%), and carbohydrate (41.6%) and moderate ash content (1.5%) (), implicating the presence of huge amount of valuable nutrients and mineral ions (CitationOuyang et al., 1998; CitationMa & Zhang, 2004).
Table 1. Proximate composition in the fruiting bodies of G. lucidum.
A review by CitationLindequist et al. (2005) indicated that many edible mushrooms exhibit a broad spectrum of bioactivities that are beneficially acting as a complementary alternative medicine (CAM). As often cited, the therapeutic efficacy of mushrooms includes antibacterial, antifungal, antimultiresistant bacterial, antiviral, immunomodulating, antitumor or antitumor adjuvant, cytostatic, immunosuppressive and antiallergic, antioxidative and anti-inflammatory, antiatherosclerogenic, hypoglycemic, hepatoprotective, and neurotrophic functions (CitationLindequist et al., 2005).
Characterization of each fraction
Following the purification process (), we obtained subsequently fractions L5–L10 (SGL) (). The highest and the lowest yields were 9.1 and 0.7% for fractions L6 and L5, respectively. L10 was only moderately yielded (3.4%) comparing to 14.7% of MSC (). Uniquely, L10 (SGL) contained a very high level of hexosamine (16.3%). The hexosamine content decreased in the order L9 (11.1%) >L8 (8.9%) >L7 (8.6%) >L5 (6.3%) >L6 (4.6%). Well known to all, the hexosamine content always is acting as a purity index of chitins (CitationCardenas & Miranda, 2004). As reference, in fungal biomass the chitin yield was ranging within 8.5–19.6% (% dry basis), exhibiting a degree of acetylation (AD) 91.0–98.7%; a level of glucosamine and ash content (% of chitin) 2.4–8.2 and 26.4–64.5%, respectively (CitationDi Mario et al., 2008).
Table 2. Comparison of composition in different fractions and the sacchachitins obtained by different processes from the fruiting bodies of G. lucidum.
Further, when simultaneously taking the protein/carbohydrate ratio (P/C ratio) into consideration, the fractions L5, L7, L8, and L9, that respectively have P/C values 0.29, 0.40, 0.32, and 0.19, were classified as the category of peptidoglycan (CitationKer et al., 2010), while fractions L6 and L10 (SGL) having P/C values 0.89 and 0.82 respectively were identified to be glycoproteins. Based on this criticism, the MSC having a hexaosamine content 6.6% (w/w) and a P/C ratio 0.35 was confirmed to be merely one kind of peptidoglycan. In fact it was not a real sacchachitin (). Obviously, all data obtained are pointing to the chitin characteristics of fraction SGL.
The MW of all fractions ranged within 1101–8632 kDa, in which fractions L6 and L7 had the lowest MW, SGL had the medium MW 8632 kDa, comparing to the highest 9756 kDa of MSC ().
In reality, the MW of fungal polysaccharides depends on the cultivation condition. In liquid fermentation with the rice spirit waste water plus glucose, CitationHsieh et al. (2005) obtained polysaccharides form G. lucidum that had MW only ranging within 10–200 kDa, being far smaller than what we have found (). As can be expected, in solid cultivation the counter gravity force required would be far greater than those needed in the liquid cultivation. Moreover, although the molecular distribution pattern in both the intracellular and extracellular portions was very similar, the latter always exhibit higher MW (CitationHsieh et al., 2005), an implication in the main role of polysaccahcride acting as the basic supporting substratum for fungal fruiting bodies.
IR spectra of the sacchachitins and their relevant sacchachitosans
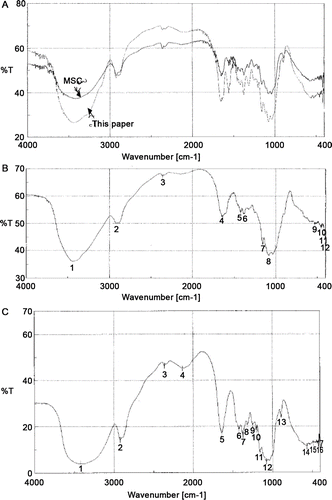
The characteristic absorption in reads (in cm–1): νO–H at 3455.7–3450.5, νN–H 3455.7–3450.5, νC–H 2932.5–2932.4, 2300–2400 unassigned, νC=O 1678.5–1678.3, δN–H amide I 1551.4–1550.4, δC–H 1413.5–1424.5, νC–C 1154.6–1152.7, νC–N 1125.9–1121.7, and νC–O 1029.5–1028.7, mostly similar to the functionalities of FTIR spectra cited for chitosan (CitationCardenas & Miranda, 2004). The strong absorption band at amide νC=O 1678.5–1678.3 cm–1 evidences the highly acetylated nature of L10 sacchachitin (; ). Similar IR spectra were found for the completely deacetylated products ( and ; ) except that the absorption at amide νC=O 1678.5–1678.3 cm–1 and amide I δN–H 1551.4–1550.4 cm–1 have completely disappeared ( and ; ).
Table 3. Assignment of IR absorption for different products and their deacetylation degree.
Figure 2. The IR spectra of sacchachitins and sacchachitosans. (A) Our product sacchachitn and MSC (the mimic CitationSu et al., 1997). (B) Our product sacchachitosan and (C) sacchachitosan from MSC. The characteristic absorption reads in (A) (at cm–1): νO–H 3455.7–3450.5, νN–H 3500–3300, νC–H 2932.5–2932.4, 2300–2400 unassigned, νC=O 1736.3–1734.6, amide I δN–H 1551.4–1550.4, δC–H 1413.5–1324.5, νC–C 1154.6–1152.7, νC–N 1125.9–1121.7, νC–O 1029.5–1028.7, δN–H 780.2–779.5, δC–H 713.3–712.3, and δC=O 690.5-690.2. Similar absorption was seen in (B) and (C), but lacking the bands νC=O 1736.3–1734.6 and the amide I bands δN–H 1551.4–1550.4.
Degree of deacetylation
The transformation of chitin to chitosan can be accomplished by either the deacetylation through the action of deacetylase (CitationCardenas & Miranda, 2004) or exhaustive alkaline hydrolysis (CitationChampagne, 2008).
The DD of the L10 sacchachitin and MSC was found to be 37.6 and 39.4%, respectively ().
In chitins, N-acetylation can facilitate the intramolecular hydrogen bonding to be formed between the intramolecular C3-OH hydroxyl to the C5-O ring oxygen across each β(1→4)-glycosidic linkage, restricting the chitin units to remain at the low-energy chair conformation and creating a rigid and linear polymer backbone (CitationChampagne, 2008). Consequently, the N-acetylation would reduce the solubility of chitin and affect the hydrogen bonding formation (Figure 5).
Pharmaceutically, poor solubility raises much limitation in clinical application of drugs. To increase the solubility in aqueous and organic solvents, chemical modification of chitin to generate new biofunctional materials is of primary interest. A very elaborate procedure includes alkaline N-deacetylation of the N-acetamido functional groups of chitin to produce chitosan. Modification of the N-acetamido groups by N-deacetylation results in functional amines that can undergo nucleophilic substitution reactions (CitationChampagne, 2008). We speculate that in a highly acetylated state, many –OH groups are orderly lined up in a state firmly bound by hydrogen bonding in the low-energy chair conformation exhibiting a rigid and linear polymer backbone (CitationChampagne, 2008). In highly deacetylated state, these –OH bonds instead are randomly exposed, and these released –OH groups no longer contact closely, resulting in reduced νO–H absorption at 3455.7–3450.5 cm–1 from %T 27 to %T 37 ( and ; ). In contrast, the MSC that originally had a %T 36.5 at νO–H 3455.7–3450.5 cm–1 retained at %T 3.5 after complete deacetylation. Such a phenomenon was also found for the δN–H bending absorption at 1640–1550 cm–1. The %T of sacchachitin (this paper) was increased, but that of MSC decreased (–; ). All of these data indicate the nonchitin character of MSC.
Table 4. %T of characteristic IR absorption bands before and after deacetylation.
Immunologically, higher DD exhibits stronger break strength and activates higher collagenase activity, and histologically, more prominent proliferation of fibroblasts (CitationMinagawa et al., 2007). CitationMinagawa et al. (2007) hypothesized the indispensable role of free amino groups in the chitin/chitosan molecules. It has been reported that chitin with a 70% DD is most effective for macrophage activation (CitationNishimura et al., 1986). Obviously, in view of practical application, these two products were in underdeacetylated forms (), and further mild enzymatic deacetylation treatment is required for transforming the low DD to high DD state.
Monosaccharide pattern of different fractions
A total of eleven monosaccharides including rhamnose, fucose, ribose, arabinose, xylose, allose, talose, mannose, galactose, glucose, and myoinositol was found in the fruiting bodies of G. lucidum (). Amazingly, ribose was almost undetected in fractions L5–L9. Allose was undetected in fractions L6–L8, and myoinositol was only absent in fraction L7. Alternatively, extremely high content of talose and mannose were present in these two sacchachitins (). The amount reached 29.41 and 33.82 mole% for talose; and 43.92 and 47.90 mole% for mannose in the L10 (SGL) and MSC, respectively (). The glucose content was high in fractions L5–L9, particularly in fraction L8, which reached 73.48 mole%. However, glucose was extremely low in these two sachachitin peparations, giving only 1.87 and 0.69 mole% respectively in L10 (SGL) and MSC (). Characteristically, these two sacchachitins contained very high levels of talose and mannose, indicating the sacchachitins are firmly linked to talomannans in G. lucidum. Conversely, these two sacchachitins consisted of relatively low concentrations of galactose and myoinositol (). Taken together, the fact pointing to the unusually low content of hexosamine in MSC (), and the unusual IR spectral change before and after deacetylated (), we suspect that the magic anti-inflammatory and wound healing effect of MSC originally reported by CitationSu et al. (1997) must have been contaminated by a huge amount of peptidoglycans coexisting in G. tsugae ().
Table 5. Pattern of monosaccharide composition in different fractions obtained from the fruiting bodies of G. lucidum.
DPPH free radical scavenging capability
At a concentration of 10 mg/mL, fractions L6, L7, and L9 revealed higher scavenging bioactivity against DPPH radicals than the other fractions (). However, at 5 mg/mL the fraction L6 only showed a higher activity at 62%, and within 0–5.0 mg/mL, all the other fractions did not show any bioactivity higher than 50%, implicating the polysaccharides in the fruiting bodies of G. lucidum, particularly, the L10 (SGL), are not feasible for scavenge of the organic radicals like DPPH. L10 (SGL) exhibited only 33% of DPPH-scavenging bioactivity even at the highest dosage of 10 mg/mL, conferring an IC33 of 10 mg/mL (the highest scavenging capability never exceeding 33%) ().
Figure 3. The scavenging capability of different polysaccharide fractions of G. lucidum on DPPH radicals (A), the superoxide anions (B), and the hydroxyl free radicals (C). Values are expressed in means ± SD (n = 3). Fractions L5–L10 are indicated in the flow chart in .
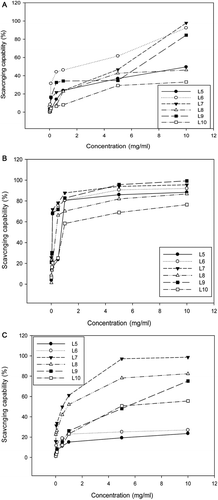
Superoxide anion scavenging capability
Nonetheless, in scavenging the water soluble superoxide anions, all the polysaccharide fractions showed activity exceeding 50% at 1.0 mg/mL. The activity of fractions L6, L7, and L9 almost reached 100% at 10 mg/mL. A plateau was soon reached for all fractions at dosages within 1.0–10 mg/mL. Nevertheless, among all factions, the fraction L10 (SGL) exhibited the lowest activity, reaching only 60 and 75% at 1.0 and 10 mg/mL and exhibiting an IC50 of 0.9 mg/mL ().
Hydroxyl free radical scavenging capability
In contrast, fractions L7 and L8 were very active against the hydroxyl free radicals, the activity exceeded 50% at 1.0 mg/mL, attaining 100 and 78% at 5.0 mg/mL, and 100 and 80% at 10.0 mg/mL, respectively (). Fraction L9 revealed a steadily increasing activity in a dose-dependent manner, and fraction L10 (SGL) was moderately active (), only exhibiting an IC50 of 4.8 mg/mL.
To summarize, all the data mentioned above indicate that these water soluble polysaccharides would be more effective in quenching the soluble oxidative species (like the superoxide anions and hydroxyl radicals) than the lipophilic radicals like DPPH.
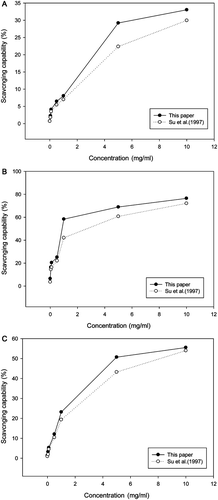
Comparison of the antioxidative capability between L10 (SGL) and MSC
The L10 sacchachitin usually exhibited higher activity than MSC in scavenging the DPPH radicals (), the superoxide anions (), and the hydroxyl radicals (). Similarly, both preparations did not show an activity >50% against the DPPH radicals (). However, to scavenge the superoxide anions in aqueous system, L10 (SGL) showed activity 59, 70, and 76%, comparing to 43, 60, and 72% for MSC at 1.0, 5.0, and 10.0 mg/mL, respectively (). In contrast, less effect was found for the hydroxyl radical system. At a dosage of 1 mg/mL, neither preparation showed any activity higher than 25%. However, L10 (SGL) exhibited 51 and 55% scavenging bioactivity and MSC showed only 42 and 54% at 5.0 and 10.0 mg/mL, respectively. As cited, the higher the DA, the less the water solubility (CitationChampagne, 2008; CitationUkai et al., 1983).
Figure 4. Scavenging capability of the two different sacchachitins isolated by different processes from the fruiting bodies of G. lucidum. For the DPPH radicals (A); for the superoxide anions (B); and for the hydroxyl free radicals (C). Values are expressed in means ± SD (n = 3).
For application, such a biofunctional polymer actually exhibits a limited processibility due to its low solubility. The highly ordered rigid structure of chitin (Supplementary Figure S1) resulting from the formation of intramolecular C3-OH hydroxyl to C5-O ring oxygen hydrogen bonds across each β(1→4)-glycosidic linkage restricts chitin units to the low-energy chair conformation, resulting in a rigid and linear polymer backbone (Supplementary Figure S2; CitationChampagne, 2008). This rigidity prevents the polymer’s complete dissolution in common organic solvents (DMSO, DMF, DCM, NMP; CitationUkai et al., 1983) and even in the aqueous solution (CitationChampagne, 2008).
As mentioned, chitin with a 70% DD is most effective for macrophage activation (CitationNishimura et al., 1986), similarly found in . L6 showed the best anti-DPPH radical capability; L7 had the best antihydroxyl free radical capability, and fractions L6, L7, and L9 were best among all in inhibiting the superoxide anions. To compare, the sacchachitin in this paper required an IC50 1.00 mg/mL for suppressing the syperoxide anions, being far better than 3.30 mg/mL of MSC ().
Table 6. Antioxidative IC50 values of different polysaccharide fractions obtained from the fruiting bodies of G. lucidum.
In conclusion, the product sacchachitin obtained by our process as fraction L10 from G. lucidum has a hexosamine content 16.3 (% w/w) that is firmly attached to talomannan. The real characteristics of sacchachitin has been evidenced by the FTIR spectral analysis and deacetylation assay. Structurally, it has a hexosamine content 16.3 (% w/w) firmly attached to a talomanna in G. lucidum. This L10 sacchachitin shows better bioactivity than MSC, a product mimic sacchachitin CitationSu et al. (1997) regarding the anti-DPPH, antihydroxyl free radical, and antisuperoxide anion capabilities.
Declaration of interest
The authors declare no conflict of interest.
Online supplementary materials are available at: http://informahealthcare.com/toc/phb
Supplementary Material
Download PDF (251.9 KB)References
- AOAC. (1995). Official Methods of Analysis, 16th ed. Washington, DC: Association of Official Analytical Chemists.
- Bensky D, Clavey S, Stoger E, Gamble A, eds. (2004). Chinese Herbal Medicine: Materia Medica, 3rd ed. Seattle, WA: Eastland Press.
- Cardenas G, Miranda SP. (2004). FTIR and TGA studies of chitosan composite films. Chil Chem Soc, 49, 291–295.
- Champagne LM. (2002–2008). The synthesis of water soluble N-acyl chitosan derivatives for characterization as antibacterial agents. Ph.D Dissertation submitted to the Graduate Faculty of the Louisiana State University and Agricultural & Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in The Department of Chemistry. (By Lakia, M.; Champagne; B. S. Xavier University of Louisiana, 2002).
- Chen XP, Chen Y, Li SB, Chen YG, Lan JY, Liu LP. (2009). Free radical scavenging of Ganoderma lucidum polysaccharides and its effect on antioxidant enzymes and immunity activities in cervical carcinoma rats. Carbohydr Polym, 77, 389–393.
- David A, ed. (1986). Mushrooms Demystified, 2nd ed. Berkeley, CA, USA: Ten Speed Press.
- Hajjaj H, Macé C, Roberts M, Niederberger P, Fay LB. (1986). Effect of 26-oxygenosterols from Ganoderma lucidum and their activity as cholesterol synthesis inhibitors. Appl Environ Microbiol, 71, 3653–3658.
- Hsieh CY, Hsu TH, Yang FC. (2005). Production of polysaccharides of Ganoderma lucidum (CCRC36021) by reusing thin stillage. Process Biochem, 40, 909–916.
- Johnson AR. (1971). Improved method of hexosamine determination. Anal Biochem, 44, 628–635.
- Ker YB, Chen KC, Chyau CC, Chen CC, Guo JH, Hsieh CL, Wang HE, Peng CC, Chang CH, Peng RY. (2005). Antioxidant capability of polysaccharides fractionated from submerge-cultured Agaricus blazei mycelia. J Agric Food Chem, 53, 7052–7058.
- Ker YB, Peng CH, Chyau CC, Peng RY. (2010). Soluble polysaccharide composition and myo-inositol content help differentiate the antioxidative and hypolipidemic capacity of peeled apples. J Agric Food Chem, 58, 4660–4665.
- Lee HJ. (1578). The Pharmacopoeia of Herbs and Medicines (B
 n-Ts
n-Ts u Gang-Moo, in Chinese).
u Gang-Moo, in Chinese). - Lee SS, Chen FD, Chang SC, Wei YH, Liu I, Chen CF, Wei RD, Chen KY, Han PW. (1984). In vivo anti-tumor effects of crude extracts from the mycelium of Ganoderma lucidum. J Chinese Oncol Soc, 5, 22–28.
- Li YQ, Wang SF. (2006). Anti-hepatitis B activities of ganoderic acid from Ganoderma lucidum. Biotechnol Lett, 28, 837–841.
- Lin MH (2001). Studies of antibacterial activity of derivatives from residue Ganoderma fruiting bodies. Master Thesis (M8802010). Graduate Institute of Medical Sciences. Taipei Medical University.
- Lin R, Jiang S, Zhang M. (1992). The determination of degree of deacetylation (in Chinese). Chem Bull, 3, 39–42.
- Lindequist U, Niedermeyer THJ, Jüli WD. (2005). The pharmacological potential of mushrooms. eCAM, 2, 285–299.
- Liu J, Kurashiki K, Shimizu K, Kondo R. (2006). Structure-activity relationship for inhibition of 5-α-reductase by triterpenoids isolated from Ganoderma lucidum. Bioorg Med Chem, 14, 8654–8660.
- Ma YH, Zhang FY. (2004). Determination of nutritive components of mycelia and fruitbody of Dictyophora indusiata. J Xan-Xi Agric Uni, 20, 389–391.
- Di Mario F, Rapanà P, Tomati U, Galli E. (2008). Chitin and chitosan from Basidiomycetes. Int J Biol Macromol, 43, 8–12.
- Minagawa T, Okamura Y, Shigemasa Y, Minami S, Okamoto Y. (2007). Effects of molecular weight and deacetylation degree of chitin/chitosan on wound healing. Carbohydr Polym, 67, 640–644.
- Moradali MF, Mostafavi H, Hejaroude GA, Tehrani AS, Abbasi M, Ghods S. (2006). Investigation of potential antibacterial properties of methanol extracts from fungus Ganoderma applanatum. Chemotherapy, 52, 241–244.
- Nishimura K, Ishihara C, Ukei S, Tokura S, Azuma I. (1986). Stimulation of cytokine production in mice using deacetylated chitin. Vaccine, 4, 151–156.
- Ouyang S, Luo Y, Liu M, Fan J, Guo X, Deng F. (1998). Analysis of amino acids, vitamins and inorganic elements in Dictyophora indusiata. Hunan Med Univ Acad Bull (Hunan Yi Ke Da Xue Xue Bao in Chinese), 23, 535–536, cont’d to 542.
- Paterson RR. (2006). Ganoderma - a therapeutic fungal biofactory. Phytochemistry, 67, 1985–2001.
- Sanodiya BS, Thakur GS, Baghel RK, Prasad GB, Bisen PS. (2009). Ganoderma lucidum: a potent pharmacological macrofungus. Curr Pharm Biotechnol, 10, 717–742.
- Su CH, Sun CS, Juan SW, Hu CH, Ke WT, Sheu MT. (1997). Fungal mycelia as the source of chitin and polysaccharides and their applications as skin substitutes. Biomaterials, 18, 1169–1174.
- Suga T, Shiio T, Maeda YY, Chihara G. (1984). Antitumor activity of lentinan in murine syngeneic and autochthonous hosts and its suppressive effect on 3-methylcholanthrene-induced carcinogenesis. Cancer Res, 44, 5132–5137.
- Ukai S, Kiho T, Hara C, Morita M, Goto A, Imaizumi N, Hasegawa Y. (1983). Polysaccharides in fungi. XIII. Antitumor activity of various polysaccharides isolated from Dictyophora indusiata, Ganoderma japonicum, Cordyceps cicadae, Auricularia auriculajudae, and Auricularia species. Chem Pharm Bull, 31, 741–744.
- Wang H, Ng TB. (2006). Ganodermin, an antifungal protein from fruiting bodies of the medicinal mushroom Ganoderma lucidum. Peptides, 27, 27–30.
- Yagi A, Nishimura H, Shida T, Nishioka I. (1986). Structure determination of polysaccharides in Aloe arborescens var. natalensis. Planta Med, 50, 213–218.
- Yang YZ, Sun CS, Juan SW, Chen PH, Chan MH, Lee LW, Su CH. (2001). Utilization of fruiting bodies from genus Ganoderma. Fungal Sci, 16, 23–32.
- Zhu XL, Chen AF, Lin ZB. (2007). Ganoderma lucidum polysaccharides enhance the function of immunological effector cells in immunosuppressed mice. J Ethnopharmacol, 111, 219–226.