Abstract
Context: A steroidal alkaloid, 4-acetoxy-plakinamine B (4APB), is a recently discovered marine natural product with inhibitory effect against acetylcholinesterase (AChE), but its mechanism of interaction with the enzyme remains to be elucidated.
Objective: The main objective was to study molecular binding mode of the compound, its interactions with catalytic subsites and molecular mechanism behind its significant inhibitory effect.
Materials and methods: All possible interactions of ligands in the binding sites were analyzed using FRED 2.1 and the OMEGA pre-generated multi-conformer library.
Results: Dipole–dipole interactions were observed between the secondary amino group of 4APB and Ser200 at a distance of 3.91 Å and also with Gly117 and Gly118. A further dipole–dipole interaction was between Arg289 and the heterocyclic nitrogen. Hydrogen bonding interactions were observed between Tyr130 and secondary amino and C-4 acetyl groups as well as between heterocyclic nitrogen and Phe288 at a distance of 3.04 Å. Hydrophobic interactions were evident between rings C/D of 4APB and with Phe288, Phe330 and Phe331. The computational studies revealed 4APB’s critical molecular interaction with amino acids of peripheral active (PAS) and anionic (AS) subsites.
Discussion: Our data provided molecular evidence for the mixed competitive inhibitory effect of 4APB. For lead optimization, structural insights revealed the N-methyl group of 4APB could be replaced by NH2 moiety to generate a more favorable hydrogen bonding with Glu199. A polar group insertion such as NH2 or OH at certain sites of the 4APB skeleton is also recommended.
Conclusion: These computational insights explained the mixed-competitive enzyme kinetic behavior of 4APB. This study outlines a strategy for designing novel derivatives of 4APB with potentially better AChE inhibitory activities through interaction at the PAS and AS sites.
Introduction
To date, over 36 million people in the world, mainly of the elderly population, are known to be affected by dementia and this number is projected to increase to 115 million by 2050 (D’Amelio & Rossini, Citation2012). By far, the most prevalent form of dementia is Alzheimer’s disease (AD) which has been shown to be associated with progressive neurodegenerative pathological changes in the basal forebrain cholinergic system as well as thalamocortical, entorhinal cortices, hippocampus and amygdala (Daulatzai, Citation2010). AD is characterized by a decline in memory and cognitive ability together with mood swing, personality and other behavioral abnormalities. Numerous reports have shown a close association between AD and loss of the cholinergic system with decreased levels of acetylcholine as well as pathological accumulation of β-amyloid (Aβ) (D’Amelio & Rossini, Citation2012; Daulatzai, Citation2010). One of the most popular approaches of slowing down the progress of deterioration in AD patients is to enhance the function of the deficient cholinergic system either by using cholinergic agonists or inhibition of the enzyme acetylcholinestrterase (AChE) that breaks down acetylcholine (Khan et al., Citation2010; Menichini et al., Citation2009; Rauter et al., Citation2007). Donepezil, galantamine, rivastigmine and tacrine are some of the common cholinesterase inhibitors that mitigate the impairment of cholinergic neurons in AD patients by slowing the degradation of acetylcholine through AChE inhibition. These drugs have limitations, however, due to either their toxic side effects (notably hepatotoxicity) or the loss of efficacy after prolonged use (Giacobini, Citation1998; Wu et al., Citation2010). The discovery of new and safer AChE inhibitors that acts through the novel mechanism is therefore urgently needed for combating AD.
The identification of several structurally diverse pharmacologically active secondary metabolites from marine life forms coupled with the well-recognized ratio of 2.43 for the marine versus the terrestrial bionetwork (Radjasa et al., Citation2011) make the ocean a huge hunting ground for exploring new lead compounds (Khan et al., Citation2010; Rauter et al., Citation2007). In this connection, the recent study by Langjae et al. (Citation2007) on unidentified sponge species of the genus Corticium was exemplary in that a novel steroidal alkaloid bearing a stigmastane skeleton, 4-acetoxy-plakinamine B (4APB, ) was discovered. Furthermore, this first known stigmastane-based alkaloid natural product has been shown to display potent AChE inhibitory activity (Langjae et al., Citation2007). To date, the mechanism of AChE inhibition by 4APB was, however, not established, and we herewith explored the molecular binding mode through computational studies. Molecular insights deduced from this study are likely to provide new directions for the discovery and development of more potent novel drug derivatives of 4APB that could potentially be used for treating AD.
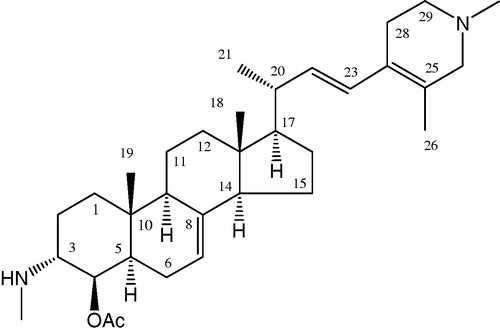
Figure 1. Chemical structure of 4-acetoxy-plakinamine B.
Materials and methods
FRED 2.1 (Khan et al., Citation2011; Martino et al., Citation2006) was used in this study to dock the OMEGA pre-generated multi-conformer library. FRED 2.1 strategy is to exhaustively dock/score all possible positions of each ligand in the binding site. The exhaustive search is based on rigid rotations and translations of each conformer within the binding site defined by a box. FRED filtered the poses ensemble by rejecting the ones that clash or does not have enough contacts with AChE enzyme. The final poses can then be scored or re-scored using one or more scoring functions. In this study, the smooth shape-based Gaussian scoring function (shapegauss) was selected to evaluate the shape complementarily between each ligand and the binding pocket. Default FRED protocol was used except for the size of the box defining the binding sites. In an attempt to optimize the docking-scoring performance, we performed exhaustive docking with shapegauss applying the “Optimization” mode. The “Optimization” mode involves a systematic solid body optimization of the top-ranked poses from the exhaustive docking. Three different boxes were explored for AChE (PDB ID: 1ACL) and three different simulations were carried out with an added value of 8 Å around the reference ligand.
Results and discussion
AChE (EC 3.1.1.7) is a complex protein of the α/β-hydrolase fold-type with an overall ellipsoid shape containing a deep and narrow groove of 20 Å deep referred to as “aromatic gorge” or “active site gorge” (). At the very bottom of the gorge, where the actual hydrolysis of acetylcholine occurs, there are four main catalytic subsites: the “Esteratic (ES)”, the “oxyanion hole (OX)”, the “Anionic (AS), also termed as choline binding subsite or hydrophobic subsite)” and the “Acyl Pocket (AP)” subsites. The ES contains the catalytic machinery of the enzyme, which is dependent on a catalytic triad of Ser200–His440–Glu327 (Harel et al., Citation1996) while the OX consists of Gly118, Gly119 and Ala201 (Khan et al., Citation2010; Szegletes et al., Citation1998; Zhang et al., Citation2002). The AS is largely comprised of aromatic residues and contains Trp84, Phe330 and Glu199, which are believed to bind with the quaternary ammonium ligands through π–cation interactions. The AP is composed of Phe288 and Phe290, and is believed to play a role in limiting the dimension of substrates that are able to enter the active site (Nisar et al., Citation2011). There is also a peripheral site (also called peripheral active site, PAS) that consists of Tyr70, Asp72, Tyr121, Tyr334 and Trp279 residues (Khan et al., Citation2010). The PAS is involved in the initial binding of Ach (prior to binding and hydrolysis by the ES) and ligand binding at this site in general could lead to changes in the allosteric conformation of the active catalytic center (Harel et al., Citation1996; Nisar et al., Citation2011; Szegletes et al., Citation1998; Zhang et al., Citation2002). Furthermore, PAS has been shown to interact with Aβ peptide and implicated in Aβ aggregation that appears to be the hallmark feature of AD. Hence, compounds interacting with both the ES and the PAS of AChE could not only alleviate cognitive deficits associated with cholinergic neuron depletion but also inhibit Aβ peptide aggregation. These so-called bivalent ligands are now being developed as a new avenue of AD therapeutic approach (Gaggeri et al., Citation2010, Citation2011; Khan et al., Citation2011; Langjae et al., Citation2007; Martino et al., Citation2006, Citation2008; Nisar et al., Citation2011). The PAS varies among AChEs and that from Torpedo californica, for example, consists of aromatic and carboxylic acid residues, Asp72, Tyr70, Tyr121, Trp279 and Phe290 (Szegletes et al., Citation1998).
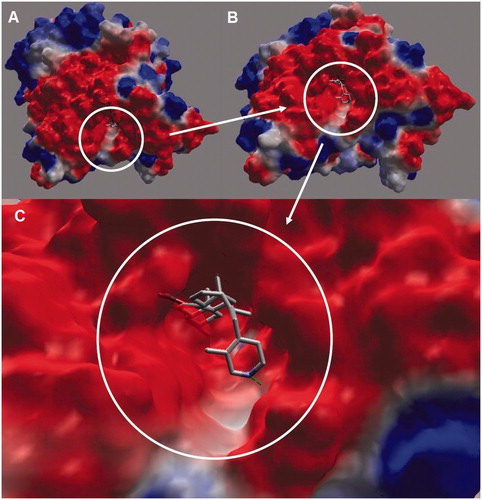
Figure 2. Electrostatic surface of AChE showing the aromatic gorge (active site) of enzyme: (A) lateral view of AChE. Encircled regions depict the aromatic gorge; (B) side view (at 90°) of AChE; (C) a closer side view of the aromatic gorge of AChE along with bound inhibitor (4APB) revealing molecular interactions between inhibitor and amino acid residues of surrounding active site of AChE. Surface colors red (aggregated negatively charged area), blue (aggregated positively charged area) and white (neutral or hydrophobic area) colors are shown.
In view of the above-discussed background, this study focuses on computational studies on 4APB that exhibited a potent inhibitory activity (IC50: 3.75 ± 1.69 μM) against AChE through mixed-competitive mode of inhibition (Langjae et al., Citation2007). Molecular docking simulations uncovered the molecular mechanism for the observed potent inhibitory effect and mode of inhibition. 4APB possesses a slightly rigid skeleton and an extended heterocyclic side chain that appears to undergo competitive molecular interactions with the ES, PAS, OX, AS and AP subsites inside the catalytic pocket of AChE ( and ). In the case of mixed competitive inhibitors, favorable molecular interactions are involved both with ES and PAS subsites of the AChE. AChE inhibitors mainly act on ES, especially through bonding interactions with Glu199 and Ser200. The secondary amino group of 4APB showed dipole–dipole interactions with Ser 200 at a distance of 3.91 Å. However, no hydrogen bonding was observed between ES and 4APB. Interestingly, Tyr130 revealed firm hydrogen bonding interactions with both the secondary amino group and the C-4 acetyl group. This interaction is partly responsible for the entrapment of the elongated skeleton of 4APB and eventually leads to blockade of ACh entry to the main catalytic site. Furthermore, both Gly117 and Gly118 (part of OX) showed dipole–dipole interactions with the secondary amino group of 4APB. In order to perform better lead optimization, structural insights revealed the fact that the N-methyl group of 4APB should be replaced by NH2 moiety to convert the dipole–dipole interactions into a relatively more favorable hydrogen bonding contact between Glu199 and the primary amino group (as a less bulky substituent than N-methyl group).
Figure 3. Molecular interactions of 4APB with selected amino acid residues inside the active site of AChE. Hydrogen atoms are omitted (except polar ones) for the purpose of clarity.
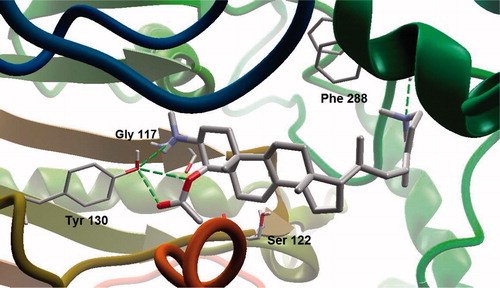
Figure 4. A closer view of the binding mode of 4APB inside the active site of AChE. Hydrogen atoms are omitted (except polar ones) for clarity. Hydrogen bonding is shown in dotted lines.
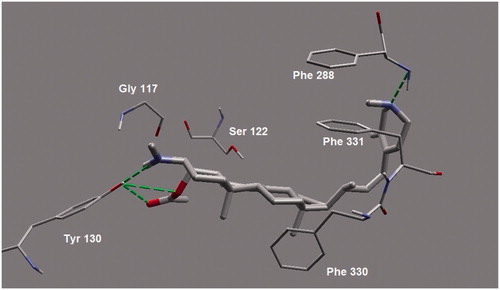
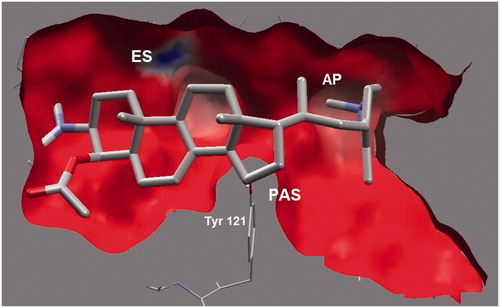
On the other side, the heterocyclic side chain of 4APB was found to be in a folded conformation. The folded conformation led to the strong and favorable hydrogen bonding (at a distance of 3.04 Å) between the heterocyclic nitrogen (N) of 4APB and Phe288 of AP, which is also another important amino acid residue of the AC subsite. Apart from hydrogen bonding, the dipole–dipole interaction was also detected between Arg289 and heterocyclic N of 4APB. In addition, the saturated and hydrophobic core of 4APB (including rings C and D) was found to be favorably interacting through the hydrophobic interaction with Phe288, Phe330 and Phe331. These simultaneous and favorable molecular contacts of 4APB with PAS clearly explain its potent inhibitory activity and mixed competitive kinetic behavior. However, no favorable contact was observed between 4APB and the most important amino acid residue of PAS subsite, Tyr121. In future lead optimization studies, a small polar group such as NH2 or OH group at C-14 of the main 4APB skeleton is recommended. Such modifications will provide a firm support via hydrogen bonding with the phenolic group of Tyr121 ().
Figure 5. A 3D-cut view showing electrostatic surface of the binding pocket. ES and AP subsites are shown as a deeper pocket while PAS is shown toward the viewer.
In conclusion, our detailed computational insight study indicated a strong potential for 4 APB to penetrate in and bind with the active subsites of AChE binding pocket. Simultaneous and favorable bonding interactions of 4APB with PAS and AS subsites provided a major clue behind its significant AChE inhibitory activity and mixed competitive kinetic profile. Based on the recommendations of the present structural insights, one could design new and better derivatives of 4APB as potential AD drugs. Such new derivatives should be subjected to both structure-based design techniques and 3D-QSAR before being considered as a candidate for clinical drug development.
Declaration of interest
Authors are thankful to Department of Pharmacy, University of Peshawar, for funding the reported research work. They also declare that there is no conflict of interest.
References
- D’Amelio M, Rossini PM. (2012). Excitability and connectivity of neuronal assemblies in Alzheimer’s disease: From animal models to human findings. Prog Neurobiol 99:42–60
- Daulatzai MA. (2010). Early stages of pathogenesis in memory impairment during normal senescence and Alzheimer’s disease. J Alzheimers Dis 20:355–67
- Gaggeri R, Rossi D, Hajikarimian N, et al. (2010). Preliminary study on TNFα-blocker activity of Amygdalus lycioides Spach extracts. Open Nat Prod J 3:20–5
- Gaggeri R, Rossi D, Collina S, et al. (2011). Quick development of an analytical enantioselective HPLC separation and preparative scale-up for the flavonoid naringenin. J Chromatogr A 1218:5414–22
- Giacobini E. (1998). Cholinesterase inhibitors for Alzheimer’s disease therapy: From tacrine to future applications. Neurochem Int 32:413–9
- Harel M, Quinn DM, Nair HK, et al. (1996). The X-ray structures of transition state analog complex reveal that molecular origin of the catalytic power of the substrate specificity of acetylcholinesterase. J Am Chem Soc 118:2340–6
- Khan I, Nisar M, Khan N, et al. (2010). Structural insights to investigate conypododiol as a dual cholinesterase inhibitor from Asparagus adscendens. Fitoterapea 81:1020–5
- Khan I, Nisar M, Shah MR, et al. (2011). Anti-inflammatory activities of taxusabietane A isolated from Taxus wallichiana Zucc. Fitoterapea 82:1003–7
- Langjae R, Bussarawit S, Yuenyongsawad S, et al. (2007). Acetylcholinesterase-inhibiting steroidal alkaloid from the sponge Corticium sp. Steroids 72:682–5
- Martino E, Collina S, Rossi D, et al. (2008). Influence of the extraction mode on the yield of hyperoside, vitexin and vitexin-2″-O-rhamnoside from Crataegus monogyna Jacq. [Hawthorn]. Phytochem Anal 19:534–40
- Martino E, Ramaiola I, Urbano M, et al. (2006). Microwave-assisted extraction of coumarin and related compounds from Melilotus officinalis (L.) pallas as an alternative to Soxhlet and ultrasound assisted extraction. J Chromatogr A 1125:147–51
- Menichini F, Tundis R, Loizzo MR, et al. (2009). Acetylcholinesterase and butyrylcholinesterase inhibition of ethanolic extract and monoterpenes from Pimpinella anisoides V Brig. (Apiaceae). Fitoterapea 80:297–300
- Nisar M, Kaleem WA, Khan I, et al. (2011). Molecular simulations probing kushecarpin A as a new lipoxygenase inhibitor. Fitoterapea 82:1008–11
- Radjasa OK, Vaske YM, Navarro G, et al. (2011). Highlights of marine invertebrate-derived biosynthetic products: Their biomedical potential and possible production by microbial associants. Bioorg Med Chem 19:6658–74
- Rauter AP, Branco I, Lopes RG, et al. (2007). A new lupene triterpenetriol and anticholinesterase activity of Salvia sclareoides. Fitoterapea 78:474–81
- Szegletes T, Mallender WD, Rosenberry TL. (1998). Non-equilibrium analysis alters the mechanistic interpretation of inhibition of acetylcholinesterase by peripheral site ligands. Biochemistry 37:4206–16
- Wu TY, Ch C-P, Jinn T-R. (2010). Alzheimer’s disease: Aging, insomnia and epigenetics. Taiwanese J Obstet Gynaecol 49:468–72
- Zhang Y, Kua J, McCammon JA. (2002). Role of the catalytic triad and oxyanion hole in acetylcholinesterase catalysis: An ab initio QM/MM study. J Am Chem Soc 124:10572–7