
Abstract
Context: Ischemia/hypoxia and reperfusion impair mitochondria and produce a large amount of reactive oxygen species (ROS), which lead to mitochondrial and brain damage. Furthermore, heme oxygenase-1 (HO-1) as a cytoprotective gene protects cells against ROS-induced cell death in ischemia–reperfusion injury. Induction of HO-1 is involved in cytoprotective effects of taxol.
Objective: We hypothesize that taxol protects cardiac myocytes possibly by preserving myocardial mitochondrial function and inducing HO-1 expression through the JNK pathway.
Materials and methods: In this project, the perfused Langendorff hearts isolated from rats were randomly divided into five groups: control, ischemic, ischemic + taxol (0.1 μM), ischemic + taxol (0.3 μM), and ischemic + taxol (1 μM). Briefly, following a 15 min equilibration period, the control group was subject to normoxic perfusion for 120 min; the ischemia group, normoxic reperfusion for 120 min after 30 min ischemia; the taxol groups, normoxic reperfusion for 120 min after 30-min ischemia with taxol (0.1, 0.3, or 1 μM). The microtubule disruption score, ROS levels, and the activity of mitochondrial electron transport chain complexes I and III were examined by using immunohistochemical methods and free radical detection kits. Western blot assay was employed to study the underlying mechanisms.
Results: After Taxol treatment (0.1 µM), the ischemic microtubule disruption score was reduced to 9.8 ± 1.9%. The study revealed that 0.1, 0.3, and 1 μM taxol reduced the level of ROS by 33, 46 and 51%, respectively (p < 0.05). In additional, 0.3 and 1 μM taxol dramatically increased the activity of mitochondrial electron transport chain complex I (99.11 ± 2.59, 103.49 ± 3.89) and mitochondrial electron transport chain complex III (877.82 ± 12.08; 907.42 ± 16.21; 914.73 ± 19.39, *p < 0.05). Additionally, phosphorylation levels of JNK1 were significantly increased in the taxol group. Furthermore, the expression level of HO-1 increased with taxol treatments, which could be inhibited by the specific inhibitor of JNK, SP600125.
Discussion and conclusion: Taxol stabilized microtubules and effectively reduced ROS levels during ischemia. It also preserved the activity of mitochondrial complexes I and III. Interestingly, taxol induced the expression of HO-1 via the JNK pathway in cardiac myocytes.
Introduction
Under normal physiological conditions, 98% of mitochondrial oxygen consumption is used to generate adenosine triphosphate (ATP), and the remaining 2% to produce oxygen free radicals (ROS). The respiratory chain complexes I and III are the main sites for ROS generation (Orr et al., Citation2003; Sedensky & Morgan, Citation2006a,Citationb; Yoshioka & Lee, 2013). Ischemia, hypoxia, and reperfusion impair the mitochondrial respiratory chain and produce a large amount of ROS. The increased amount of ROS causes oxidative damage in the mitochondrial membrane to induce irreversible cell damage (Boengler et al., Citation2007; Petrosillo et al., Citation2005). Oxidative stress has a pivotal role in ventricular arrhythmia (Kevin et al., Citation2005; Petrosillo et al., Citation2003; Shah et al., Citation2005; Ter & Boyden, Citation2007; Yang et al., Citation1995). Additionally, mitochondrial damage plays an important role in many vascular diseases including hypertension, stroke, heart failure, and cardiac ischemia/reperfusion injury (Halestrap, Citation2006; Rosca & Hoppel, 2010).
Microtubules are important components of the cytoskeleton, the disruption to microtubules can be induced by oxidative stress. Previous studies found that paclitaxel (Taxol®), a well-known killer for cancer cells (Orr et al., Citation2003), is a type of “microtubule stabilizer” that attenuated ischemia–reperfusion injury in cardiac myocytes (Cao et al., Citation2011). Additionally, taxol protects cultured adult rat cardiomyocytes against the oxidative stress induced by H2O2 (Lesnefsky & Hoppel, Citation2003; Turrens, Citation2003).
Heme oxygenase-1 (HO-1) degrades heme into iron, biliverdin, and carbon monoxide (CO) (Akamatsu et al., Citation2004). This enzyme is cytoprotective and can be used therapeutically to protect against the ischemia–reperfusion injury (I–R injury) (Zhang et al., Citation2002). Several studies have confirmed that increase expression of HO-1 correlates with improved survival and organ function in the brain, kidney, liver, heart, and lungs (Amersi et al., Citation1999; Choi et al., Citation2004; Fujita et al., Citation2001). Recently, it was reported that HO-1 regulation is mediated with the mitogen-activated protein kinases (MAPKs) including ERK, JNK, and p38 MAPK pathways (Elbirt et al., 1999; Otterbein et al., Citation2000; Zhang et al., Citation2014). RIPC protected the liver from I-R by induction of HO-1/p38-MAPK-dependent autophagy (Wang et al., Citation2014a,Citationb). Interestingly, JNK, an apoptotic MAPK, was shown to up-regulate HO-1 (Choe et al., Citation2014). In this study, we investigated whether taxol, as a microtubule stabilizer, protects heart against ischemia-reperfusion possibly from induction of HO-1 gene expression via JNK phosphorylation.
Materials and methods
Animals
Male adult Sprague–Dawley rats (n = 75; 230–260 g) were obtained from the Animal Laboratory of Nanjing Medical University (Nanjing, China). All animals were maintained in accordance with the Guide for Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication number 85-23, revised 1996) as well as the Policy of the Animal Care and Use Committee of Nanjing Medical University.
Model of myocardial ischemia–reperfusion
Rats were killed by stunning and cervical dislocation. Each heart was removed rapidly and placed in ice-cold Krebs–Henseleit (K–H) buffer. The heart was mounted onto a Langendorff apparatus and perfused with K–H buffer gassed with 95% O2/5% CO2, equilibrated at pH 7.35–7.45 at a constant pressure of 75 mmHg and maintained at 37 °C for 20 min (de Wyse et al., Citation2001; Diego et al., Citation2008). These conditions represent normoxic superfusion.
Ischemia was induced by ligation of the coronary artery. The left main coronary artery was ligated for 30 min. Coronary flow rate was monitored before and during ligation, with successful ligation indicated by a 25% reduction in coronary flow (Cao et al., Citation2011). Rats were divided into five groups (n = 15 per group) at random: control; ischemic; ischemic + taxol (0.1 μM); ischemic + taxol (0.3 μM); and ischemic + taxol (1 μM). Following a 15 min equilibration period, the control group was subjected to normoxic perfusion for 120 min; the ischemia group, normoxic reperfusion for 120 min after 30 min ischemia; the Taxol group, normoxic reperfusion for 120 min after 30 min ischemia with taxol (0.1, 0.3, or 1 μM). Taxol was dissolved in dimethyl sulfoxide (DMSO), added to the perfusate, and diluted into different concentrations (0.1, 0.3, and 1 μM).
After perfusion, left ventricular muscle was removed to make frozen sections. Microtubules were observed by immunohistochemical methods.
Western blot assay
Animals were anesthetized with sodium pentobarbital (100 mg/kg IP). Myocytes were stored in a Tyrode solution containing 1 mM CaCl2 and maintained at room temperature. Rats were divided into three groups at random: ischemic, ischemic + taxol (1 μM), and ischemic + colchicine (1 μM, Sigma-Aldrich, St. Louis, MO).
For the pharmacological inhibition of signaling pathways experiment, in three groups, SP600125 (1 µM, a JNK inhibitor, Sigma-Aldrich, St Louis, MO), SB203580 (10 µM, a p38 MAPK inhibitor, Sigma-Aldrich, St Louis, MO), and U0125 (10 µM, a MEK1 inhibitor, Cell Signaling, Beverly, MA) were added into perfusion solutions.
After incubation, tissues were washed in PBS and lysed in RIPA lysis buffer supplemented with protease inhibitor cocktail (Roche, Mannheim, Germany). Total protein was quantified using a BCA Protein Assay Kit (Beyotime, Jiangsu, China), and an equal amount of whole cell lysates were resolved by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Eschborn, Germany). The blots were blocked in BSA (5% w/v in PBS + 0.1% Tween 20) for 1 h at room temperature. The primary antibodies were then used according to the instructions of the manufacturer. Antibodies against HO-1, Tubulin, and GAPDH were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against p-p38, p38, p-JNK, JNK, p-ERK, and ERK were purchased from Abcam (Cambridge, MA). The appropriate secondary antibodies were used at 1:1000–1:2000 (v/v) dilutions in PBS + 0.1% Tween 20 for 1 h at room temperature, and the signals were detected using ECL kit (Thermo Scientific Pierce, Rockford, IL).
Measurement of microtubule disruption
Microtubule disruption measurement was described in previous studies (Sato et al., Citation1993). Briefly, myocardial tissues were perfused with a periodate-lysine-paraformaldehyde fixative at the end of the perfusion period. Subsequently, left ventricles were rinsed successively in PBS solutions. Frozen sections were generated and processed for double staining of microtubules and actin filaments. Some sections were incubated with a monoclonal β-tubulin antibody (1:300 dilution; Abcam, Cambridge, UK) and incubated in fluorescein isothiocyanate-labeled sheep anti-mouse immunoglobulin (1:300 dilution; Abcam, Cambridge, MA). Other sections were incubated with rhodamine-phalloidin (1:70 dilution; Cytoskeleton, Inc., Denver, CO) for 20 min at room temperature and rinsed in PBS. Finally, these sections were examined under a fluorescence microscope (BX51; Olympus, Tokyo, Japan).
Measurement of ROS and activities of oxidative enzymes
ROS and activities of oxidative enzymes were tested using their respective kits. Frozen sections were generated using a cryostat (CM 3050; Leica, Wetzlar, Germany) and stained with 2′,7′-dichlorofluorescein diacetate (DCFH-DA) from a Tissue Reactive Oxygen Species Assay kit (Genmed Scientifics Inc., Arlington, MA). DCFH-DA is cleaved intracellularly by non-specific esterases to form DCFH, which is oxidized further by ROS to form the highly fluorescent 2′,7′-dichlorofluorescein (DCF). Fresh-frozen cross-sections were stained, rinsed, mounted, and observed using fluorescence microscopy with excitation at 490 nm and emission at 520 nm (BX51; Olympus, Center Valley, PA).
To study ROS qualitatively, left ventricles from each group were homogenized in PBS with a PT 10/35 Polytron system (Brinkman, Westbury, NY) after perfusion. A Bicinchoninic Acid Protein Assay kit (Merck, Berlin, Germany) was used to determine protein content. Homogenates were stained with DCFH-DA from the Tissue Reactive Oxygen Species Assay kit (Merck, Berlin, Germany). DCFH-DA is oxidized by ROS to form the highly fluorescent DCF. Samples were placed in 96-well plates, and the rate of fluorescence was measured after a 20 min incubation period at 37 °C with an excitation frequency of 488 nm and an emission frequency of 520 nm.
Some homogenized solutions were centrifuged at 5000 × g for 10 min at 4 °C, and the supernatant was used for various assays. The activity of xanthine oxidase, NADPH oxidase (NOX) activity, specific oxidase activity, and the superoxide dismutase (SOD) assay were tested according to protocols of the manufacturer.
Mitochondria were isolated as described previously (Lee et al., Citation2005; Li et al., Citation2009; Skobel & Kammermeier, Citation1997). The enzyme activity assay of NADH CoQ reductase (mitochondrial complex I) and ubiquinol ferricytochrome C oxidoreductase (mitochondrial complex III) was carried out according to the instruction manual of the Tissue Mitochondrial Complex I and Complex III Assay kit (Genmed Scientifics Inc., Shanghai, China).
Statistical analyses
Results are expressed as mean ± SE. One-way ANOVA was conducted to evaluate the one-way layout data. All the histogram was evaluated by performing GraphPad Prism, version 4.0 (GraphPad Software, San Diego, CA). Statistical analyses were performed using Stata. 11.0 (SAS Inc., Cary, NC). p < 0.05 indicated statistical significance.
Results
Taxol preserves microtubule structure and induces the expression of tubulin during ischemia
The microtubule disruption score was increased to 28.8 ± 6.7% in the ischemia group, as compared with 3.2 ± 0.9% the control group (p < 0.05). After Taxol treatment (0.1 µM), the disruption score was reduced to 9.8 ± 1.9%. However, the actin filaments were intact even in cells with microtubule disruption () (n = 10 per group). Additionally, we observed that the expression level of tubulin increased with taxol treatments but decreased with colchicine treatments ().
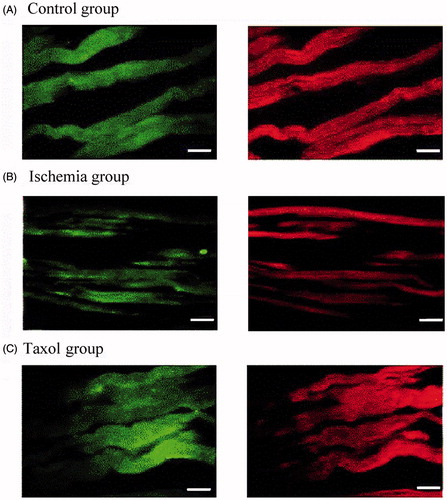
Figure 1. Microtubule morphology. (A) Microtubule structure is integral and no fracture is observed in the control group. (B) In the ischemia group, microtubule continuity is interrupted, and microtubule fracture can be seen. (C) In the 0.1 μM taxol group, microtubule structure is integral but disrupted (n = 10 per group). Magnification ×400; bar, 40 μm.
Taxol reduces ROS levels during ischemia
Consistently, ROS levels in cardiac myocytes were increased during ischemia. Taxol significantly reduced this increase (). 0.1, 0.3, and 1 μM taxol reduced the level of ROS by 33, 46, and 51%, respectively (n = 15 per group) (p < 0.05) ().
Figure 2. Taxol reduces the levels of reactive oxygen species (ROS) during ischemia. (A) Representative qualitative assessment of ROS (×400 magnification). Bar, 40 μm. (B) ROS level. Values are the mean ± SE (n = 15 per group). *p < 0.05 versus the control group; Δp < 0.05 versus the control group and the ischemia group; ∇p < 0.05 versus the control group, the ischemia group, and taxol (0.1 and 0.3 μM) groups.
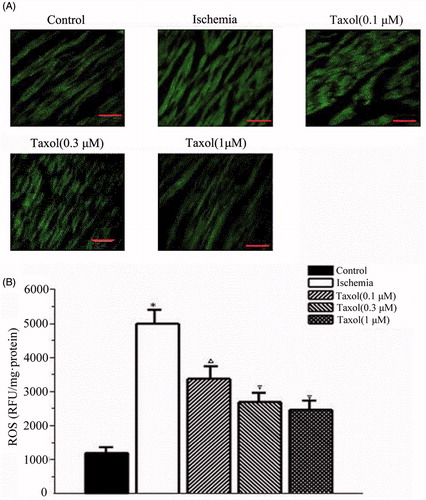
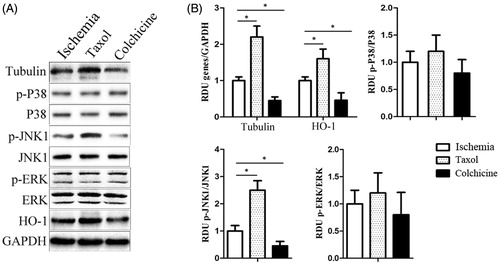
Figure 3. The expression of tubulin and HO-1 genes and the activity of all three subfamilies of MAPKs (ERK, JNK, and p38) in cardiomyocytes cells with taxol or colchicine treatment. (A) Representative western blot of tubulin and HO-1 gene expression and the activity of p-p38, p-JNK, and p-ERK in the ischemia group, the taxol group, and the colchicine group. (B) The histogram is the mean of densitometric analysis showing relative density units (RDU) of the western blot signals for genes normalized to control. All data represent mean ± SE (n = 10 per group). *p < 0.05 by Student’s t-test.
Taxol increases the activity of mitochondrial electron transport chain complexes I and III during ischemia
The enzyme activities of mitochondrial electron transport chain complexes I and III in cardiac myocytes were reduced during ischemia when compared with those of the control group (p < 0.05). In contrast, taxol increased the activity of mitochondrial electron transport chain complex I at 0.3 and 1 μM and complex III at 0.1, 0.3, and 1 μM (n = 15 per group) ().
Table 1. Effects of taxol on the activities of oxidative enzymes (n = 15 per group).
Taxol induced the expression of HO-1 genes and activated JNK pathway in cardiac myocytes against I-R injury
HO-1 was markedly induced by taxol treatments during ischemia (). Additionally, among three members of MAPKs (ERK, JNK, and p38), only JNK1 phosphorylation level was significantly induced in the taxol group and reduced in the colchicine group (n = 10 per group) (). These results suggest that taxol protects cardiomyocyte cells from I–R injury through increased expression of HO-1 possibly via the JNK1 signaling induction.
JNK activated by taxol induced the expression of HO-1
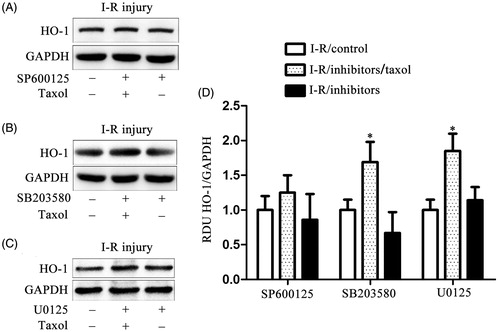
Three MAPK inhibitors, U0125 (a MEK1 inhibitor), SB203580 (a p38 MAPK inhibitor), and SP600125 (a JNK inhibitor) were used to investigate if JNK activated by taxol regulated HO-1 expression (n = 10 per group). In , we found both U0125 and SB2035580 did not alter taxol-induced HO-1 expression. In contrast, SP600125 significantly inhibited taxol-induced HO-1 expression. Our results confirmed that taxol induced HO-1 expression through JNK activation.
Figure 4. Taxol increases the expression of HO-1 gene via activating JNK in cardiomyocytes cells. (A) Representative western blot of HO-1 gene expression in cardiomyocytes cells incubated with U0125 (10 μM). (B) Representative western blot of HO-1 gene expression in cardiomyocytes cells incubated with SB203580 (10 μM). (C) Representative western blot of HO-1 gene expression in cardiomyocytes cells incubated with SP600125 (1 μM). (D) The quantification of the HO-1 gene expression for the western blot assay. All data represent mean ± SE (n = 10 per group). *p < 0.05 by Student’s t-test.
Discussion and conclusion
Consistent to previous reports, we found that taxol as a microtubule stabilizer could effectively reduce ROS levels during I–R injury. ROS reduction could result from either inhibiting ROS production or inducing anti-oxidants. Since mitochondrial complexes I and III are major sources to generate cellular ROS (Turrens, Citation2003), in this study, we investigated and demonstrated that taxol markedly preserved the activity of these two enzymes during ischemia. Additionally, we also investigated whether taxol induced HO-1 during ischemia since HO-1 could significantly increase the resistance of the ischemic brain to oxidative stress (Yoo et al., Citation2014). Overexpression of HO-1 was also shown to protect from I-R injury (Amersi et al., Citation1999; Choi et al., Citation2004; Zhang et al., Citation2002). As expected, taxol indeed induced the expression of HO-1. MAPK cascades were reported to regulate HO-1 expression. It was demonstrated that I-R-induced HO-1 expression was involved in upstream MAPK pathways and there was a intricate relationship among the MAPK subfamilies, I–R injury (Zhang et al., Citation2002), and HO-1 expression (Guan et al., Citation2014; Jiang et al., Citation2014; Lal et al., Citation2014; Sun et al., Citation2014; Wang et al., 2014; Zhang et al., Citation2014). In this study, we found that as in cancer cells, taxol in cardiac myocytes also activated JNK that was proved to induce cell death (Bhalla et al., Citation1993; Choe et al., Citation2014; Engels et al., Citation2008; Ling et al., Citation2004; Rowinsky & Donehower, Citation1995). Interestingly enough, in cardiac myocytes, activation of JNK mediated taxol-induced HO-1 expression. SP600125, a specific inhibitor of JNK, could significantly inhibit taxol-induced HO-1 expression. This may partially explain why taxol protects but does not kill cardiac myocytes cells during I–R injury.
Declaration of interest
The authors do not have any financial relationship with the granting authority. The authors report that they have no conflicts of interest. This work was supported by the Jiangsu Provincial Natural Science Fund (BK2012100).
References
- Akamatsu Y, Haga M, Tyagi S, et al. (2004). Heme oxygenase-1-derived carbon monoxide protects hearts from transplant associated ischemia reperfusion injury. FASEB J 18:771–2
- Amersi F, Buelow R, Kato H, et al. (1999). Upregulation of heme oxygenase-1 protects genetically fat Zucker rat livers from ischemia/reperfusion injury. J Clin Invest 104:1631–9
- Bhalla K, Ibrado AM, Tourkina E, et al. (1993). Taxol induces internucleosomal DNA fragmentation associated with programmed cell death in human myeloid leukemia cells. Leukemia 7:563–8
- Boengler K, Gres P, Dodoni G, et al. (2007). Mitochondrial respiration and membrane potential after low-flow ischemia are not affected by ischemic preconditioning. J Mol Cell Cardiol 43:610–15
- Cao HM, Wang Q, You HY, et al. (2011). Stabilizing microtubules decreases myocardial ischaemia-reperfusion injury. J Int Med Res 39:1713–9
- Choe YJ, Lee SY, Ko KW, et al. (2014). Nutlin-3 induces HO-1 expression by activating JNK in a transcription-independent manner of p53. Int J Oncol 44:761–8
- Choi BM, Kim YM, Jeong YR, et al. (2004). Induction of heme oxygenase-1 is involved in anti-proliferative effects of paclitaxel on rat vascular smooth muscle cells. Biochem Biophys Res Commun 321:132–7
- de Diego C, Pai RK, Chen F, et al. (2008). Electrophysiological consequences of acute regional ischemia/reperfusion in neonatal rat ventricular myocyte monolayers. Circulation 118:2330–37
- Elbirt KK, Whitmarsh AJ, Davis RJ, Bonkovsky HL. (1998). Mechanism of sodium arsenite-mediated induction of heme oxygenase-1 in hepatoma cells. Role of mitogen-activated protein kinases. J Biol Chem 273:8922–31
- Engels B, Dahm P, Jennewein S. (2008). Metabolic engineering of taxadiene biosynthesis in yeast as a first step towards Taxol (Paclitaxel) production. Metab Eng 10:201–206
- Fujita T, Toda K, Karimova A, et al. (2001). Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat Med 7:598–604
- Guan J, Li H, Lv T, Chen D, et al. (2014). Bone morphogenic protein-7 contributes to cerebral ischemic preconditioning induced-ischemic tolerance by activating p38 mitogen-activated protein kinase signaling pathway. Inflammation 37:1289–96
- Halestrap AP. (2006). Calcium, mitochondria and reperfusion injury: A pore way to die. Biochem Soc Trans 34:232–7
- Jiang M, Li J, Peng Q, et al. (2014). Neuroprotective effects of bilobalide on cerebral ischemia and reperfusion injury are associated with inhibition of pro-inflammatory mediator production and down-regulation of JNK1/2 and p38 MAPK activation. J Neuroinflammation 11:167
- Kevin LG, Novalija E, Stowe DF. (2005). Reactive oxygen species as mediators of cardiac injury and protection: The relevance to anesthesia practice. Anesth Analg 101:1275–87
- Lal H, Ahmad F, Parikh S, Force T. (2014). Troponin I-interacting protein kinase: A novel cardiac-specific kinase, emerging as a molecular target for the treatment of cardiac disease. Circ J 78:1514–9
- Lee CF, Liu CY, Hsieh RH, Wei YH. (2005). Oxidative stress-induced depolymerization of microtubules and alteration of mitochondrial mass in human cells. Ann NY Acad Sci 1042:246–54
- Lesnefsky EJ, Hoppel CL. (2003). Ischemia–reperfusion injury in the aged heart: Role of mitochondria. Arch Biochem Biophys 420:287–97
- Li J, Xiao J, Liu Y, Zhang G, et al. (2009). Mitochondrial benzodiazepine receptors mediate cardioprotection of estrogen against ischemic ventricular fibrillation. Pharmacol Res 60:61–7
- Ling X, Bernacki RJ, Brattain MG, Li F. (2004). Induction of survivin expression by taxol (paclitaxel) is an early event, which is independent of taxol-mediated G2/M arrest. J Biol Chem 279:15196–203
- Orr GA, Verdier-Pinard P, McDaid H, Horwitz SB. (2003). Mechanisms of taxol resistance related to microtubules. Oncogene 22:7280–95
- Otterbein LE, Bach FH, Alam J, et al. (2000). Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med 6:422–8
- Petrosillo G, Ruggiero FM, Di Venosa N, Paradies G. (2003). Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: Role of reactive oxygen species and cardiolipin. FASEB J 17:714–16
- Petrosillo G, Di Venosa N, Ruggiero FM, et al. (2005). Mitochondrial dysfunction associated with cardiac ischemia/reperfusion can be attenuated by oxygen tension control. Role of oxygen-free radicals and cardiolipin. Biochim Biophys Acta 1710:78–86
- Rosca MG, Tandler B, Hoppel CL. (2013). Mitochondria in cardiac hypertrophy and heart failure. J Mol Cell Cardiol 55:31–41
- Rowinsky EK, Donehower RC. (1995). Paclitaxel (taxol). N Engl J Med 332:1004–14
- Sato H, Hori M, Kitakaze M, et al. (1993). Reperfusion after brief ischemia disrupts the microtubule network in canine hearts. Circ Res 72:361–75
- Sedensky MM, Morgan PG. (2006a). Mitochondrial respiration and reactive oxygen species in C. elegans. Exp Gerontol 41:957–67
- Sedensky MM, Morgan PG. (2006b). Mitochondrial respiration and reactive oxygen species in mitochondrial aging mutants. Exp Gerontol 41:237–45
- Shah M, Akar FG, Tomaselli GF. (2005). Molecular basis of arrhythmias. Circulation 112:2517–29
- Skobel E, Kammermeier H. (1997). Relation between enzyme release and irreversible cell injury of the heart under the influence of cytoskeleton modulating agents. Biochim Biophys Acta 1362:128–34
- Sun L, Chen C, Jiang B, et al. (2014). Grb2-associated binder 1 is essential for cardioprotection against ischemia/reperfusion injury. Basic Res Cardiol 109:420
- Ter Keurs HE, Boyden PA. (2007). Calcium and arrhythmogenesis. Physiol Rev 87:457–506
- Turrens JF. (2003). Mitochondrial formation of reactive oxygen species. J Physiol 552:335–44
- Wang C, Chen K, Xia Y, et al. (2014a). N-Acetylcysteine attenuates ischemia–reperfusion-induced apoptosis and autophagy in mouse liver via regulation of the ROS/JNK/Bcl-2 pathway. PloS One 9:e108855
- Wang Y, Shen J, Xiong X, et al. (2014b). Remote ischemic preconditioning protects against liver ischemia-reperfusion injury via heme oxygenase-1-induced autophagy. PloS One 9:e98834
- Wyse DG, Friedman PL, Brodsky MA, et al. (2001). Life-threatening ventricular arrhythmias due to transient or correctable causes: High risk for death in follow-up. J Am Coll Cardiol 38:1718–24
- Yang CS, Tsai PJ, Chou ST, et al. (1995). The roles of reactive oxygen species and endogenous opioid peptides in ischemia-induced arrhythmia of isolated rat hearts. Free Radic Biol Med 18:593–98
- Yoo SJ, Nakra NK, Ronnett GV, Moon C. (2014). Protective effects of inducible HO-1 on oxygen toxicity in rat brain endothelial microvessel cells. Endocrinol Metab (Seoul) 29:356–62
- Yoshioka J, Lee RT. (2014). Thioredoxin-interacting protein and myocardial mitochondrial function in ischemia-reperfusion injury. Trends Cardiovasc Med 24:75–80
- Zhang GM, Wang Y, Li TD, et al. (2014). Post-conditioning with gradually increased reperfusion provides better cardioprotection in rats. World J Emerg Med 5:128–34
- Zhang X, Bedard EL, Potter R, et al. (2002). Mitogen-activated protein kinases regulate HO-1 gene transcription after ischemia-reperfusion lung injury. Am J Physiol Lung Cell Mol Physiol 283:L815–29