
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Context Dioscorea bulbifera L. (Dioscoreaceae) has been used in a traditional Thai longevity medicine preparation. Isolation of inhibitors from natural products is a potential source for continuous development of new HIV-1 integrase (IN) inhibitors.
Objective The objective of this study is to isolate the compounds and evaluate their anti-HIV-1 IN activity, as well as to predict the potential interactions of the compounds with an IN.
Materials and methods The ethyl acetate and water fractions (1–100 μg/mL) of Dioscorea bulbifera bulbils were isolated and tested for their anti-HIV-1 IN activity using the multiplate integration assay (MIA). The interactions of the active compounds with IN were investigated using a molecular docking method.
Results and discussions The ethyl acetate and water fractions of Dioscorea bulbifera bulbils afforded seven compounds. Among these, allantoin (1), 2,4,3′,5′-tetrahydroxybibenzyl (2), and 5,7,4′-trihydroxy-2-styrylchromone (5) were isolated for the first time from this plant. Myricetin (4) exhibited the most potent activity with an IC50 value of 3.15 μM, followed by 2,4,6,7-tetrahydroxy-9,10-dihydrophenanthrene (3, IC50 value= 14.20 μM), quercetin-3-O-β-d-glucopyranoside (6, IC50 value = 19.39 μM) and quercetin-3-O-β-d-galactopyranoside (7, IC50 value = 21.80 μM). Potential interactions of the active compounds (3, 4, 6, and 7) with the IN active site were additionally investigated. Compound 4 showed the best binding affinity to IN and formed strong interactions with various amino acid residues. These compounds interacted with Asp64, Thr66, His67, Glu92, Asp116, Gln148, Glu152, Asn155, and Lys159, which are involved in both the 3′-processing and strand transfer reactions of IN. In particular, galloyl, catechol, and sugar moieties were successful inhibitors for HIV-1 IN.
Introduction
Human immunodeficiency virus (HIV) is a retrovirus that causes acquired immune deficiency syndrome (AIDS). The long-term toxicity, adverse effects, and rapid emergence of drug resistance have become serious problems in treating HIV-1 infections (Asres et al. Citation2005). In recent years, the HIV-1 integrase (IN) has been a very attractive target for developing novel anti-HIV drugs because it is one of the essential enzymes used for replication of HIV and it has no known counterpart in humans. Currently, there are only three HIV-1 IN inhibitors (raltegravir, elvitegravir, and dolutegravir) approved by the Food and Drug Administration (Clercq Citation2009; Saag Citation2012). Therefore, continuous development of new HIV-1 inhibitors is needed. Isolation of potential inhibitors from natural products is a potential source for such purposes.
IN is a key enzyme involved in the replication cycle of retrovirus. It catalyses two reactions referred to as 3′-processing and strand transfer that are responsible for integration of the transcribed viral DNA into the host chromosome. The structure of a full length IN is composed of 288 amino acid residues that fold into three structural and functional domains. The N-terminal domain (residues 1–49) contains HHCC zinc binding which promotes enzyme multimerisation. The catalytic core domain (residues 50–212) contains the flexible loop which comprises residues 141–148. This domain also contains the catalytic triad (Asp64, Asp116, and Glu152) which are known as the DDE motif. This motif coordinates with two divalent metal ions (Mg2+ or Mn2+) that are crucial for the catalytic activity. The C-terminal domain (residues 213–288) has non-specific DNA binding activity (Asante-Appiah and Skalka Citation1997; Greenwald et al. Citation1999; Luca et al. Citation2003).
Dioscorea bulbifera L. (Dioscoreaceae) is a medicinal plant generally used in traditional Indian and Chinese medicine for the treatment of sore throat, gastric cancer, carcinoma of the rectum, and goiter (Ghosh et al. Citation2012). In Thailand, this plant is known as Wan Phra Chim and has been used as a traditional longevity preparation. Extracts of bulbils have exhibited various pharmacological properties, such as antibacterial (Teponno et al. Citation2006), antihyperglycemic, antidyslipidemic (Ahmed et al. Citation2009), analgesic, anti-inflammatory (Mbiantcha et al. Citation2011), and antitumour activities (Wang et al. Citation2012). Many phytochemical studies have demonstrated that clerodane diterpenoids, flavonoids, steroidal saponins, and sapogenins are present as the major compounds of D. bulbifera (Gao et al. Citation2002; Liu et al. Citation2010, Citation2011; Tapondjou et al. Citation2013).
For our continuing search for new HIV-1 IN inhibitors from natural sources, the ethyl acetate and water fractions of D. bulbifera, that were previously shown to have potent activity against HIV-1 IN, were selected for further study on anti-HIV-1 IN activity. This study isolates the active compounds and tests them for anti-HIV-1 IN activity, as well investigating the interactions of the active compounds with HIV-1 IN using a molecular-docking technique.
Materials and methods
Plant materials
The bulbils of D. bulbifera were collected from Uttaradit province, Thailand, in 2011. The plant was identified by a botanist of the Forest Herbarium, Department of National Parks, Wildlife and Plant Conservation, Thailand. The voucher specimen (SKP 062040201) was deposited in the Faculty of Pharmaceutical Sciences, Prince of Songkla University.
Enzyme and chemicals
The HIV-1 IN protein was kindly provided by Dr. Robert Craigie, National Institute of Health, Bethesda, MD. This enzyme was expressed in Escherichia coli and purified according to the method described by Goldgur et al. (Citation1999).
Oligonucleotide substrates
Oligonucleotides of long-terminal repeated bases from donor DNA (LTRD) and from target-substrate DNA (TS) were purchased from QIAGEN Operon, Alameda, CA, and stored at −25 °C before use. The sequence of the biotinylated LTRD and its unlabelled complement were 5′-biotin-ACCCTTTTAGTCAGTGTGGAAAATCTCTAGCAGT-3′ and 3′-GAAAATCAGTCACACCTTTTAGAGATCGTCA-5′, respectively. While those of the target substrate DNA (digoxigenin-labelled target DNA, TS-1) and its 3′-labelled complement were 5′-TGACCAAGGGCTAATTCACT-digoxigenin and digoxigenin-ACTG GTTCCCGATTAAGTGA-5′ (TS-2), respectively.
Extraction and isolation of compounds from D. bulbifera extract
Dried bulbil powders of D. bulbifera (1.7 kg) were macerated three consecutive times with ethanol at room temperature and concentrated under reduced pressure to obtain the ethanol extract (258.0 g). This extract was successively partitioned with hexane, chloroform, ethyl acetate and water to yield a chloroform (108.15 g), ethyl acetate (44.44 g) and water fraction (105.18 g), respectively. In this extract, there was no residue from the hexane fraction.
A bioassay-guided isolation of active compounds possessing anti-HIV-1 IN activity was conducted. In our previous study, we had reported that the anti-HIV-1 IN activity of isolated compounds from the chloroform fraction possessed the highest activity (IC50 value = 5.42 μg/mL). In this study, we selected the ethyl acetate (IC50 value = 6.49 μg/mL) and water fractions (IC50 value = 9.12 μg/mL) () which exhibited high inhibitory activity for further purification of the active compounds by chromatographic techniques.
Table 1. Anti-HIV-1 integrase activityTable Footnotea of the EtOH extract and fractions from Dioscorea bulbifera.
The ethyl acetate fraction (30.00 g) was separated by vacuum liquid chromatography (VLC) using silica gel as the stationary phase and eluted by a step-gradient elution starting from mixtures of chloroform and ethyl acetate (30–100% v/v) and followed by mixtures of ethyl acetate and methanol (10–50% v/v) to obtain six pooled major fractions (A–E), based on TLC analysis.
Fraction A (3.18 g) was further separated by VLC and eluted with chloroform/methanol (0-50% v/v) to give five pooled subfractions (A1–A5). Subfraction A2 was further purified by recrystallisation in methanol to provide compound 1 as a pale brown amorphous solid (28.9 mg, 0.0963% w/w). Subfraction A4 was further subjected to silica gel column chromatography and initially eluted with 20% ethyl acetate in chloroform, followed by an increasing polarity obtained with ethyl acetate to obtain five subfractions (A4/1–A4/5). Subfraction A4/2 was purified by Sephadex LH-20 (Sigma, St. Louis, MO) using methanol as eluent to give compound 2 as a brown powder (12.7 mg, 0.0423% w/w) and compound 3 as a brown powder (37.4 mg, 0.1247% w/w). Subfraction A4/4 was also purified by Sephadex LH-20 using methanol as eluent to give compound 4 as a yellow powder (9.5 mg, 0.0317% w/w).
Fraction B (3.20 g) was isolated by VLC and eluted with ethyl acetate/methanol (0–50% v/v) to afford five subfractions (B1–B5). Subfraction B2 was further chromatographed by column chromatography over silica gel and washed with a gradient of chloroform and methanol to obtain five subfractions (B2/1–B2/5). Subfraction B2/1 was purified by Sephadex LH-20 and eluted with methanol/chloroform (90:10) to give compound 5 as a yellow powder (4.9 mg, 0.0163% w/w).
For the water fraction (20.00 g), it was suspended in water and fractionated on a Diaion HP-20 column using H2O and methanol as eluents. The methanol subfraction was further isolated through an RP-18 column and eluted with H2O/methanol (50:50) to give six subfractions (W1–W6). Subfraction W4 was purified by Sephadex LH-20 using methanol as the eluent to afford compound 6 as a yellow powder (5.6 mg, 0.0280% w/w). Subfraction W5 was purified by preparative TLC using chloroform/methanol (70:30) to give compound 7 as a yellow powder (3.3 mg, 0.0165% w/w).
The chemical structures of the isolated compounds 1–7 were identified by EIMS, 1D and 2D NMR and were confirmed by comparison with the NMR spectral data in the literatures (Santos et al. Citation2003; Guvenalp & Demirezer Citation2005; Sripathi et al. Citation2011; Rashed et al. Citation2012).
Identifications of compounds 1–7
Allantoin (1): brown amorphous, UV (MeOH) λmax: 276 nm. 1H-NMR (DMSO-d6, 500 MHz): δ 5.24 (1H, dd, J = 8.0, 1.2 Hz, H-3), 5.75 (2H, s, H-4′), 6.87 (1H, d, 8.0 Hz, H-3′), 8.02 (1H, s, H-2′), 10.50 (1H, s, H-1′). 13C-NMR (DMSO-d6, 125 MHz): δ 62.5 (C-3), 156.8 (C-1), 157.4 (C-3), 173.6 (C-4).
2,4,3′,5′-Tetrahydroxybibenzyl (2): brown amorphous, UV (MeOH) λmax: 223, 281 nm. 1H-NMR (CD3OD, 500 MHz): δ 2.66 (2H, m, H-α), 2.68 (2H, m, H-β), 6.08 (1H, t, J = 2.2 Hz, H-4′), 6.12 (2H, d, J = 2.2 Hz, H-2′, H-6′), 6.48 (1H, dd, J = 8.0, 2.0 Hz, H-5), 6.60 (1H, d, J = 2.0 Hz, H-3), 6.65 (1H, d, J = 8.0 Hz, H-6). 13C-NMR (CD3OD, 125 MHz): δ 38.2 (C-α), 39.4 (C-β), 101.1 (C-4′), 108.0 (C-2′, C-6′), 116.2 (C-3), 116.5 (C-6), 120.7 (C-5), 134.9 (C-1), 144.2 (C-4), 145.6 (C-2), 145.9 (C-1′), 159.2 (C-1′, C-5′).
2,4,6,7-Tetrahydroxy-9,10-dihydrophenanthrene (3): brown amorphous, UV (MeOH) λmax: 222, 275, 303 nm. 1H-NMR (CD3OD, 500 MHz): δ 2.56 (2H, m, H-9), 2.58 (2H, m, H-10), 6.19 (1H, d, J = 2.4 Hz, H-1), 6.23 (1H, d, J = 2.4 Hz, H-3), 6.59 (1H, s, H-5), 7.89 (1H, s, H-8). 13C-NMR (CD3OD, 125 MHz): δ 30.3 (C-9), 32.2 (C-10), 102.7 (C-3), 107.7 (C-1), 115.3 (C-4a), 115.4 (C-5), 116.6 (C-8), 126.7 (C-4b), 130.6 (C-8a), 141.9 (C-10a), 143.6 (C-6, C-7), 156.3 (C-2), 156.9 (C-4).
Myricetin (4): yellow powder, UV (MeOH) λmax: 223, 254, 371 nm. 1H-NMR (DMSO-d6, 500 MHz): δ 6.17 (1H, d, J = 1.9 Hz, H-6), 6.35 (1H, d, J =5 1.9 Hz, H-8), 7.22 (2H, s, H-2′, H-6′), 9.16 (2H, s, 3′, 5′-OH), 12.47 (1H, s, 5-OH).
5,7,4′-Trihydroxy-2-styrylchromone (5): yellow powder, UV (MeOH) λmax: 370 nm. 1H-NMR (DMSO-d6, 500 MHz): δ 6.16 (1H, d, J = 1.9 Hz, H-6), 6.28 (1H, s, H-3), 6.42 (1H, d, 1.9 Hz, H-8), 6.83 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.92 (1H, d, J = 16.2 Hz, H-α), 7.56 (2H, d, J = 8.6 Hz, H-2′, H-6′), 7.58 (1H, d, J = 16.2 Hz, H-β), 12.91 (1H, s, 5-OH).
Quercetin-3-O-β-d-glucopyranoside (6): yellow powder, UV (MeOH) λmax: 216, 255, 362 nm. 1H-NMR (CD3OD, 500 MHz): δ 6.19 (1H, d, J = 2.0 Hz, H-6), 6.38 (1H, d, J = 2.0 Hz, H-8), 6.86 (1H, d, J = 8.5 Hz, H-5′), 7.58 (1H, dd, J = 8.5, 2.2 Hz, H-6), 7.70 (1H, d, J = 2.2 Hz, H-2′), 5.23 (1H, d, J = 7.8 Hz, H-1″), 3.23-3.71 (sugar moiety, 6H, m, H-2″ to H-6″). 13C-NMR (CD3OD, 125 MHz): δ 62.5 (C-6″), 71.2 (C-4″), 75.7 (C-2″), 78.1 (C-5″), 78.3 (C-3″), 94.7 (C-8), 99.8 (C-6), 104.3 (C-10), 105.7 (C-1″), 116.0 (C-2′), 117.5 (C-5′), 123.1 (C-6′), 123.2 (C-1′), 135.6 (C-3), 145.9 (C-3′), 149.8 (C-4′), 158.4 (C-2), 159.0 (C-9), 163.0 (C-5), 166.0 (C-7), 179.5 (C-4).
Quercetin-3-O-β-d-galactopyranoside (7): yellow powder, UV (MeOH) λmax: 218, 255, 365 nm. 1H-NMR (CD3OD, 500 MHz): δ 6.20 (1H, d, J = 2.0 Hz, H-6), 6.40 (1H, d, J = 2.0 Hz, H-8), 6.87 (1H, d, J = 8.5 Hz, H-5′), 7.59 (1H, dd, J = 8.5, 2.2 Hz, H-6′), 7.83 (1H, d, J = 2.2 Hz, H-2′), 5.16 (1H, d, J = 7.8 Hz, H-1″), 3.41–3.84 (sugar moiety, 6H, m, H-2″ to H-6″). 13C-NMR (CD3OD, 125 MHz): δ 61.9 (C-6″), 70.0 (C-4″), 73.1 (C-2″), 77.2 (C-5″), 75.1 (C-3″), 94.7 (C-8), 99.9 (C-6), 105.6 (C-10), 105.4 (C-1″), 116.2 (C-2′), 117.8 (C-5′), 122.9 (C-6′), 123.2 (C-1′), 135.8 (C-3), 145.8 (C-3′), 149.9 (C-4′), 158.3 (C-2), 158.8 (C-9), 162.6 (C-5), 166.0 (C-7), 178.7 (C-4).
Assay of HIV-1 IN inhibitory activity
The inhibitory activity of HIV-1 IN by MIA was determined according to the method described by Tewtrakul et al. (Citation2002). Briefly, a mixture (45 μL) composed of 12 μL of IN buffer [containing 150 mM 3-(N-morpholino) propane sulfonic acid, pH 7.2 (MOPS), 75 mM MnCl2, 5 mM dithiothritol (DTT), 25% glycerol, and 500 μg/mL bovine serum albumin], 1 μL of 5 pmol/mL digoxigenin-labelled target DNA and 32 μL of sterilised water was added into each well of a 96-well plate. Subsequently, 6 μL of sample solution in DMSO and 9 μL of a 1/5 dilution of the IN enzyme was added to each well and incubated at 37 °C for 80 min. After washing the wells three times with 270 μL PBS, 100 μL of 500 mU/mL alkaline phosphatase (AP)-labelled anti-digoxigenin antibody was added and incubated at 37 °C for 1 h. The plates were washed again three times with 270 μL washing buffer containing 0.05% Tween 20 in PBS and then another three times with 270 μL PBS. Then, AP buffer (150 μL) containing 100 mM Tris-HCl (pH 9.5), 100 mM NaCl, 5 mM MgCl2 and 10 mM p-nitrophenyl phosphate was added to each well and incubated at 37 °C for 1 h. Finally, the absorbance in each well was measured with a microplate reader at a wavelength of 405 nm. A control composed of a reaction mixture of 50% DMSO and IN enzyme, while a blank was buffer-E containing 20 mM MOPS (pH 7.2), 400 mM potassium glutamate, 1 mM ethylenediaminetetraacetate disodium salt, 0.1% Nonidet-P 40, 20% glycerol, 1 mM DTT and 4 M urea without the IN enzyme. Suramin, an HIV-1 IN inhibitor, was used as a positive control. The % inhibition against HIV-1 IN was calculated as follows:
where OD is the absorbance detected from each well.
Molecular docking method
Ligand setup
The three-dimensional structure of compounds was modelled with the HyperChem Professional 8.0 program (Hypercube Inc., Gainesville, FL). Each structure was geometrically optimised using the semi-empirical PM3 method. Subsequently, Gasteiger charges were assigned using the AutoDockTools program (Hypercube Inc., Gainesville, FL).
Integrase setup
The X-ray crystallographic structure of the HIV-1 IN catalytic core domain (residues 56–209), in complex with Mg2+ and 1-(5-chloroindol-3-yl)-3-hydroxy-3-(2H-tetrazol-5-yl)-propenone (5CITEP) was obtained from the Protein Data Bank (PDB code 1QS4). The IN structure was prepared for docking by selecting only monomer A which is the only monomer that co-crystallised with 5CITEP, an inhibitor. All water molecules and the 5CITEP were removed while the Mg2+ at the active site was maintained. The missing residues in the loop at positions 141–144 were incorporated and the polar hydrogen atoms were added to this chain (Vajragupta et al. Citation2005). The second Mg2+ was placed in the same relative position according to the two metal structures of the Prototype Foamy Virus IN (PDB code 3OYA), a structural homolog of HIV-1 IN (Krishnan and Engelman Citation2012).
Docking
All docking calculations were performed using the AutoDock 4.2 program (Hypercube Inc., Gainesville, FL). A grid box with a dimension of 60 × 60 × 60 Å3 and a grid spacing of 0.375 Å, which is large enough for a free rotation of a ligand, was built and placed at the location of the co-crystallised inhibitor (5CITEP) to cover the active site of HIV-1 IN. The Lamarckian genetic algorithm (LGA) method was used with the following parameters. The number of GA runs was 100, the population size was 150, the maximum number of energy evaluations was increased to 2 500 000 per run and the maximum number of generations in the genetic algorithm was increased to 100 000. All other docking parameters were set at their default values.
A cluster analysis was used to group similar conformations. For each compound, all 100 independent conformations were clustered into groups with a criterion of the root mean-square deviation (RMSD) of less than 2 Å. The best-docked conformation was selected from a cluster with the lowest binding energy and containing the greatest number of conformations (Healy et al. Citation2009). The amino acid residues within 6 Å of the ligand in the IN active site were chosen for the analysis of hydrogen bond interaction.
Statistical analysis
For statistical analysis, the results of the anti-HIV-1 IN activity were expressed as a mean ± SEM from four determinations. The IC50 values were calculated using the Microsoft Excel program.
Results and discussion
The ethyl acetate and water fractions of D. bulbifera bulbils were fractionated by chromatographic techniques. Five compounds (1–5) were isolated from the ethyl acetate fraction and two compounds (6 and 7) were obtained from the water fraction. Their structures were identified as allantoin (1), 2,4,3′,5′-tetrahydroxybibenzyl (2), 2,4,6,7-tetrahydroxy-9,10-dihydrophenanthrene (3), myricetin (4), 5,7,4′-trihydroxy-2-styrylchromone (5), quercetin-3-O-β-d-glucopyranoside (6), and quercetin-3-O-β-d-galactopyranoside (7). It is of interest that compounds 1, 2, and 5 were isolated for the first time from this plant. Compound 1 had been previously found in Dioscorea batatas Decne. (Dioscoreaceae), Dioscorea pseudojaponica Yamamoto (Fu et al. Citation2006), Dioscorea opposita Thunb. and Dioscorea japonica Thunb. (Yoon et al. Citation2008). Compound 5 had been isolated from the bark of Platanus acerifolia (Aiton) Willd. (Plantanaceae) (Yang et al. Citation2013) whereas compound 2, identified as bibenzyl, had been synthesised by catalytic palladium on carbon (Pd/C) hydrogenation of trans-2,4,3′,5′-tetrahydroxystilbene (Likhitwitayawuid et al. Citation2006).
The anti-HIV-1 IN activities of compounds 1–7 were determined by the MIA method and compared with suramin as the positive control (IC50 value = 2.30 μM, ). Myricetin (4) exhibited the most potent activity with an IC50 value of 3.15 μM, followed by 2,4,6,7-tetrahydroxy-9,10-dihydrophenanthrene (3, IC50 value = 14.20 μM), quercetin-3-O-β-d-gluco pyranoside (6, IC50 value = 19.39 μM), and quercetin-3-O-β-d-galactopyranoside (7, IC50 value = 21.80 μM), whereas all other isolated compounds were inactive (IC50 value > 100 μM). It was interesting to note that the activity of myricetin was comparable with that of the positive control. The result indicated that myricetin (4), whose structure contains a galloyl moiety possessed a higher activity than the other compounds. This implied that the galloyl moiety was crucial for its most effective inhibitory effect. This is in agreement with the previous study, in which compounds containing a galloyl moiety often had a high potency as HIV-1 IN inhibitors (Jiang et al. Citation2010). In addition, the catechol and sugar moieties contained in compounds 3, 6, and 7 were also believed to be another reason for their activity ().
Figure 1. The structures of compounds isolated from D. bulbifera bulbils (1: Allantoin; 2: 2,4,3′,5′-Tetrahydroxybibenzyl; 3: 2,4,6,7-Tetrahydroxy-9,10-dihydrophenanthrene; 4: Myricetin; 5: 5,7,4′-Trihydroxy-2-styrylchromone; 6: Quercetin-3-O-β-d-glucopyranoside; 7: Quercetin-3-O-β-d-galactopyranoside).
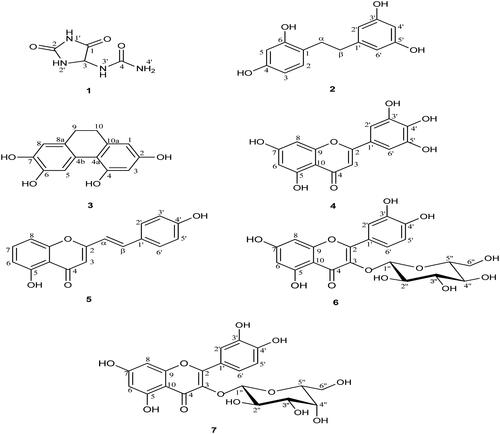
Table 2. Anti-HIV-1 integrase activityTable Footnotea of pure compounds from Dioscorea bulbifera L. bulbils.
In order to investigate the potential interactions of the active compounds with the IN enzyme, a molecular-docking technique was employed. The docking results showed that all four active compounds bound to the IN with similar patterns, i.e., close to the catalytic triad, flexible loop, and Mg2+. Compound 4, that had the galloyl moiety and possessed the most potent activity (IC50 value = 3.15 μM), was found to be located close to the catalytic triad. It exhibited the best binding affinity to IN in terms of having the lowest binding energy (−5.68 kcal/mol, ). In addition, compound 4 interacted with various amino acid residues. The interactions were formed by seven hydrogen bonds with Thr66, His67, Asp116, Glu152, Asn155, and Lys159; especially it interacted strongly with two residues of the catalytic triad (). This docking result was related to its inhibitory effect as measured by the MIA.
Figure 2. Molecular docking of active compounds with HIV-1 IN. The ribbon models show the backbone of HIV-1 IN catalytic domain with all hydrogen bonding residues are shown as stick models and coloured by heteroatom. Hydrogen bond interactions are shown as red dash lines and represent bond length in angstrom (Å). Mg2+ ions are shown as purple balls. Compound 3 (A), compound 4 (B), compound 6 (C), and compound 7 (D).
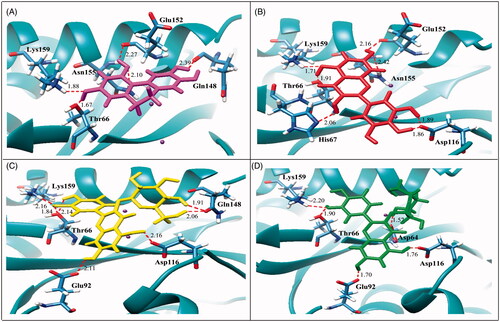
Table 3. The binding energy and H-bonding residues of active compounds with HIV-1 IN.
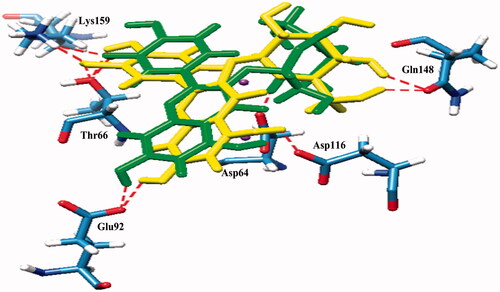
Compound 3 which contained a catechol moiety was also found to place close to the catalytic triad as well as to the flexible loop and possessed strong interactions (IC50 value = 14.20 μM). It formed five hydrogen bonds with Thr66, Gln148, Glu152, Asn155, and Lys159 with binding energy at −4.55 kcal/mol (). Compounds 6 and 7 were identified as flavonoid glycosides whose structures had catechol and sugar moieties. They exhibited activity against the HIV-1 IN with IC50 values of 19.39 and 21.80 μM, respectively. The docking study showed that compounds 6 and 7 had different binding energies (−5.24 and −4.03 kcal/mol, respectively) while the overall binding conformation and binding location were similar. Compound 6 formed seven hydrogen bonds with Thr66, Glu92, Asp116, Gln148, and Lys159 (), whereas compound 7 formed only five hydrogen bonds with Asp64, Thr66, Glu92, Asp116, and Lys159 (), thereby compound 7 (IC50 value= 21.80 μM) exhibited lower activity than that of compound 6 (IC50 value = 19.39 μM). According to , a slight difference was observed in the orientation of their sugar moiety. For compound 6, it was directly pointed to the flexible loop and interacted tightly with the two hydrogen bonds of Gln148 while the sugar moiety of compound 7 formed a strong interaction with Asp64, a catalytic residue 1.5 Å distant but without any interaction with the flexible loop.
Figure 3. Superimposed structures of the docked conformations of compound 6 (yellow) and compound 7 (green) with amino acid residues of IN (stick models coloured by heteroatom) and Mg2+ (purple balls).
Results from the molecular docking study supported the high inhibitory effect of the four active compounds because they could establish hydrogen bonds with Asp64, Thr66, His67, Glu92, and Asp116, the amino acid residues participating in the 3′-processing reaction. They also interacted with Gln148, Glu152, Asn155, and Lys159, the amino acid residues involved in the strand transfer reaction (Dayam & Neamati Citation2004) that resulted in an inhibition of HIV-1 IN activity. Therefore, the predicted interactions of the active compounds with the IN enzyme by the docking study clearly confirmed their inhibitory potency toward the IN catalytic reaction.
Conclusions
From the present study, seven compounds were isolated from the ethyl acetate and water fractions of D. bulbifera bulbils and this is the first report of compounds 1, 2, and 5 isolated from this plant. These results provide a strong basis for the use of D. bulbifera for AIDS treatment. Moreover, the active compounds inhibited the catalytic reaction of IN by blocking the amino acid residues involved in the 3′-processing and strand transfer reactions. The structural models of the interactions at the IN active site can provide a useful guide for further designs for structure-based drugs and development of more effective inhibitors of HIV-1 IN.
Funding information
The authors are grateful to the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission, and the Thailand Research Fund (TRF, RSA 5680012) for financial support.
Acknowledgements
The authors thank R. Craigie, National Institute of Health (NIH), Bethesda, MD, for providing an HIV-1 IN enzyme and the Faculty of Pharmaceutical Sciences, Prince of Songkla University for providing laboratory facilities. Thanks also to Dr Brian Hodgson for assistance with the English.
Disclosure statement
The authors report that they have no conflicts of interest.
References
- Ahmed Z, Chishti MZ, Johri RK, Bhagat A., Gupta KK, Ram G. 2009. Antihyperglycemic and antidyslipidemic activity of aqueous extract of Dioscorea bulbifera tubers. Diabetol Croat. 38:63–72.
- Asante-Appiah E, Skalka AM. 1997. Molecular mechanisms in retrovirus DNA integration. Antiviral Res. 36:139–156.
- Asres K, Seyoum A, Veeresham C, Bucar F, Gibbonds S. 2005. Naturally derived anti-HIV agents. Phytother Res. 19:557–581.
- Clercq ED. 2009. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int J Antimicrob Agents 33:307–320.
- Dayam R, Neamati N. 2004. Active site binding modes of the beta-diketoacids: a multi-active site approach in HIV-1 integrase inhibitor design. Bioorg Med Chem. 12:6371–6381.
- Fu YC, Ferng LHA, Huang PY. 2006. Quantitative analysis of allantoin and allantoic acid in yam tuber, mucilage, skin and bulbil of the Dioscorea species. Food Chem. 94:541–549.
- Gao H, Kuroyanagi M, Wu L, Kawahara N, Yasuno T, Nakamura Y. 2002. Antitumor-promoting constituents from Dioscorea bulbifera L. in JB6 mouse epidermal cells. Biol Pharm Bull. 25:1241–1243.
- Ghosh S, Ahire M, Patil S, Jabgunde A, Dusane MB, Joshi BN, Pardesi K, Jachak S, Dhavale DD, Chopade BA. 2012. Antidiabetic activity of Gnidia glauca and Dioscorea bulbifera: potent amylase and glucosidase inhibitors. J Evid Based Complement Altern Med. 2012:1–9.
- Goldgur Y, Craigie R, Cohen GH, Fujiwara T, Yoshinaga T, Fujishita T, Sugimoto H, Endo T, Murai H, Davies DR. 1999. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: a platform for antiviral drug design. Proc Natl Acad Sci USA. 96:13040–13043.
- Greenwald J, Le V, Butler SL, Bushman FD, Choe S. 1999. The mobility of an HIV-1 integrase active site loop is correlated with catalytic activity. Biochemistry 38:8892–8898.
- Guvenalp Z, Demirezer O. 2005. Flavonol glycosides from Asperula arvensis L. Turk J Chem. 29:163–169.
- Healy EF, Sanders J, King PJ, Robinson Jr WE. 2009. A docking study of l-chicoric acid with HIV-1 integrase. J Mol Graph Model 27:584–589.
- Jiang F, Chen W, Yi K, Wu Z., Si Y, Han W, Zhao Y. 2010. The evaluation of catechins that contain a galloyl moiety as potential HIV-1 integrase inhibitors. Clin Immunol. 137:347–356.
- Krishnan L, Engelman A. 2012. Retroviral integrase proteins and HIV-1 DNA integration. J Biol Chem. 287:40858–40866.
- Likhitwitayawuid K, Sornsute A, Sritularak B, Ploypradith P. 2006. Chemical transformations of oxyresveratrol (trans-2,4,3',5'-tetrahydroxystilbene) into a potent tyrosinase inhibitor and a strong cytotoxic agent. Bioorg Med Chem Lett. 16:5650–5653.
- Liu H, Chou GX, Guo YL, Ji LL, Wang JM, Wang ZT. 2010. Norclerodane diterpenoids from rhizomes of Dioscorea bulbifera. Phytochemistry 71:1174–1180.
- Liu H, Tsim KWK, Chou GX, Wang JM, Ji LL, Wang ZT. 2011. Phenolic compounds from the rhizomes of Dioscorea bulbifera. Chem Biodivers. 8:2110–2116.
- Luca LD, Pedretti A, Vistoli G, Barreca ML, Villa L, Monforte P, Chimirri A. 2003. Analysis of the full-length integrase-DNA complex by a modified approach for DNA docking. Biochem Biophys Res Commun. 310:1083–1088.
- Mbiantcha M, Kamanyi A, Teponno RB, Barreca ML, Villa L, Monforte P, Chimirri A. 2011. Analgesic and anti-inflammatory properties of extracts from the bulbils of Dioscorea bulbifera L. var sativa (Dioscoreaceae) in mice and rats. Evid Based Complement Alternat Med. 2011:1–9..
- Rashed K, Zhang XJ, Luo MT, Zheng YT. 2012. Anti-HIV-1 activity of phenolic compounds isolated from Diospyros lotus fruits. Phytopharmacology 3:199–207.
- Saag MS. 2012. New and investigational antiretroviral drugs for HIV infection: mechanisms of action and early research findings. Top Antivir Med. 20:162–167.
- Santos CMM, Silva AMS, Cavaleiro JAS. 2003. Synthesis of new hydroxy-2-styrylchromones. Eur J Org Chem. 2003:4575–4585.
- Sripathi SK, Gopal P, Lalitha P. 2011. Allantoin from the leaves of Pisonia grandis R.Br. Int J Pharm Life Sci. 2:815–817.
- Tapondjou LA, Jenett-Siems K, Bottger SMelzig MF. 2013. Steroidal saponins from the flowers of Dioscorea bulbifera var. sativa. Phytochemistry 95:341–350.
- Teponno RB, Tapondjou AL, Gatsing D, Djoukeng JD, Abou-Mansour E, Tabacchi R, Tane P, Stoekli-Evans H, Lontsi D. 2006. Bafoudiosbulbins A, and B, two anti-salmonellal clerodane diterpenoids from Dioscorea bulbifera L. var sativa. Phytochemistry 67:1957–1963.
- Tewtrakul S, Nakamura N, Hattori M, Fujiwara T, Supavita T. 2002. Flavanone and flavonol glycosides from the leaves of Thevetia peruviana and their HIV-1 reverse transcriptase and HIV-1 integrase inhibitory activities. Chem Pharm Bull. 50:630–635.
- Vajragupta O, Boonchoong P, Morris GM, Olson AJ. 2005. Active site binding modes of curcumin in HIV-1 protease and integrase. Bioorg Med Chem Lett. 15:3364–3368.
- Wang JM, Ji LL, Branford-White CJ, Wang ZY, Shen KK, Liu H, Wang ZT. 2012. Antitumor activity of Dioscorea bulbifera L. rhizome in vivo. Fitoterapia 83:388–394.
- Yang CH, Yang Y, Liu JH. 2013. Platachromone A–D: cytotoxic 2-styrylchromones from the bark of Platanus x acerifolia (Aiton) Willd. Phytochem Lett. 6:387–391.
- Yoon KD, Yang MH, Chin YW, Park JH, Kim J. 2008. Determination of allantoin in Dioscorea rhizoma by high performance liquid chromatography using cyano columns. Nat Prod Sci. 14:254–259.